

Abstract
Proliferating cell nuclear antigen (PCNA) assumes an indispensable role in supporting cellular DNA replication and repair by organizing numerous protein components of these pathways via a common PCNA-interacting sequence motif called a PIP-box. Given the multifunctional nature of PCNA, the selective inhibition of PIP-box–mediated interactions may represent a new strategy for the chemosensitization of cancer cells to existing DNA-directed therapies; however, promiscuous blockage of these interactions may also be universally deleterious. To address these possibilities, we utilized a chemical strategy to irreversibly block PIP-box–mediated interactions. Initially, we identified and validated PCNA methionine 40 (M40) and histidine 44 (H44) as essential residues for PCNA/PIP-box interactions in general and, more specifically, for efficient PCNA loading onto chromatin within cells. Next, we created a novel small molecule incorporating an electrophilic di-chloro platinum moiety that preferentially alkylated M40 and H44 residues. The compound, designated T2Pt, covalently cross-linked wild-type but not M40A/H44A PCNA, irreversibly inhibited PCNA/PIP-box interactions, and mildly alkylated plasmid DNA in vitro. In cells, T2Pt persistently induced cell cycle arrest, activated ATR-Chk1 signaling and modestly induced DNA strand breaks, features typical of cellular replication stress. Despite sustained activation of the replication stress response by the compound and its modestly genotoxic nature, T2Pt demonstrated little activity in clonogenic survival assays as a single agent, yet sensitized cells to cisplatin. The discovery of T2Pt represents an original effort directed at the development of irreversible PCNA inhibitors and sets the stage for the discovery of analogues more selective for PCNA over other cellular nucleophiles.
Keywords: PCNA, protein-protein interaction, chemotherapy, small molecule, irreversible inhibitor, platinum complex
Graphical Abstract
1. Introduction
Proliferating cell nuclear antigen (PCNA) is a homotrimeric protein that organizes numerous components of the DNA replication and repair machinery by functioning as a clamp platform 1–3 that encircles and slides along DNA 2–4. In higher eukaryotes, many DNA replication and repair proteins interact with PCNA using a common PCNA-binding motif called a PCNA-interacting protein box (PIP-box) 2. PCNA accommodates these interactions via a cavity located underneath an interdomain connecting loop on the outer face of the PCNA clamp 2, 4. Given the ubiquitous role PCNA adopts in supporting DNA replication and repair pathways 2 and neutrophil survival 5, it was rationalized that an inhibitor targeting the PIP-box binding cavity of PCNA may impair these cellular processes and render cancer cells more susceptible to existing DNA damage-based therapies 6, 7. Conversely, it is also a reasonable concern that the functional inactivation of the PIP-box binding cavity may be exceedingly deleterious, prohibiting the therapeutic utility of the approach.
Few methods are currently available for investigating the functional consequences of rapidly inactivating essential protein-protein interactions such as PIP-box–mediated binding events. RNA interference has emerged as a leading technology in proof-of-concept studies designed to validate the suitability of a protein target for therapeutic inhibition; however, the technology is not applicable for analyzing specific protein-protein interactions since it entirely erases the protein target 8. Although the technique is not especially rapid, site-directed mutagenesis provides an ideal starting point for defining the functional relevance of specific amino acid residues within a target protein. Numerous studies have successfully identified the sequence requirements of the PIP-box within PCNA binding partners, and these studies have yielded a characteristic consensus sequence 9. In contrast, very little is known of the structural elements located within the PIP-box binding cavity of PCNA itself that are required for binding. We sought to identify these elements and initiated our studies with methionine 40 (M40) and histidine 44 (H44), two nucleophilic residues lining the PIP-box binding cavity of PCNA.
2. Material and Methods
2.1. Materials
All chemicals were purchased from Sigma Chemical Co. (St. Louis, MO) unless stated otherwise. All chemical compounds assayed were prepared as 10 mM DMSO solutions, except cisplatin was prepared as a 5 mM solution in 0.9% aqueous sodium chloride. T2AA was prepared as previously described 6. Micro Bio-Spin 6 chromatography columns and 10× TBS solution were obtained from Bio-Rad (Hercules, CA), an APC BrdU Flow Kit was purchased from BD Pharmingen (San Jose, CA), and 20× MOPS/SDS-PAGE Running Buffer was obtained from Teknova (Hollister, CA). Halt Phosphatase Inhibitor Cocktail was from Thermo Scientific (Waltham, MA). EconoTips were from New Objective (Woburn, MA), and propidium iodide was purchased from Roche (Mannheim, Germany). Biotin-alkyne, 4× LDS Sample Buffer, 10× Sample Reducing Agent, Novex 4–12% Bis-Tris Gels, Alexa Fluor 488 goat α-rabbit antibody, iBlot Gel Transfer Nitrocellulose Stacks, Opti-MEM medium, Lipofectamine 2000, and a GeneArt Site-Directed Mutagenesis System were from Life Technologies (Carlsbad, CA). Rabbit α-Chk1 (pSer345) antibody, mouse α-PCNA PC10 antibody, α-mouse IgG HRP-linked secondary antibody, rabbit α-β-actin (13E5) HRP-linked antibody, and a streptavidin-HRP conjugate were obtained from Cell Signaling Technology (Beverly, MA). Goat α-rabbit IRDye 800CW secondary antibody was from LI-COR Biosciences (Lincoln, NE). A FlowCellect H2A.X DNA Damage Kit, c4 and c8 Zip Tips, and Amicon Ultra-0.5 Centrifugal Filter Devices were purchased from Millipore (Billerica, MA). A Micro BCA Protein Assay Kit, Halt Protease Inhibitor Cocktail, Supersignal Pico West chemiluminescent substrate, SuperBlock Blocking Buffer, and Restore Stripping Buffer were obtained from Thermo Scientific (Waltham, MD). Vectashield mounting medium was from Vector Laboratories (Burlingame, CA). A 5-carboxyfluorescein–labeled PL peptide (SAVLQKKITDYFHPKK)9 was synthesized by the Hartwell Center for Bioinformatics & Biotechnology, St Jude Children’s Research Hospital. Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). The pEGFP-PCNA plasmid was originally obtained from David Gerlich 10 via Addgene (Cambridge, MA), and a T7-PCNA plasmid encoding full-length human PCNA was provided by Toshiki Tsurimoto (Kyusyu University, Japan) 11.
2.2. Synthesis of T2Pt (8)
All commercial reagents were used without further purification, and the solvents were dried using a dry solvent system (Glass Contour Solvent Systems, SG Water USA). Reactions requiring the exclusion of air were carried out under an atmosphere of dry nitrogen in oven dried glassware. All reactions were monitored by thin-layer chromatography (TLC) carried out on EMD Chemicals silica gel 60-F 254 coated glass plates and visualized using UV light (254 nm). Analysis by LC-MS was performed by using an XBridge C18 column run at 1 mL/min and using gradient mixtures of (A) water (0.05% TFA) and (B) methanol. Low-resolution mass spectra (ESI) were collected on a Waters Micromass ZQ in positive-ion mode. Flash-chromatography was performed on a Biotage SP4 chromatography system using Biotage Flash + KP SIL pre-packed columns, and the solvent mixture in brackets was used as eluent. Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker Avance II NMR spectrometer at 400 MHz for 1H NMR spectra. Chemical shifts (ppm) are reported relative to TMS or the solvent peak. Signals are designated as follows: s, singlet; d, doublet; dd, doublet of doublet; t, triplet; q, quadruplet; m, multiplet. Coupling constants (J) are shown in Hertz.
2.2.1. (S)-tert-butyl (1-hydroxy-3-(4-(4-hydroxyphenoxy)-3,5-diiodophenyl)propan-2-yl)carbamate (2)
A solution of lithium borohydride (2 M in THF, 3 mL, 6 mmol) was added to a two-neck flask flushed with nitrogen and containing 1,4-dioxane (5 mL). Chlorotrimethylsilane (1.53 mL, 12.0 mmol) was added dropwise to the reaction mixture, and the solution was stirred under nitrogen for 20 min. The white opaque solution was cooled to 0°C, and (S)-3,5-diiodo-2-amino-3-[4-(4-hydroxyphenoxy)phenyl]propanoic acid (1) (1.05 g, 2.00 mmol) was added in one portion. The reaction mixture was stirred overnight allowing the solution to reach room temperature. Additional lithium borohydride (2 M in THF, 3 mL, 6 mmol) and chlorotrimethylsilane (1.53 mL, 12.0 mmol) were added to the reaction mixture and stirred for an additional 24 h. The reaction mixture was slowly poured into a flask containing 2 g of ice, using THF to rinse the reaction flask. The solution was stirred at 0°C and the pH adjusted to 9–10 using 5 M sodium hydroxide. A solution of bis(tert-butyl)carbonate (564 μL, 2.43 mmol) in THF (6 mL) was added, and the reaction mixture was stirred for 3 h allowing the solution to reach room temperature. The reaction mixture was acidified with 2 M hydrochloric acid and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and concentrated. The crude product was purified by flash column chromatography (Biotage SP4, 40+M column, eluting with hexanes/ethyl acetate, 20–100% gradient) to give an off-white solid (834 mg, 68% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.05 (s, 1H), 7.71 (s, 2H), 6.70–6.58 (m, 3H), 6.47 (d, J = 8.9 Hz, 2H), 4.73 (t, J = 5.5 Hz, 1H), 3.61–3.47 (m, 1H), 3.37–3.31 (m, 1H), 3.29–3.21 (m, 1H), 2.80 (dd, J = 13.5, 3.9 Hz, 1H), 2.41 (dd, J = 13.3, 10.2 Hz, 1H), 1.29 (s, 9H).
2.2.2. (S)-tert-butyl (1-(3,5-diiodo-4-(4-((4-methoxybenzyl)oxy)phenoxy)phenyl)-3-hydroxypropan-2-yl)carbamate (3)
1-(Bromomethyl)-4-methoxybenzene (144 μL, 0.988 mmol) and potassium carbonate (372 mg, 2.69 mmol) were added to a solution of compound 2 (549 mg, 0.898 mmol) in DMF (3 mL) and stirred overnight at room temperature. Additional 1-(bromomethyl)-4-methoxybenzene (55 μL, 0.45 mmol) and potassium carbonate (185 mg, 1.35 mmol) were added to the reaction mixture, which was stirred at room temperature for an additional 3.5 h. Saturated aqueous ammonium chloride (10 mL) was added to the reaction mixture, and extracted with ethyl acetate (3 × 10 mL). The ethyl acetate layers were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated. The crude product was purified by flash column chromatography (Biotage SP4, 40+M column, eluting with hexanes/ethyl acetate, 0–50% gradient) to give a white solid (291.5 mg, 44.4% yield). 1H NMR (400 MHz, chloroform-d) δ 7.72 (s, 2H), 7.34 (d, J = 8.5 Hz, 2H), 7.26 (s, 1H), 6.90 (t, J = 8.5 Hz, 4H), 6.70 (d, J = 9.1 Hz, 2H), 4.93 (s, 2H), 4.81 (d, J = 6.5 Hz, 1H), 3.81 (s, 3H), 3.71 (d, J = 11.0 Hz, 1H), 3.66–3.57 (m, 1H), 2.80 (d, J = 7.1 Hz, 2H), 2.12 (s, 1H), 1.58 (s, 1H), 1.44 (s, 9H).
2.2.3. (S)-tert-butyl (1-azido-3-(3,5-diiodo-4-(4-((4-methoxybenzyl)oxy)phenoxy)phenyl)propan-2-yl)carbamate (4)
DBU (270 μL, 1.79 mmol) and DPPA (386 μL, 1.79 mmol) were added to a solution of compound 3 (873 mg, 1.19 mmol) in DMF (12 mL). The solution was stirred under nitrogen and heated to 100°C for 18 h. The reaction mixture was cooled, added to water (50 mL), and extracted with ethyl acetate (3 × 50 mL). The combined organic layers were washed with saturated brine, dried over sodium sulfate, filtered, and concentrated. The crude product was purified by flash column chromatography (Biotage SP4, 40+M column, eluting with hexanes/ethyl acetate, 0–80% gradient) to give a white solid (409 mg, 45% yield). 1H NMR (400 MHz, chloroform-d) δ 7.70 (s, 2H), 7.34 (d, J = 8.5 Hz, 2H), 7.26 (s, 1H), 6.93–6.86 (m, 4H), 6.70 (d, J = 9.0 Hz, 2H), 4.93 (s, 2H), 4.69 (d, J = 8.3 Hz, 1H), 3.92 (s, 1H), 3.82 (s, 3H), 3.45 (qd, J = 12.4, 4.2 Hz, 2H), 2.85–2.64 (m, 2H), 1.44 (s, 9H).
2.2.4. (S)-tert-butyl (1-amino-3-(3,5-diiodo-4-(4-((4-methoxybenzyl)oxy)phenoxy)phenyl)propan-2-yl)carbamate (5)
Triphenylphosphine (40.4 mg, 0.154 mmol) was added to a solution of compound 4 (107 mg, 0.141 mmol) in THF (1.6 mL) and water (161 μL). After stirring at room temperature for 18 h, the reaction mixture was concentrated and purified by flash column chromatography (Biotage SP4, 25+S column, eluting with DCM/methanol/28% ammonia aq., 100:0:0 to 85:14.8:0.2 gradient) to give a pale brown solid (89.7 mg, 87.0% yield). 1H NMR (400 MHz, chloroform-d) δ 7.70 (s, 1H), 7.35 (d, J = 8.6 Hz, 1H), 6.90 (t, J = 8.9 Hz, 2H), 6.70 (d, J = 9.0 Hz, 1H), 4.93 (s, 1H), 4.06–3.55 (m, 2H), 2.72 (br s, 1H), 1.59–1.10 (m, 7H).
2.2.5. (S)-4-(4-(2,3-diaminopropyl)-2,6-diiodophenoxy)phenol (6)
Triethylsilane (65 μL, 0.41 mmol) was added to a solution of compound 6 (75.5 mg, 0.103 mmol) in DCM (585 μL) at 0°C. TFA (650 μL) was added to the reaction mixture and stirred for 1 h at 0°C and 1.5 h at room temperature. The solution was concentrated and then treated with 2 M sodium hydroxide (3 mL) and ethyl acetate (3 × 3 mL). The ethyl acetate layers were combined, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and concentrated. The crude product was dissolved in aqueous 1 M hydrochloric acid (3 mL) and washed with ethyl acetate (3 mL). The aqueous layer was basified with aqueous 2 M sodium hydroxide and then extracted with ethyl acetate (3 mL). The ethyl acetate layer was concentrated to give a pale yellow solid (38.3 mg, 72.6% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.81 (s, 2H), 6.69 (d, J = 8.9 Hz, 2H), 6.55 (d, J = 8.9 Hz, 2H), 3.00 (br s, 1H), 2.84–2.66 (m, 2H), 2.61–2.50 (m, 2H), 1.91 (s, 1H).
2.2.6. Dichloro((S)-4-(4-(2,3-diaminopropyl)-2,6-diiodophenoxy)phenol)platinum(II) (7)
Potassium tetrachloroplatinate(II) (17 mg, 41 μmol) was added to a solution of compound 6 (20 mg, 39 μmol) and 1 M hydrochloric acid (81 μL, 81 μmol) in water (382 μL). The reaction mixture was stirred for 20 min and then basified with 6 M sodium hydroxide (32 μL, 193 μmol). The homogeneous solution was stirred overnight at room temperature. Saturated aqueous ammonium chloride (400 μL) and ethyl acetate (400 μL) were added to the reaction mixture. A precipitate formed and collected at the interface of the two liquid phases after a short time in a centrifuge. The aqueous layer was removed, and the precipitate was washed with saturated aqueous ammonium chloride (400 μL), water (2 × 400 μL), and saturated brine (400 μL). The suspended precipitate in the organic layer was concentrated to give a pale brown solid (17 mg, 57% yield).
2.2.7 Chloro(dimethylsulfoxide)((S)-4-(4-(2,3-diaminopropyl)-2,6-diiodophenoxy)phenol)platinum(II) chloride (T2Pt, 8)
Compound 7 was dissolved with DMSO to generate a 10 mM stock solution of 8. Purity and DMSO complexation were verified by LC-MS (Figure S1). 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 7.83–7.73 (m, 2H), 6.67 (d, J = 8.1 Hz, 2H), 6.53 (d, J = 8.1 Hz, 2H), 5.58–5.38 (m, 2H), 5.31–5.09 (m, 2H), 2.94–2.69 (m, 3H), 2.31 (d, J = 9.3 Hz, 1H), 2.18 (d, J = 6.4 Hz, 1H).
2.3. Synthesis of azide-tagged T2Pt (N3-T2Pt)
See Supporting Information (Scheme S1 and Figures S2–S3).
2.4. Site-directed mutagenesis
PCNA mutant plasmids were generated from T7-PCNA or pEGFP-PCNA expression vectors using a GeneArt Site-Directed Mutagenesis System according to the manufacturer’s instructions. A M40A mutant template was generated using the oligonucleotides 5′ GTGTAAACCTGCAGAGCGCAGACTCGTCCCACGTCTC and 5′ GAGACGTGGGACGAGTCTGCGCTCTGCAGGTTTACAC, while a H44A mutant template was produced using the oligonucleotides 5′ GAGCATGGACTCGTCCGCAGTCTCTTTGGTGCAG and 5′ CTGCACCAAAGAGACTGCGGACGAGTCCATGCTC. A M40A/H44A double PCNA mutant was also generated from the M40A template using oligonucleotides 5′ GTGTAAACCTGCAGAGCGCAGACTCGTCCGCAGTCTC and 5′ GAGACTGCGGACGAGTCTGCGCTCTGCAGGTTTACAC. The sequences of mutated plasmids were subsequently validated by Sanger DNA sequencing performed by the Hartwell Center. Recombinant PCNA proteins were prepared as described previously 6.
2.5. Fluorescence polarization
A fluorescence polarization (FP) assay was performed as previously described 6. Briefly, a 5-carboxyfluorescein–labeled PL peptide (10 nM) was mixed with 0–333 nM solutions of either wild-type or mutant PCNAs in FP buffer (35 mM HEPES, pH 7.4, 10% glycerol, and 0.01% Triton X-100) at room temperature. FP was measured immediately on an EnVision plate reader (PerkinElmer Life Sciences, Waltham, MA) using 485 nm excitation (20 nm bandpass) and 535 nm emission (20 nm bandpass) filters and a 510 nm dichroic mirror. For competitive displacement assays, 5-carboxyfluorescein–labeled PL peptide (10 nM) was mixed with 100 nM recombinant human PCNA in FP buffer, and the complex was exposed to 0–100 μM solutions of either T2AA or T2Pt at room temperature. At defined time points, the FP of each sample was measured as described above.
2.6. Crosslinking of PCNA by T2Pt
Recombinant wild-type and mutant PCNAs (1 μg) were pretreated with N3-T2Pt (Figure S2) as indicated in 35 mM HEPES, pH 7.4 (40 μL), for 2 h at room temperature. Biotin-alkyne (25 μM), TCEP (1 mM), and copper (II) sulfate (1 mM) were subsequently added to each sample and incubated for 1 h at room temperature. All samples were denatured in 1× LDS Sample Buffer and 1× Sample Reducing Agent at 85°C for 10 min prior to loading onto a Novex 4–12% Bis-Tris Gel. Samples were subjected to SDS-PAGE at 200 V for 50 min in MOPS buffer and electrotransferred to a nitrocellulose membrane. The membrane was blocked using SuperBlock Blocking Buffer overnight at 4°C and then washed with TBS-Tween 20 (0.05%) for 10–15 min three times. The membrane was subsequently exposed to 1:2000 streptavidin-HRP conjugate for 1 h at room temperature, and the washes were repeated as above. The membrane was treated with SuperSignal West Pico substrate, and a chemiluminescent image of the membrane was taken using an ImageQuant LAS4000 imaging system (GE Healthcare Bio-sciences, Uppsala, Sweden). Membranes were subsequently stripped with Restore Stripping Buffer for 15 min at room temperature, washed as above, re-blocked with SuperBlock Blocking Buffer for 40–60 min at room temperature, washed again, and probed overnight at 4°C with 1:2000 α-PCNA mouse antibody. A chemiluminescent image of the membrane was taken as described above after treatment with 1:2000 α-mouse HRP-linked secondary antibody.
2.7. Cell culture
All cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained at sub-confluent levels in Dulbecco’s modified Eagle’s medium (Mediatech, Inc., Manassas, VA) supplemented with 10% fetal bovine serum (Thermo Scientific, Waltham, MD) at 37°C in a humidified 5% CO2 atmosphere. Unless otherwise stated, cells were typically seeded into 6-well cluster plates at 100,000 cells per well and allowed to attach overnight prior to treatment the following day. Each specific treatment schedule is defined in the relevant Figure legend.
2.8. Plasmid transfection, chromatin staining, confocal microscopy, and immunoblotting
On the day after seeding onto coverslips, U2OS cells were transfected with 4 μg of either wild-type pEGFP-PCNA or M40A pEGFP-PCNA plasmid using Opti-MEM media and Lipofectamine 2000 according to the manufacturer’s instructions. Approximately 20 h after transfection, cells were processed in preparation for analysis by either confocal microscopy or immunoblotting. In preparation for confocal microscopy, cells were pre-extracted with ice-cold CSK buffer (100 mM NaCl, 3 mM MgCl2, 10 mM HEPES, pH 7.4, 300 mM sucrose, 0.5% Triton X-100, and 1× Halt Protease Inhibitor Cocktail) for 2 min on ice and then fixed with 4% paraformaldehyde for 12 min at room temperature. Samples were blocked in 3% FBS in PBS, pH 7.4, for 20 min at room temperature, washed three times with PBS, pH 7.4, and then mounted onto glass slides in VectaShield containing 1 μg/mL DAPI. The stained cells were examined on a TE2000 microscope equipped with a C2 confocal and a 20×/0.8 NA Plan Apo objective (Nikon, Tokyo, Japan). Images were taken and processed using Nikon NIS-Elements and Photoshop 7 software (Adobe Systems, San Jose, CA). For immunoblotting, transfected cells were harvested, washed with 1 mM PMSF in PBS, pH 7.4, and lysed in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, and 1× Halt Protease Inhibitor Cocktail for 10 min on ice. Lysates were sonicated to release chromatin-bound protein and then assayed for protein concentration using a Micro BCA Protein Assay Kit according to the manufacturer’s instructions. Approximately 13 μg of each lysate was analyzed for PCNA expression by SDS-PAGE followed by immunoblotting as described above using 1:1000 α-PCNA mouse antibody.
2.9. Protein mass spectrometric analysis
Recombinant human PCNA (13.3 μM) was reacted with 0–50 μM T2Pt in 35 mM HEPES, pH 7.4, for 2 h at ambient temperature. Following the reaction, samples were filtered into 10 mM ammonium acetate by gel filtration (Micro Bio-Spin 6 chromatography column) to remove free T2Pt. Samples were subsequently desalted using a reverse phase c4 or c8 Zip Tip and eluted into 50% acetonitrile, 2% formic acid. The eluent was ionized by static nanospray using EconoTips on a Micromass LCT mass spectrometer [Waters (Milford, MA)] operating in positive mode. The resultant charge envelope was deconvoluted using MaxEnt 1 algorithm of MassLynx V 4.0 sp 4 software. A mass error of 1 Da for every 10,000 Da was permissible using this mass spectrometer.
2.10. Electrophoretic band shift assay
Supercoiled pEGFP-PCNA plasmid (500 ng) was reacted with cisplatin or T2Pt (0–50 μM) in 35 mM HEPES, pH 7.4, at 37°C overnight. Samples were subjected to electrophoresis through 0.8% agarose gels containing 0.5 μg/mL ethidium bromide for 5 h at 50 V in 1× TAE buffer and then visualized and photographed under UV illumination.
2.11. Alkaline comet assay
U2OS cells were initially seeded and allowed to attach overnight prior to treatment with either cisplatin or T2Pt (0–50 μM) for 5–7 h. Compound-containing media was then removed, the samples washed once with PBS and then released into compound-free media for a further 17 h. Cells were subsequently harvested and then subjected to the alkaline comet assay as described previously 12. Briefly, cells were suspended in molten Type VII low gelling temperature agarose following harvest and allowed to set on glass slides. Cells were lysed in gels with ice-cold lysis buffer [100 mM EDTA, 2.5 M NaCl, 10 mM Tris-HCl (pH 10.5), 1% Triton X-100] for 1 h and then subsequently washed with ice-cold Milli-Q water for 15 min × 4. Samples were submerged in chilled 1× alkali buffer (300 mM sodium hydroxide, 1 mM EDTA) for 1 h at 4°C and then subjected to electrophoresis at 30 V for 30 min at 4°C. Neutralization buffer [500 mM Tris–HCl (pH 7.5)] was added to each slide and then removed with 2 × 10 min PBS (pH 7.4) washes. Each sample was stained twice with 2.5 μg/mL propidium iodide for 5 min each, rinsed with Milli-Q water and visualized using epi-fluorescence microscopy. DNA damage was visually measured using the scoring system described previously.12 Approximately 100 cells were scored per sample.
2.12. Cell cycle analysis by flow cytometry
HeLa cells were seeded into 60 mm dishes (100,000 per dish) and allowed to attach overnight. The following day, samples were exposed to 100 μM T2Pt, 40 μM T2AA, or DMSO continuously for 24 h. The cells were washed twice with PBS and then released into compound-free medium. At defined time points after release, samples were processed using an APC BrdU Flow Kit according to the manufacturer’s instructions and then analyzed using an LSR Fortessa flow cytometer (BD Biosciences, San Jose, CA) equipped with a 488-nm laser line which was used for sample excitation. Fluorescence emission was collected using the FL5 channel. Ten thousand events were collected for each sample and gated using forward- versus side-scatter to eliminate cell debris and doublets. The data were analyzed using FlowJo version 7.6.5 software (Tree Star, San Carlos, CA).
2.13. Immunocytochemical detection of pSer345 Chk1 and γH2AX by flow cytometry
Following detachment, cells were fixed with 4% paraformaldehyde in PBS for 10–15 min at room temperature, and the samples were washed with 1× TBS. Cells were then permeabilized with TFX (1× TBS, 4% fetal bovine serum, and 0.1% Triton X-100) for 10 min on ice and then exposed to 1:200 α-Chk1 (pSer345) rabbit antibody in TFX for 2 h at room temperature. After the primary antibody was removed using two washes with TFX, samples were probed with 1:500 goat α-rabbit Alexa 488 secondary antibody in TFX for 30 min at room temperature away from light. Unbound secondary antibody was removed with a TFX wash, and the samples were incubated with 2.5 μg/mL propidium iodide and RNase in TFX for 30 min at room temperature. For γH2AX staining, samples were harvested and processed using the FlowCellect H2A.X DNA Damage Kit according to the manufacturer’s instructions. All samples were analyzed by flow cytometry using an LSR Fortessa flow cytometer as described for cell cycle analysis with one exception: green and red fluorescence was collected using FL1 and FL3 channels, respectively.
2.14 Immunoblotting for pSer345 Chk1
Immunoblotting for pSer345 Chk1 was performed as described for the analysis of EGFP-PCNA expression with some modifications. Lysis buffer was supplemented with 1× Halt Phosphatase Inhibitor Cocktail and the nitrocellulose membrane was initially exposed to 1:500 α-Chk1 (pSer345) rabbit antibody. Primary antibody was subsequently detected with a 1:10,000 goat α-rabbit IRDye 800CW secondary antibody using a LI-COR Odyssey infrared imaging system. The membrane was subsequently stripped, re-probed with 1:1000 α-β-actin (13E5) HRP-linked antibody and a chemiluminescent image of the membrane taken as described above.
2.15. Clonogenic survival assay
U2OS cells (300 cells/well) were initially allowed to settle in 6-well cluster plates overnight prior to treatment with 0 – 300 nM cisplatin as indicated. After 24 h, T2Pt was added. Another 24 h after T2Pt addition, the culture medium was replaced with fresh compound-free medium, and cells allowed to expand into colonies for a further 6 days. Each treatment was performed in triplicate. The cells were gently rinsed twice with PBS, fixed with 3.7% formaldehyde, and stained with 0.5% crystal violet. Colonies containing more than approximately 40 cells were scored. The clonogenic survival rate for each condition was expressed relative to the average number of colonies that received the vehicle control (0.9% sodium chloride and DMSO) which was normalized to a value of 1.
3. Results
3.1. PCNA M40 and H44 residues are essential for PIP-box–mediated interactions in vitro
Based on the structure of the PIP-box binding cavity of human PCNA 1, it was anticipated that methionine 40 (M40) and histidine 44 (H44), both suspended on a small loop overhanging the cavity, may be key residues for PIP-box–mediated binding events. In an initial effort to address this idea, PCNA alanine mutants were generated at each of these residues and their relative affinities for a representative PIP-box–containing PL peptide9 was evaluated by an FP assay. Wild-type PCNA was bound strongly by the PIP-box–containing PL peptide, consistent with a reasonably low dissociation constant (Kd ~100 nM) reported for the interaction 9. In contrast and quite strikingly, mutant M40A PCNA completely failed to interact with the PL peptide while binding of the peptide by a H44A mutant was also significantly attenuated relative to wild-type PCNA (Figure 1). An M40A/H44A PCNA double mutant also completely failed to bind the PL peptide (Figure 1), further highlighting the importance of these residues in facilitating PIP-box-mediated interactions. It should be noted that a more rigorous analysis of absolute binding affinities was not feasible given the excessively high concentrations of each mutant required to attain saturating binding conditions. Correspondingly, it is emphasized that the data presented in Figure 1 reflect only the relative affinities of each mutant for the PIP-box-containing PL peptide.
Figure 1.

PCNA M40 and H44 residues are essential for PIP-box-mediated interactions. The fluorescence polarization (mP) of samples containing fluorescently labeled PL peptide (10 nM) is presented as a function of PCNA concentration as indicated. Error bars represent the standard deviation of triplicate experiments. WT: wild-type.
3.2. PCNA M40 is essential for efficient chromatin loading of PCNA
Given the importance of M40 in mediating the PCNA-PL peptide interaction in vitro, it was subsequently rationalized that the residue may assume a functionally significant role within cells. To verify a functional role for PCNA M40, GFP-fusions of either wild-type or M40A PCNA were exogenously expressed in U2OS cells. Following removal of non-chromatin-bound protein by pre-extraction, cell samples were analyzed for chromatin-bound GFP-PCNA by confocal microscopy (Figure 2A). Cells expressing GFP-fused wild-type PCNA (Figure 2A) showed a pattern of chromatin-bound PCNA foci consistent with those described in previous reports13, suggesting that the wild-type fusion protein was successfully loaded onto DNA. It is noteworthy that only a minor fraction of cells were positive for wild-type PCNA tagged with GFP (Figure 2A), a feature that may be attributed to two complementary factors. First, not all cells take up the vector upon transfection. Second, and more importantly, the fraction of cells in S phase in an asynchronous population at any given point is typically low. GFP-PCNA loaded onto chromatin in S phase is resistant to the pre-extraction method applied to the cells, while unloaded GFP-PCNA is typically removed through washing, leaving the small population of GFP-positive cells evident in Figure 2A. Unlike wild-type GFP-PCNA, chromatin-bound M40A PCNA was barely detectable (Figure 2A), indicating that the mutant was not efficiently loaded onto chromatin, despite expression levels that were similar to those of GFP-tagged wild-type PCNA (Figure 2B). This observation suggests that the M40A substitution abolishes fundamental functions of the protein and led us to hypothesize that the functional deactivation of PCNA may be achieved by selective alkylation of M40.
Figure 2.

The M40A mutation of PCNA impairs chromatin loading within cells. (A) U2OS cells were initially transfected with an EGFP–wild-type PCNA or EGFP-M40A PCNA construct, pre-extracted to remove all non-chromatin-bound proteins, and imaged for GFP. DAPI was used as a nuclear counterstain. (B) The expression level of each GFP fusion presented in (A) as assayed by immunoblotting. Endogenous PCNA served as a loading control.
3.3. The rational generation of T2Pt, a chemical agent that preferentially alkylates PCNA via M40 and H44
The thyroid hormone T3 and its structural analogue T2AA are non-peptide small molecules that bind reversibly to the PIP-box binding cavity of PCNA6, 7. T2AA efficiently and reversibly suppressed DNA synthesis in cells, most likely via inhibition of the PCNA/DNA polymerase δ PIP-box interaction. Moreover, T2AA and its analogues inhibited translesion DNA synthesis, an important DNA damage tolerance mechanism in cells 6, 14. In a PCNA/T3 co-crystal structure 6, it was noted that the amino acid moiety of T3 was juxtaposed with the functionally important M40 and H44 residues of PCNA. Accordingly, we surmised that a T2AA analogue bearing an appropriately positioned electrophilic moiety may promote the generation of persistent, covalent bonds with the nucleophilic M40 and H44 residues of PCNA. Few organic electrophiles conventionally utilized for bioconjugation (such as alkyl halides and Michael acceptors) alkylate methionine and histidine residues within a physiological environment. In an effort to circumvent these restrictions, we selected an inorganic electrophilic moiety capable of strong coordination by methionine and histidine, namely platinum 15. We have generated a T2AA analogue, T2Pt, by substituting the aminoalcohol moiety of T2AA for a cisplatin-like structure (Scheme 1 and Figure 3). An azide variant of T2Pt, designated N3-T2Pt, was also generated (Scheme S1) by incorporating an additional azide tag at a non-pharmacophore position that was based on the PCNA/T3 co-crystal structure (Figures 3 and S3). T2Pt and N3-T2Pt were insoluble in either aqueous buffers or ethanol, yet readily dissolved in DMSO. The co-ordination of cisplatin by DMSO has previously been described and may reduce its DNA reactivity 16 while increasing its reactivity with the histidine residues of protein 17. Correspondingly, the solvation of T2Pt in DMSO was expected to increase the selectivity of T2Pt for PCNA H44 over DNA. The co-ordination of T2Pt by DMSO following solvation was confirmed via the identification of a mono-DMSO-T2Pt complex by mass spectrometry (Figures S1 and S2).
Scheme 1.

Synthetic route of T2Pt (8) and chemical structure of T2AA (9).
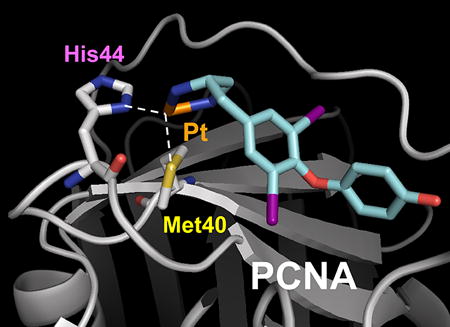
Figure 3.
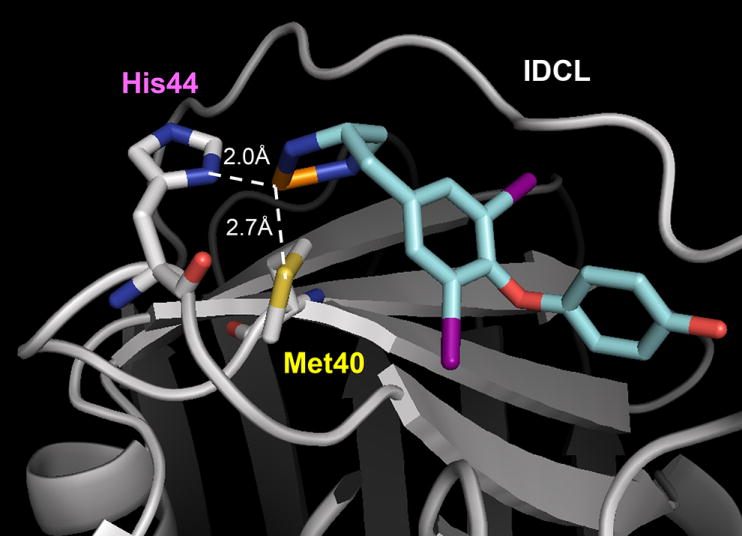
The rational design of T2Pt. A hypothetical model of the PCNA/T2Pt interaction. Platinum, orange; nitrogen, blue; sulfur, yellow; oxygen, red; iodine, violet. IDCL: interdomain connecting loop. The structure of T2Pt (cyan) was docked to the PIP-box binding cavity of PCNA (gray) using the PCNA/T3 complex 6, in which projections of M40 and H44 residues were optimized to adopt the octahedral platinum coordination (while dotted line). DMSO and a chloride atom coordinating the platinum atom were omitted for clarity.
The PCNA/T2Pt interaction was initially characterized using a competitive FP assay. A mixture of recombinant wild-type PCNA and fluorescently tagged PIP-box–containing PL peptide 9 was titrated with either T2Pt or T2AA. Unlike T2AA, T2Pt showed a very potent and time-dependent displacement of PL peptide from PCNA (Figure 4B). Whereas the IC50 of T2AA essentially remained unchanged (Figure 4A), the apparent IC50 of T2Pt proceeded to decrease by a factor of ~10-fold throughout the time course (Figure 4B), a characteristic typical of a covalent interaction 18. Similar reactivity was also demonstrated by N3-T2Pt (data not shown). In an effort to identify the site of PCNA/T2Pt cross-linking, recombinant M40A and H44A single PCNA mutants and a M40A/H44A double PCNA mutant were prepared by site-directed mutagenesis. The PCNAs were treated with N3-T2Pt and conjugated with a biotin-alkyne probe via click chemistry, and the products were subjected to SDS-PAGE followed by blotting using a streptavidin probe. Unlike wild-type PCNA, which readily reacted with N3-T2Pt, both single mutants showed attenuated bonding by N3-T2Pt, implicating both M40 and H44 as sites of modification. Consistent with this, the double mutant essentially failed to accommodate bonding by the N3-T2Pt beyond background (Figure 4C).
Figure 4.

T2Pt forms an irreversible complex with PCNA. The fluorescence polarization (mP) of samples containing fluorescently labeled PL peptide (10 nM) and PCNA (100 nM) were measured at defined time points after the addition of either T2AA (A) or T2Pt (B) (0 – 100 μM). Whereas the IC50 of T2AA essentially remained unchanged throughout the time course (IC50 ~1.5 μM), the apparent IC50 of T2Pt proceeded to decrease to ~0.1 μM through 4 h. Error bars represent the standard deviation of triplicate experiments. (C) Recombinant human wild-type or mutant PCNAs (0.9 μM) were reacted with N3-T2Pt (0, 2.5, 5, and 10 μM as denoted by an open triangle) for 2 h at room temperature. Samples were conjugated to a biotin-alkyne probe using a copper catalyst and then analyzed by probing with a streptavidin-HRP conjugate as described in the Materials and Methods. Membranes were subsequently stripped and re-probed with α-PCNA antibody as indicated.
To further verify the irreversible nature of the T2Pt-PCNA complex, recombinant wild-type PCNA was reacted with an excess of T2Pt and subsequently subjected to mass spectrometric analysis (Figures 5A and 5B). The PCNA/T2Pt reaction generated new peaks that were consistent with a PCNA/T2Pt species. The stoichiometry of the reaction suggested that one T2Pt was associated with each PCNA molecule, indicating a site-specific unimolecular reaction with PCNA. The PCNA/T2Pt species remained intact after removal of free T2Pt for up to 20 h (data not shown), suggesting that the interaction was irreversible and covalent in character. Inactivity of the M40A/H44A double mutant was also verified by mass spectrometry (Figures 5C and 5D), in which no peak shift was observed under the same treatment conditions utilized for wild-type PCNA. Collectively, these results strongly suggest that T2Pt site-selectively alkylated PCNA via M40 and H44 (Figure 3).
Figure 5.

Representative mass spectra of products generated by reacting human recombinant PCNA in the presence or absence of 50 μM T2Pt for 2 h at room temperature. A. Wild type PCNA, no treatment (observed mass, 28770 Da; predicted mass, 28769 Da). B. Wild type PCNA and 50 μM T2Pt. The peaks were consistent with a reaction involving the displacement of chlorine atoms from T2Pt and platinum coordination by DMSO (observed mass, 29552 Da; predicted mass, 29552 Da). C. M40A/H44A PCNA, no treatment (observed mass, 28646 Da; predicted mass, 28643 Da). D. M40A/H44A PCNA and 50 μM T2Pt. Few peaks distinct from free M40A/H44A PCNA were detected.
3.4. T2Pt demonstrates modest DNA reactivity
Cisplatin is a well-established anticancer therapeutic that generates stable covalent DNA adducts within cells, a property that is widely considered to underpin its significant biological activity19. The electrophilic platinum moiety of T2Pt (Scheme 1) is structurally analogous to cisplatin and may promote the generation of stable T2Pt-DNA adducts, much like cisplatin itself. To verify this in vitro, an electrophoretic band shift assay was used. Covalent bonding of supercoiled plasmid DNA by cisplatin typically renders the cisplatin-plasmid complex more tightly compact and yields a dose-dependent increase in the complex’s electrophoretic mobility (Figure 6A) 20. T2platin also induced a dose-dependent increase in the mobility of plasmid DNA during electrophoresis (Figure 6A), although plasmid compaction by the novel compound was less efficient relative to cisplatin. Nonetheless, the T2platin-induced plasmid shift indicates that the novel compound may covalently interact with DNA. An alkaline comet assay was next applied to evaluate DNA damage induction by T2Pt within cells. Notably, T2Pt was reasonably membrane-permeable (Table S1), thereby permitting its use in cellular assays. T2Pt dose-dependently induced DNA strand breaks in cells, however the levels were rather modest and attenuated relative to cisplatin at equimolar doses (Figure 6B).
Figure 6.

T2Pt demonstrates modest DNA reactivity. (A) Supercoiled plasmid DNA was reacted overnight at 37°C with various concentrations of cisplatin or T2Pt as indicated. Samples were then subjected to fractionation through agarose gels containing ethidium bromide by electrophoresis and photographed under UV illumination. (B) U2OS cells were initially exposed to 0–50 μM cisplatin or T2Pt for 7 h and then allowed to recover for 17 h in compound-free medium. Cells were then harvested and subjected to an alkaline comet assay as described in the Materials and Methods. DNA damage was assessed visually and each column represents the mean level of damage in arbitrary units. Error bars represent the standard error of the mean while n indicates number of cells scored.
3.5. T2Pt elicits a persistent cellular DNA damage response
Given the relatively mild induction of DNA strand breaks by T2Pt (Figure 6B), it was rationalized that T2platin may elicit a DNA damage response. To investigate the potential nature of such a response, the recovery of cells from T2Pt- or T2AA-induced cell cycle arrest was analyzed by flow cytometry. T2Pt induced a sustained cell cycle arrest in late S/G2 phase while cells treated with the reversible PCNA inhibitor T2AA readily moved from accumulation within early S phase upon release from the compound (Figure 7A). The inhibition of DNA replication is often coupled with phosphorylation of the histone variant H2AX at Ser139 (designated γH2AX), a cellular marker of replication stress 21. In accordance with this, cells were exposed to T2Pt either continuously for 24 h or pulsed for 7 h and allowed to recover in T2Pt-free medium for 17 h. Immunostaining for γH2AX established that T2Pt robustly induced γH2AX within cells in either treatment schedule. A distinctive feature of T2Pt-induced γH2AX was its persistence at relatively high levels following T2Pt withdrawal. In sharp contrast, the level of damage within cells largely recovered from pulse treatment with the reversible PCNA inhibitor T2AA (Figure 7B).
Figure 7.

T2Pt induces persistent cell cycle arrest and DNA replication stress. (A) The cell cycle distribution of HeLa cells upon release from 24 h of exposure to either T2Pt (100 μM) or T2AA (40 μM). The phosphorylation of H2AX Ser139 (B) or Chk1 Ser345 (C) in U2OS cells treated with either T2Pt (25 μM) or T2AA (25 μM) continuously for 24 h or pulsed for 7 h followed by 17 h recovery in compound-free medium. Following treatment, cells were fixed, processed for the immunocytochemical detection of phosphorylation of H2AX at Ser139 or Chk1 at Ser345 and analyzed by flow cytometry. Solid bars represent the area arbitrarily defined as positive for the phosphorylation status of each protein. (D) U2OS cells (330,000 per well) were initially seeded into six well cluster plates and allowed to attach overnight. Following treatment as described in (C), cells were harvested and analyzed for the expression of pSer345 Chk1 and β-actin by immunoblotting as described in the Materials and Methods section. Densitometry quantification was performed by dividing band intensity of the pSer345 Chk1 by that of β-actin for each lane, and values normalized to DMSO control are indicated as ratio.
The induction of γH2AX by T2Pt strongly implicated the involvement of at least one kinase in the replication stress response. Ataxia telangiectasia mutated and rad3-related (ATR) is a master kinase known to phosphorylate H2AX at Ser139 and functions in the replication stress response pathway 22–24. Consistent with this notion, T2Pt-treated cells exhibited intense phosphorylation at Ser345 of checkpoint kinase 1 (Chk1), an ATR substrate and a classic hallmark of ATR activation and the DNA replication stress response 22, 25. Furthermore, the level of Chk1 phosphorylation at Ser345 persisted after the release of cells from T2Pt, while Chk1 phosphorylation levels within T2AA-treated cells recovered by ~50% in identical conditions (Figure 7C). Collectively, these results suggest that T2Pt induced replication stress and enforced a subsequent late S/G2 phase block by persistently activating the ATR-Chk1 signaling axis.
3.6. T2Pt is not cytotoxic as a single agent, yet sensitizes cells to cisplatin
Finally, to determine how persistent chemical inhibition of PCNA function may be detrimental to cells, we assayed T2Pt for cytotoxicity. Unexpectedly, T2Pt showed little effect as a single agent in a clonogenic survival assay (Figure 8A) at a concentration in which persistent γH2AX induction and ATR/Chk1 activation were observed (Figure 7C). This suggests that persistent inhibition of the PCNA/PIP-box interactions was not cytotoxic in isolation. Previously, we established that the chemical inhibition of PCNA/PIP-box interactions sensitized U2OS cells to cisplatin 6, 14. Similarly, T2Pt predisposed U2OS cells to enhanced cisplatin cytotoxicity despite its inactivity as a single agent (Figure 8B).
Figure 8.

As a single agent, T2Pt shows little cytotoxicity yet sensitizes cells to cisplatin. U2OS cells were cultured with either (A) 0–30 μM T2Pt for 24 h and then allowed to expand into colonies in compound-free medium for a further 6 days, or (B) 0–300 nM cisplatin for 24 h, followed by incubation with DMSO or 30 μM T2Pt for another 24 h. Cells were allowed to expand into colonies in fresh culture medium for a further 6 days. Samples were then assayed for clonogenic survival as detailed in the Materials and Methods. The survival rate was reported as 1 for the average of number colonies that received the vehicle control. Error bars represent the standard deviation of triplicate experiments.
4. Discussion
M40 lines part of the PIP-box binding cavity of PCNA, and its substitution for alanine has deleterious implications for binding by a PIP-box–bearing peptide (Figure 1), suggesting that the mutation may locally disrupt interactions with residues of the PIP-box motif, which generally adopts a conserved 310 helix hydrophobic plug 26. The importance of M40 in PIP-box recognition may also be exemplified by mutational analyses27 of its immediate neighboring residue, aspartic acid 41 (D41). Among 29 individual PCNA mutations, a D41A PCNA point mutant was uniquely unable to stimulate both in vitro DNA synthesis by polymerase δ and the ATPase activity of RF-C, the protein complex chiefly responsible for loading the PCNA clamp onto DNA 27. An ensuing study 28 established that D41 of PCNA was also critical for its loading onto a plasmid DNA template by RF-C in vitro. Consistent with this observation, the GFP-tagged M40A PCNA mutant was inefficiently loaded onto chromatin within living eukaryotic cells (Figure 2A), implicating an important functional role for M40 in chromatin loading. It is presently unclear whether the M40 residue makes direct contact with RF-C or other factors that are required for loading PCNA onto chromatin within living cells, although it was noted that D41 most likely directly interacts with the RF-C loading complex, at least in vitro28. The M40A mutant has potential as a molecular tool for the functional dissection of the role of the PIP-box binding cavity in the recruitment of PCNA binding partners. Moreover, the mutant holds promise as a tool and for the identification of novel PIP-box containing PCNA-binding molecules by differential proteomics 11.
The identification of PCNA M40 and H44 as functionally relevant in PIP-box recognition indicated that the residues may be valid targets for the generation of a PIP-box binding inhibitor. Coupled with their functional significance, the relatively nucleophilic nature of both M40 and H44 presented a rare opportunity for the generation of a selective small molecule inhibitor capable of a covalent, irreversible interaction with PCNA. Functionally relevant nucleophilic amino acid residues have emerged as attractive targets for covalent modification, particularly in the development of therapeutic inhibitors and activity-based probes 18, 29, 30. Very few chemical agents are capable of covalent interaction with methionine residues in an intracellular environment, although it is well established that platinum bonds avidly with the sulfur atom of methionine to generate a particularly strong coordination complex 31. Favorably, platinum is coordinated also by the imidazole ring of histidine 15, and it is likely that T2Pt is coordinated by both M40 and H44 PCNA residues since only the M40A/H44A double PCNA mutant completely failed to interact with PCNA (Figure 4C and 5D). The robust nature of the PCNA/T2Pt interaction was typified by detection of the complex following immunoblotting (Figure 4C), a technique that involves the application stringent conditions that protein denaturation. The survival of the T2Pt/PCNA complex, despite the application of these conditions, suggests that the PCNA/T2Pt complex is likely to endure the relatively mild conditions of the cellular milieu.
PCNA assumes an indispensable role in supporting DNA replication and is therefore an obvious requirement for smooth S phase traverse and cell cycle progression. 2, 9 It follows that the inhibition of PCNA-mediated functions would be anticipated impair these biological processes. Consistent with this notion, the onset of cell cycle arrest following T2Pt exposure (Figure 7A) correlated with the induction of γH2AX (Figure 7B). ATR phosphorylates H2AX at Ser139 and functions at the apex of the replication stress response pathway, particularly in response to extended stretches of single-stranded DNA generated by arrested replication forks 22–24. T2Pt-treated cells exhibited intense phosphorylation at Ser345 of Chk1 (Figure 7C), an ATR substrate and a hallmark of ATR activation and the DNA replication stress response 22, 23. Significantly, ATR-mediated phosphorylation of H2AX in response to replication stress stimulated by hydroxyurea, a well-established DNA replication inhibitor, occurs exclusively in S and G2 phases 21, the very same phases in which cells were persistently arrested after T2Pt release (Figure 7A). The persistent nature of the replication stress response induced by T2Pt, but not by the reversible inhibitor T2AA (Figure 7C), complements its enduring arrest of the cell cycle (Figure 7A) and sustained induction of DNA damage after release (Figures 7B) and is entirely consistent with the generation of an irreversible T2Pt/PCNA complex within cells.
It is also a reasonable argument that the T2Pt-induced replication stress response evident in Figure 7B and 7C may be a manifestation of direct DNA damage by the compound (Figure 6) as opposed to inhibition of PCNA-mediated functions. It is difficult to discern between the relative involvement of each potential mechanism given the intimate link shared by PCNA function and DNA metabolism. It is noteworthy, however, that the compound induced modest levels of DNA damage relative to cisplatin (Figure 6), a feature that may be attributable in part to platinum complexation of T2Pt by DMSO. It has been reported that the coordination of cisplatin by DMSO completely eliminated cytotoxicity demonstrated by the drug 32, possibly due to a loss in DNA reactivity. Moreover, T2Pt demonstrated little activity as a single agent in clonogenic survival assays (Figure 8A), a result that is inconsistent with the extremely toxic nature of DNA-directed agents in general.33 Rather, it was observed when cells were challenged with cisplatin-induced DNA damage prior to T2Pt exposure that the novel compound significantly impaired clonogenic survival (Figure 8B), a feature that is perhaps a reflection of the inhibition of PCNA-mediated DNA repair by T2Pt.
5. Conclusion
The requirement of platinum for interaction with M40 clearly restricts the design of a second generation of irreversible PCNA/PIP-box inhibitors; however, it may be possible to substitute the platinum moiety of T2Pt with a functional group capable of covalent interaction with H44 alone, an approach that has been applied in the development of a protein-protein interaction inhibitor 34. Inhibitors of this nature may avoid bonding with DNA, improve selectivity for PCNA and therefore provide a second generation of analogues potentially useful for probing the rapid inactivation of PIP-box-mediated PCNA binding events.
Supplementary Material
Acknowledgments
Funding Sources
The American Lebanese Syrian Associated Charities (ALSAC) and American Cancer Society Research Scholar Grant #RSG CDD-120969 (to N.F.). Microscopy images were acquired at the Cell & Tissue Imaging Center in St. Jude Children’s Research Hospital which is supported by ALSAC and NCI P30 CA021765-34.
We thank Dr. Scott Perry for flow cytometry support; Dr. Jennifer Peters for assistance with confocal microscopy; Dr. Akira Inoue for technical advice and helpful discussions; Mr. Kiran Kodali for protein mass spectrometry; Mr. Sotaro Kikuchi, Mr. Keita Kawarazaki (Yokohama City University), and Dr. Hiroshi Hashimoto (University of Shizuoka) for attempting structural characterization of PCNA/T2Pt complex; Drs. David Gerlich and Toshiki Tsurimoto for plasmids; and Mr. David Galloway for grammatical advice and preparation of this manuscript.
Abbreviations
- APC
allophycocyanin
- ATR
ataxia telangiectasia mutated and rad3-related
- BCA
bicinchoninic acid
- Chk1
checkpoint kinase 1
- CSK buffer
cytoskeleton buffer
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- DPPA
diphenylphosphoryl azide
- EDTA
ethylenediaminetetraacetic acid
- FP
fluorescence polarization
- γH2AX
phosphorylation of histone H2AX at Ser139
- HATU
2-(1H-7-azabenzo[d][1,2,3]triazol-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- HRP
horse radish peroxidase
- IDCL
interdomain connecting loop
- LDS
lithium dodecyl sulfate
- MOPS
3-morpholinopropane-1-sulfonic acid
- N3-T2Pt
chloro(dimethylsulfoxide)((S)-N-(3-azidopropyl)-5-(4-(2,3-diaminopropyl)-2,6-diiodophenoxy)-2-hydroxybenzamide)platinum(II) chloride
- PCNA
proliferating cell nuclear antigen
- PIP-box
PCNA-interacting protein box
- PL peptide
Polo-Ligase peptide
- PMSF
phenylmethylsulfonyl fluoride
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- T2AA
T2 amino alcohol
- T2Pt
chloro(dimethylsulfoxide)((S)-4-(4-(2,3-diaminopropyl)-2,6-diiodophenoxy)phenol)platinum(II) chloride
- TBS
Tris-buffered saline
- TCEP
tris(carboxyethyl)phosphine
- TEA
triethylamine
- TES
triethylsilane
- THF
tetrahydrofuran
- TFA
trifluoroacetic acid
Footnotes
Conflict of interest statement: None declared.
References
- 1.Gulbis JM, Kelman Z, Hurwitz J, O’Donnell M, Kuriyan J. Cell. 1996;87:297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- 2.Moldovan GL, Pfander B, Jentsch S. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Paunesku T, Mittal S, Protic M, Oryhon J, Korolev SV, Joachimiak A, Woloschak GE. Int J Radiat Biol. 2001;77:1007–1021. doi: 10.1080/09553000110069335. [DOI] [PubMed] [Google Scholar]
- 4.Naryzhny SN. Cell Mol Life Sci. 2008;65:3789–3808. doi: 10.1007/s00018-008-8305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witko-Sarsat V, Mocek J, Bouayad D, Tamassia N, Ribeil JA, Candalh C, Davezac N, Reuter N, Mouthon L, Hermine O, Pederzoli-Ribeil M, Cassatella MA. J Exp Med. 2010;207:2631–2645. doi: 10.1084/jem.20092241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Punchihewa C, Inoue A, Hishiki A, Fujikawa Y, Connelly M, Evison B, Shao Y, Heath R, Kuraoka I, Rodrigues P, Hashimoto H, Kawanishi M, Sato M, Yagi T, Fujii N. J Biol Chem. 2012;287:14289–14300. doi: 10.1074/jbc.M112.353201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoue A, Kikuchi S, Hishiki A, Shao Y, Heath R, Evison BJ, Actis M, Canman CE, Hashimoto H, Fujii N. J Biol Chem. 2014;289:7109–7120. doi: 10.1074/jbc.M113.520429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss WA, Taylor SS, Shokat KM. Nat Chem Biol. 2007;3:739–744. doi: 10.1038/nchembio1207-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kontopidis G, Wu SY, Zheleva DI, Taylor P, McInnes C, Lane DP, Fischer PM, Walkinshaw MD. Proc Natl Acad Sci U S A. 2005;102:1871–1876. doi: 10.1073/pnas.0406540102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 11.Ohta S, Shiomi Y, Sugimoto K, Obuse C, Tsurimoto T. J Biol Chem. 2002;277:40362–40367. doi: 10.1074/jbc.M206194200. [DOI] [PubMed] [Google Scholar]
- 12.Collins AR. Mol Biotechnol. 2004;26:249–261. doi: 10.1385/MB:26:3:249. [DOI] [PubMed] [Google Scholar]
- 13.Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, Zink D, Cardoso MC. J Cell Biol. 2000;149:271–280. doi: 10.1083/jcb.149.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Actis M, Inoue A, Evison B, Perry S, Punchihewa C, Fujii N. Bioorg Med Chem. 2013;21:1972–1977. doi: 10.1016/j.bmc.2013.01.022. [DOI] [PubMed] [Google Scholar]
- 15.Hohage O, Sheldrick WS. J Inorg Biochem. 2006;100:1506–1513. doi: 10.1016/j.jinorgbio.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Fischer SJ, Benson LM, Fauq A, Naylor S, Windebank AJ. Neurotoxicology. 2008;29:444–452. doi: 10.1016/j.neuro.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 17.Tanley SW, Schreurs AM, Kroon-Batenburg LM, Meredith J, Prendergast R, Walsh D, Bryant P, Levy C, Helliwell JR. Acta Crystallogr D Biol Crystallogr. 2012;68:601–612. doi: 10.1107/S0907444912006907. [DOI] [PubMed] [Google Scholar]
- 18.Singh J, Petter RC, Baillie TA, Whitty A. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 19.Kelland L. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 20.Cohen GL, Bauer WR, Barton JK, Lippard SJ. Science. 1979;203:1014–1016. doi: 10.1126/science.370979. [DOI] [PubMed] [Google Scholar]
- 21.Ward IM, Chen J. J Biol Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 22.Osborn AJ, Elledge SJ, Zou L. Trends Cell Biol. 2002;12:509–516. doi: 10.1016/s0962-8924(02)02380-2. [DOI] [PubMed] [Google Scholar]
- 23.Cimprich KA, Cortez D. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chanoux RA, Yin B, Urtishak KA, Asare A, Bassing CH, Brown EJ. J Biol Chem. 2009;284:5994–6003. doi: 10.1074/jbc.M806739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA. Cell Cycle. 2010;9:662–669. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 26.Bruning JB, Shamoo Y. Structure. 2004;12:2209–2219. doi: 10.1016/j.str.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda K, Morioka H, Imajou S, Ikeda S, Ohtsuka E, Tsurimoto T. J Biol Chem. 1995;270:22527–22534. doi: 10.1074/jbc.270.38.22527. [DOI] [PubMed] [Google Scholar]
- 28.Zhang G, Gibbs E, Kelman Z, O’Donnell M, Hurwitz J. Proc Natl Acad Sci U S A. 1999;96:1869–1874. doi: 10.1073/pnas.96.5.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Speers AE, Adam GC, Cravatt BF. J Am Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 30.Gushwa NN, Kang S, Chen J, Taunton J. J Am Chem Soc. 2012;134:20214–20217. doi: 10.1021/ja310659j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zimmermann T, Zeizinger M, Burda JV. J Inorg Biochem. 2005;99:2184–2196. doi: 10.1016/j.jinorgbio.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 32.Yi YW, Bae I. DNA Repair (Amst) 2011;10:1084–1085. doi: 10.1016/j.dnarep.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hurley LH. Nat Rev Cancer. 2002;2:188–200. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- 34.Fujii N, Haresco JJ, Novak KA, Stokoe D, Kuntz ID, Guy RK. J Am Chem Soc. 2003;125:12074–12075. doi: 10.1021/ja035540l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.