

Abstract
Formation of arterial vasculature, here termed arteriogenesis, is a central process in embryonic vascular development as well as in adult tissues. While the process of capillary formation, angiogenesis, is relatively well understood, much remains to be learned about arteriogenesis. Recent discoveries point to the key role played by vascular endothelial growth factor receptor 2 (VEGFR2) in control of this process and to newly identified control circuits that dramatically influence its activity. The latter can present particularly attractive targets for a new class of therapeutic agents capable of activation of this signaling cascade in a ligand-independent manner, thereby promoting arteriogenesis in diseased tissues.
Keywords: Angiogenesis, arteries, vascular biology, animal model cardiovascular disease, vascular endothelial growth factor
Introduction
Arteriogenesis is a complex set of events that involve interactions among various cell types and signaling circuits. Recent studies have revealed many details of these processes, but much remains to be learned about how these vessels form and how arterial identity is acquired. One of the challenges has been in defining the term arteriogenesis itself1, in part because so little about its biology is known. In developmental biology the term is used to describe formation of the arterial vessel network from the primary vascular plexus. This includes endothelial arterial fate specification, recruitment of smooth muscle cells (SMCs) and formation of the arterial vessel wall, and growth and branching of the forming arterial tree. The branching process also leads to formation of artery-to-artery connections, termed collaterals.
In adult setting the term arteriogenesis refers to the formation of new (usually collateral) arteries after occlusion of an arterial trunk. Their origin is debated: one school of thought attributes their formation exclusively to the enlargement of a pre-existing collateral network, while the other allows for de novo formation of new arterial vessels by means of capillary arterialization1. Semantics aside, these differences have potentially important clinical applications, as stimulation of enlargement of an existing artery may be quite different from induction of new artery formation.
Recent studies suggested that there are fundamental molecular differences between arterial and other vasculatures and that arterial fate is determined early in the course of development, in some settings even before the onset of blood circulation2, 3. Yet very little is known about arterial fate specification in the adult.
The importance of clear understanding of arteriogenesis cannot be overestimated. Given the paucity of success in the therapeutic angiogenesis field over the last decade and realization that it is arterial and not the capillary growth that is the key to restoring effective circulation to compromised tissues, “therapeutic arteriogenesis” has emerged as a new concept4. One driver, then, behind the desire to understand these events is the need for better therapeutic strategies to stimulate arterial growth that could benefit patients with a variety of illnesses compromised by defective or impaired arterial circulation. It is also becoming increasingly clear that many disorders of arterial circulation such as arterio-venous malformations (AVMs), aneurysms and the like have genetic origins rooted in arteriogenic signaling cascades5, 6. Thus, better understanding of arterial specification and arterial conduit formation promises to have a significant diagnostic and therapeutic impact in a variety of important disease conditions.
Arteriogenesis in development
Early studies identified the key requirement for VEGF/VEGFR2 signaling during vascular development. The loss of a single allele of Vegfa is sufficient to induce early embryonic (~E8.5) lethality due to failure of primitive vasculature formation7, 8. A homozygous disruption of Vegfr2, the principal signaling VEGF receptor in endothelial cells, induces a similar phenotype9.
Arterial differentiation is thought to occur in angioblasts exposed to higher VEGF concentrations, while less exposed cells acquire venous fate. In mice, removal of nerve-derived VEGF in the embryonic skin prevents arterial differentiation of primitive vessels10, 11. In zebrafish embryos, VEGF expression is induced by the morphogen sonic hedgehog (Shh)12. Angioblasts close to Shh expressing tissues receive high VEGF concentrations and subsequently differentiate into arterial cells that form the dorsal aorta. Angioblasts located further away differentiate as veins but can be converted into arterial cells by Shh or VEGF overexpression12, 13.
In addition to VEGF, other pathways may be involved in arterial fate specification. The TGFβ superfamily receptor Activin receptor-like kinase 1 (ALK1) is predominantly expressed in arterial cells14. Heterozygous mutations in human ALK1 cause hereditary hemorrhagic telangiectasia (HHT), a disease characterized by focal AVMs in various tissues15. Alk1-deficient vessels lack certain arterial genes, including Jagged1, UNC5B and ephrinB2, suggesting that defective arterial differentiation could contribute to AVM formation16–18. Furthermore, the Alk1 ligand BMP919–21 induces expression of Jagged1, UNC5B and EphrinB2 in a Smad-dependent manner, thereby linking Smad signaling to arterial fate specification22, 23.
Embryonic endothelial cells are not committed to the arterial fate: some dorsal aorta cells become incorporated into veins, and this fate switch is accompanied by the loss of arterial and gain of venous gene expression24, 25. Likewise, grafting of embryonic quail arteries and veins showed that arterial cells can colonize veins of the host and vice-versa, again accompanied by switch in gene expression26. Thus, reprogramming of arterial and venous endothelial cells occurs during normal development and can be induced experimentally. Factors governing fate switch are poorly understood, but may involve involve repulsion between cells expressing ephrinB2 and its receptor EphB4 that trigger segregation of vein-fated endothelial cells from arteries24, 27.
Blood flow further contributes to arterial-venous specification and differentiation. In chick embryos, ligation of the extraembryonic artery induces a profound vascular remodeling and morphological and genetic transformation of arteries into veins and vice-versa28, 29. In adult vasculature, positioning of a venous conduit in an artery leads to the loss of venous genes such as EphB4 without increased expression of Ephrin-B2, Dll4 or other markers of arterial identity30.
Taken together, current evidence suggests that embryonic endothelial cells exhibit a significant degree of plasticity with respect to arterial-venous differentiation that is lost later in development. The switch in arterial and venous identity may be facilitated by a signaling system where threshold levels of morphogens such as VEGF-A activate arterial gene expression, while lowering VEGF-A input reverses the gene expression program to a venous one. Better understanding of arterial programming is relevant in clinical settings where vessels of different identity are grafted together, such as during bypass surgery or dialysis treatment. Changes in the transplanted vessels after grafting and the significant risk of graft failure involved in these therapies suggests a limited degree of plasticity in adults that could be improved by manipulation of pathways driving arterial-venous differentiation31.
In contrast to the endothelial arterial identity acquisition, how SMCs in the arteries acquire identity remains poorly understood. Mature arterial tubes are surrounded by multiple concentric SMC rings, which are themselves surrounded by an adventitial layer of fibroblasts embedded in a collagen matrix. The size and pattern of the SMC layer depends on the arterial diameter and is developmentally controlled in a vessel-specific manner, with small diameter resistance arteries surrounded by one or two SMC layers, while larger diameter arteries can have a dozen or more SMC layers. How SMC assembly is controlled at the cellular and molecular levels is largely unknown but clinically important, since dysregulation of SMC development causes cardiovascular diseases such as aortic aneurysm, atherosclerosis and pulmonary hypertension32.
SMCs are derived from multiple embryonic tissue sources33, 34. Cellular and molecular mechanisms leading to arterial wall formation have been recently investigated in the pulmonary arteries in mice35. Notch signaling is critically involved in SMC development, in addition to its role in arterial endothelial specification (see below). Among the 4 mammalian Notch receptors, Notch3 is the isoform most prominently expressed in arterial SMCs. It is required for arterial SMC development following activation by Jagged-1 on arterial endothelial cells.36–38 Mutations in Notch3 cause cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), a hereditary disease of the cerebral arteries that causes stroke and vascular dementia.
A few genes specifically expressed in arterial, but not venous SMCs are known. Intriguingly, these control development of sympathetic nerves and arterial innervation, which controls blood supply to organs. The glial-derived neurotrophic factor (GDNF) family member artemin, and the neurotrophins Nerve growth factor (NGF) and neurotrophin-3 (NT-3) are expressed in embryonic arterial SMCs, and inactivation of the genes encoding these molecules in mice leads to defects in sympathetic axon growth and extension along the arterial vasculature39, 40. Postnatal arterial SMCs acquire the expression of the axon guidance molecule netrin-1, which is required for innervation of arterial SMCs by sympathetic neurons41.
Arteriogenesis in disease
Abnormal arterial development can lead to several inherited disorders, including aortic arch malformations, aneurisms and arterio-venous malformations (AVMs). In adults, a number of vasoocclusive diseases, ranging from atherosclerotic coronary, peripheral and cerebral arterial disease to arteriosclerosis (transplant-related and unrelated) and systemic and pulmonary hypertension, among others, all reduce arterial blood flow to healthy tissues. This can result in outright cell death and organ damage as happens in myocardial infarction or stroke or may lead to a chronic impairment of various organs function as happens, for example, in pulmonary arterial hypertension, peripheral artery disease or chronic stable angina. In all of these cases, restoration of blood inflow would be of considerable therapeutic benefit.
The current clinical strategies for treatment of vasoocclusive diseases entail either some form of mechanical revascularization (e.g. stenting, bypass grafting) or medications designed to limit end-organ oxygen demand (e.g. beta-blockers in patients with chronic stable angina). Despite numerous attempts, no strategy has evolved that allows restoration of arterial inflow either by reversing disease processes that lead to arterial narrowing and occlusion or by creating new arterial conduits.
Early studies using various angiogenic growth factors, mostly VEGF and FGFs, failed due to a number of reasons including poor understanding of biology, inadequate delivery methodologies and problems with clinical assessment of benefits42. Recent advances in our understanding of the biology of arteriogenesis, discussed below, offer clues to what went wrong and provide a foundation for the development of different therapeutic strategies.
In discussion of the failure of the initial rounds of clinical trials and the literature dealing with arteriogenesis, it is important to consider animal models used to assess arteriogenesis in adult tissues. Almost all studies utilize a hindlimb ischemia model in which arteriogenesis is induced by femoral artery ligation43. A number of animal species, ranging from mice to pigs have been used in this fashion. Flow recovery in this model occurs by a combination of recruitment and expansion of pre-existing collaterals and by de novo arteriogenesis. Genetic factors play a major role in this response both in animals44–46 and in patients47 in a not yet fully defined manner48. The vast majority of studies using this model are performed in young and disease-free animals, with the important limitation that both age49 and disease states such as diabetes50, 51 and hypercholesterolemia52 impair arteriogenic response. The other important model is the ameroid occluder model usually carried out in young pigs. Unlike the rodent models, most arterial growth in this setting is of the de novo variety and the gradual occlusion of a one of the main coronary trunks by the ameroid better mimics arteriogenesis in chronic CAD or PAD.
In addition to the importance of arteriogenesis in adults with vasoocclusive diseases, abnormal vascular development, and in particular, abnormal arterio-venous fate specification can lead to a number of other important illnesses including AVM and CCMs.
Genetics of arterial collateral circulation
Key insights into arteriogenic signaling came from studies of genetic differences in the extent of collateral circulation in rodents and people46, 47, 53–55. Numerous genes affecting native collateral density have been identified including CD4456, chloride intracellular channel-457, gap junction proteins connexin-3758 and connexin-4028, PECAM-159, NFkB60, Delta-like-4 (Dll4)61, HIF2α62 and RGS563 among others (Table 1). Broadly speaking, these fall into three distinct categories: genes affecting endothelial ERK activation and Delta-Notch signaling, genes affecting shear stress and SMC G-protein signaling and genes affecting monocyte/macrophage recruitment and “inflammatory” response. Roles of a number of other genes have not been established.
Table 1.
Genetics of arteriogenic defects
| Gene | Molecular mechanism | Biological processes |
|---|---|---|
| Synectin | EC VEGFR2 trafficking/ERK activation | Arterial branching and lumen size |
| Myosin-VI | EC VEGFR2 trafficking/ERK activation | Arterial branching and lumen size |
| Nrp1 | EC VEGFR2 trafficking/ERK activation | Arterial branching and lumen size |
| PTP1b | VEGFR2 trafficking/ERK activation | Arterial branching and lumen size |
| Dll4 | EC Delta-Notch signaling | Arterial branching and maturation |
| HIF2α | EC Delta-Notch signaling | Arterial branching and maturation |
| NFkB | EC Delta-Notch signaling | Arterial branching and maturation |
| Connexin-37 | gap junctions/EC-EC communication | Arterial branching and remodeling |
| Connexin-40 | gap junctions/EC-EC communication | Arterial branching and remodeling |
| PECAM-1 | shear stress signaling | Arterial branching, lumen size |
| Shc | shear stress signaling | Arterial fate and remodeling |
| eNOS | SMC G protein signaling | Arteriolar density/vascular regression |
| RGS5 | SMC G protein signaling | Arterial fate, branching and remodeling |
| ICAM-1 | monocytes recruitment | perivascular ‘inflammation” |
| Mac-1 | monocytes recruitment | perivascular “inflammation” |
| MCP1 | monocytes recruitment | perivascular “inflammation” |
| CD44 | monocytes recruitment | perivascular “inflammation” |
| PHD2 | monocytes recruitment | perivascular “inflammation” |
| NFkB | monocytes recruitment | perivascular “inflammation” and Dll4 signaling |
| CD73 | monocytes recruitment | perivascular “inflammation” |
| HUR | monocyte VEGF-A stability | monocyte VEGF production |
| Ang2 | inflammatory response modulation | perivascular “inflammation” |
| CD180 | inflammatory response modulation | perivascular “inflammation” |
| ADAMS 17 | vascular stabilization | vessel stabilization |
| Clic4 | unknown | unknown |
| NPY | unknown | unknown |
| Canq1 locus | unknown | unknown |
| BMX/ETK | unknown | unknown |
Clic4: chloride intracellular channel-4
NPY: Neuropeptide Y
VEGF signaling is clearly central to these differences. Thus, the extent of collateral density in mouse strains correlates with the level of VEGF-A expression45 while in the rat repetitive coronary occlusions model anti-VEGF antibody treatment significantly reduces collateral growth64. Furthermore, a reduction in endothelial VEGF signaling input seen in synectin null, myosin-VI null and Nrp1cyto mice also correlates with reduced collateral formation65–68. It is further interesting to speculate that reduced arteriogenesis and/or collateral extent in some patient populations may be also due to the same. In particular, patients with diabetes mellitus, a population with well-established arteriogenic defects69, demonstrate a dramatic reduction in VEGFR2 expression and activation despite increased VEGF-A expression70 while exhibiting a ligand-independent receptor activation71.
Molecular mechanisms of arteriogenesis
Growth of the arterial vasculature requires coordinate action of a number of cell types including endothelial cells, smooth muscle cells, pericytes and various auxiliary cells such as monocyte-derived macrophages, neurons and others that regulate this process. These will be discussed in turn.
Cellular controls of arteriogenesis: endothelial cells
Endothelial cells play a critical role in developmental arteriogenesis as they establish the arterial identity of the forming vessel and form a backbone of what later becomes an artery. They play an equally important, if less understood, role in adult arteriogenesis. In both cases VEGF-A appears to be the major driver.
VEGF-A binding to VEGFR2 activates a number of intracellular signaling cascades including MAPK (ERK1/2), PI3K/Akt, Src and Rac among others72, 73. Of these, VEGF-dependent ERK activation is particularly important in vascular development as a knock-in of a VEGFR2 mutant carrying a single amino acid mutation in this site (Y1175F) results in a failure of vasculature development that is virtually indistinguishable from Vegfr2 knockout74. Furthermore, ERK activation is critical to arteriogenesis as mice mutants carrying mutations that reduce VEGF-dependent ERK activation65, 67, 68, 75 demonstrate reduced formation of arterial but not venous vasculature (Table 2). In contrast, stimulation of endothelial ERK signaling results in exuberant arteriogenesis76–78 (Table 2).
Table 2.
Endothelial ERK activation and arterial morphogenesis.
|
Cellular controls of arteriogenesis: smooth muscle cells
While developmental arteriogenesis is clearly driven by the endothelium with SMCs coming into play later when a tube with arterial fate specification has already been established, the situation in adult arteriogenesis is more complex and less well understood. In part, this is due to the existence of two distinct types of adult arteriogenesis- adaptive growth, a term that refers to enlargement of pre-existing arterial collaterals, and de novo arteriogenesis, the process of capillary bed arterialization1. These have been difficult to distinguish experimentally and it is likely that smooth muscle cells play a much larger role in the former than in the latter.
Remodeling of the pre-existing collaterals is thought to be driven by biomechanical factors including shear-stress-dependent activation of eNOS signaling leading to their dilatation28, 79–81. This in turn leads to increased circumferential wall stress that then stimulates growth and expansion of the media layer largely due to SMC proliferation and hypertrophy82. SMC proliferation entails a switch in SMC phenotype from contractile to proliferative that can be activated directly by mechanical stresses as well as by nitric oxide. The critical role played by NO is demonstrated by a markedly reduced arteriogenesis and vessel rarefication in eNOS null mice83, 84 albeit the latter may also be due to the regulatory role of NO in regulation of angiopoietin-2 release from endothelial cells85. At the same time, dysregulation of G protein signaling in SMC as, for example, seen with a knockout of RGS5 also affects arteriogenesis63.
Capillary arterialization86 is a very poorly understood process that involves expansion of the capillary bed, change in endothelial cell fate and acquisition of the medial layer. This is largely driven by endothelial cells87 but details are murky. A recent study demonstrated that thymosin-β4 stimulation of myocardin-related transcription factor-A (MRTF-A) in endothelial cells promotes capillary growth and pericyte maturation88 thereby expanding the microcirculation bed that can then undergo arterialization. This, in turn, may augment flow the pre-existing collaterals in the more proximal parts of the arterial tree thereby reducing peripheral resistance and enhancing perfusion of ischemic territories.
Cellular control of arteriogenesis: extravascular cells
Given the key role VEGF-A plays in arteriogenesis, its source is an important question. During embryonic development, nerves serve as an important source of VEGF-A10, 11. Whether nerves contribute to VEGF production in adult arteriogenesis has not been clearly established. While endothelial and smooth muscle cells have the ability to secrete VEGF, this is largely restricted to a hypoxic environment. As arteriogenesis takes place in tissues with normal oxygen content, it is unlikely that these cells are the main source of VEGF. A substantial body of literature points to the role of blood (monocyte)-derived macrophages in arteriogenesis.
Early studies suggested that macrophage-secreted FGF1 or FGF2 are the key arteriogenic factors89. However, mice deficient in FGF1, FGF2 or FGF5 do not demonstrate any arteriogenic defects and FGF signaling appears to be more important in maintenance of the vasculature than in its formation.90 On the other hand, macrophages with reduced VEGF-A expression demonstrate defective adult arteriogenesis,91 while expansion of macrophage population due to, for example, PHD2 haploinsufficiency, significantly increases developmental and adult arteriogenesis92. Similarly, activating macrophage HIF-1α expression promotes while suppressing it inhibits, adult arteriogenesis85.
Blood monocyte levels also correlate with collateral growth93. Thus, mice lacking MCP-1 receptor CCL2 demonstrate decreased arteriogenesis53 while MCP-1-driven increase in macrophage numbers at the site of arteriogenesis stimulates it94, 95. Taken together, these data point to macrophages as the key source of VEGF during arteriogenesis.
Regulation of endothelial ERK signaling
As foregoing discussions illustrate, VEGFR2-driven endothelial ERK activation is critical to formation of arterial vasculature and to regulation of branching extent and lumen size. Indeed, ERK plays a central role during branching morphogenesis96–98. Thus manipulation of this signaling pathway may be of direct benefit for therapeutic arteriogenesis. The two principle themes that emerge from studies of VEGFR2-specific ERK activation is the role of the receptor’s endocytosis and trafficking and cross-talk with cellular signaling cascades. These will be discussed in turn.
Regulation of ERK activation: VEGFR2 endocytosis and trafficking
Early studies of VEGFR2 signaling suggested that its internalization via a clathrin-dependent endocytic pathway is critical to its ability to signal99. Subsequent experiments refined this concept. The endothelial deletion of one of VEGFR2 interacting proteins, EphrinB2, leads to a complete lack of VEGFR2 endocytosis following VEGF-A binding100. One consequence of this is the lack of ERK1/2 activation by VEGF-A. But VEGFR2 endocytosis by itself is not sufficient for a full ERK activation. Upon entering the cytoplasm via a clathrin-dependent endocytic pathway, VEGFR2 is found in Rab5+ early endosomes68, 99, shuttled to EEA1+ endosomes, and then either recycled to the plasma membrane or delivered to lysosomes for cargo degradation. The movement to the EEA1+ compartment occurs via a protein complex that includes another VEGF-A165 receptor, neuropilin-1, as well as synectin and myosin-VI. As Rab5+ VEGFR2-containing endosomes traffic through the cytoplasm, they come in a close contact with an endoplasmic reticulum protein tyrosine phosphatase 1b (PTP1b) that specifically dephosphorylates VEGFR2 Y1175 site thus leading to decreased ability of the receptor to bind PLCγ and activate ERK signaling (Fig 1)67, 101. Any disruption of this complex (e.g. knockout of synectin or myosin-VI or knock-in of a Nrp1 mutant lacking its synectin biding site) increases the length of time VEGFR2 spends near PTP1b thus increasing dephosphorylation of Y1715 site and reducing ERK activation. The consequence of this is the reduction in the number of arterioles and arteriolar branching (Fig 2).
Figure 1.

Intracellular trafficking of VEGFR2.
VEGFR2 endocytosis and Neuropilin-1/synectin/myosin-VI-dependent trafficking. VEGF-A binding to VEGFR2 and Nrp1 creates a multiprotein complex and induces its endocytosis. A Nrp1-dependent trafficking that proceeds in a synectin/myosin-VI-dependent manner leads to VEGR2-driven activation of ERK signaling in EEA1+ endosomes.
Figure 2.
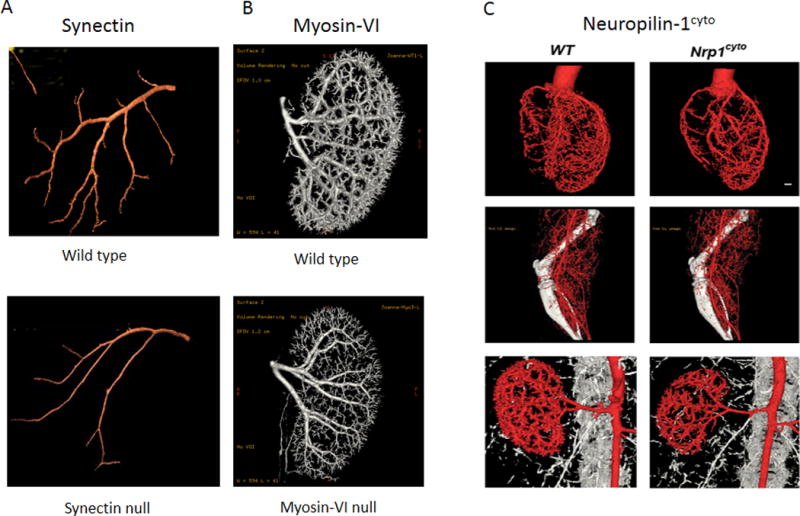
Arteriogenic defects associated with delayed intracellular VEGFR2 trafficking.
A. Micro-CT images of mouse coronary arteries from wild-type (top) and Synectin null (bottom mice. B. Micro-CT of mouse renal arterial circulation from wild type (top) and myosin-VI null (bottom) mice. C. Micro-CT images of mouse heart (top), hindlimb (middle) and renal (bottom) arterial circulations from mice carrying a deletion of the Nrp1 cytoplasmic domain. Adapted from Lanahan et al Dev Cell 2010 and Lanahan et al Dev Cell 2013.
The phosphatase thus becomes a potential target for therapeutic interventions aimed to stimulate arteriogenesis. Indeed, suppression of PTP1b activity restores full ERK activation and normal arteriogenesis in synectin null mice68. Genetically, this has been confirmed by crossing mice with endothelial-specific PTP1b knockout onto the synectin null strain, which fully restores abnormal arteriogenic phenotype in the latter75.
Regulation of ERK activation: Raf1-centered cross-talk
VEGF activates ERK via a VEGFR2-dependent activation of PLCγ that in turn activates the Raf1-MEK-ERK cascade. Despite the apparent simplicity of this pathway, it turned out to be elaborately regulated.
Raf1 activity, which is critical to VEGF-dependent ERK activation, is controlled via a series of phosphorylation events that can affect different parts of the molecule. Activation of MEK involves dephosphorylation of the inhibitory Raf1 Ser259 site and phosphorylation of the activating Ser338 site that result in activation of Raf1 kinase activity and subsequent phosphorylation of MEK102–104 (Fig 3A). The key event is Raf1 Ser259 dephosphorylation as the site is phosphorylated under normal conditions, thereby repressing MEK-ERK activation104. While there is a debate as to the nature of the kinase(s) that phosphorylates Raf1 on this site, recent evidence points to LATS1 (a key member of the Yap/Hippo signaling pathway) although PKA and PKC have been also implicated104, 105 (Fig 3A). Particularly interesting is the suggestion of Yap/Hippo- MAPK crosstalk. Two key factors here are the aforementioned LATS1 phosphorylation of Raf1 Ser259 that makes Raf1 unable to phosphorylate MEK, and high Raf1 affinity for phosphorylated MST2 (Hippo) that reduces its availability to phosphorylate MEK104, 106. The later event is subjected to regulation by Akt that likely explains previously reported Akt/ERK cross-talk (Fig 3B)77, 107.
Figure 3.
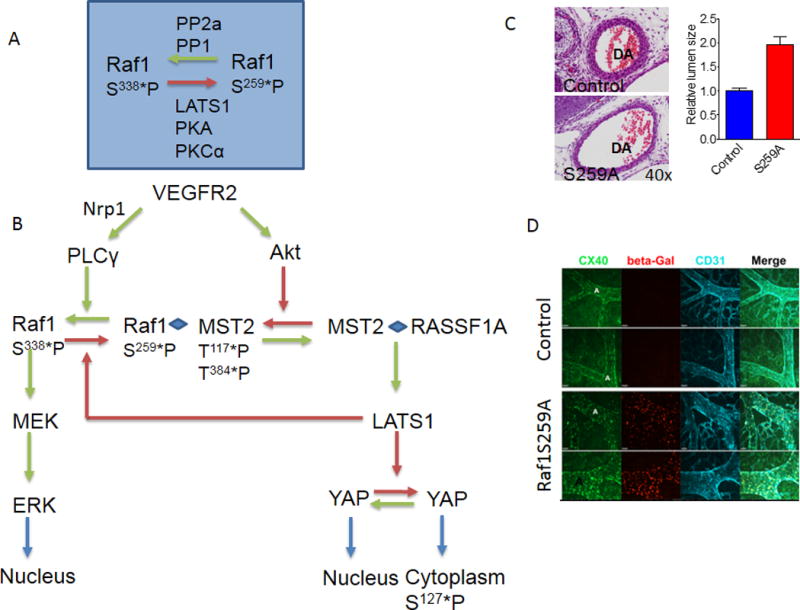
Inhibition-dependent regulation of VEGFR2-driven ERK activation
A. A schematic of Raf1 phosphorylation control. Dephosphorylation of Ser259 allows phosphorylation of Ser338 and activation of Raf1 kinase activity. B. Raf1 regulation of ERK, AKT and MST2 pathways cross-talk. VEGFR2-induced activation of Akt leads to MST2 phosphorylation and promotes formation of Raf1-MST2 complex that maintains Raf1 in an inactive (Ser259-phosphorylated) state. Dephosphorylation of Raf1S259 site shifts MST2 to the RASSF1A complex thereby activating LATS1 that subsequently acts on YAP. At the same time this allows phosphorylation of Raf1Ser338 thereby activating MEK/ERK signaling. Modified, in part, from104.
C. Increase dorsal aorta diameter in Raf1S29A (bottom) compared to WT (top) mice. D. Extra-embryonic vasculature in control (wild type) and Raf1SA259 mouse embryos. Note a marked increase in arterial size. Cx40-Connexin-40. Panels C and D adapted from78
Thus, phosphorylation of Raf1 Ser259 sites simultaneously regulated both Raf/MEK/ERK and Hippo pathways. This Raf1-dependent regulation of the Hippo pathway is particular interesting in the light of recent reports linking Hippo activation to early vascular development108, angiogenesis109 and arteriogenesis110.
In agreement with these studies, endothelial expression of the Raf1 mutant Raf1S259A that is resistant to phosphorylation of this site results in constitutive increase of ERK phosphorylation in the absence of VEGF stimulation. Analysis of endothelial gene expression in this setting demonstrated increased expression of virtually all arterial fate markers. Consistent with these observations, endothelial activation of Raf1S259A expression during early development resulted in excessive development of arterial at the expense of venous circulation78. The arteries in the mutant mice were characterized by excessive branching and larger than normal lumen diameters (Fig 3C,D). Of note, human Raf1 mutations associated with decreased Raf1 Ser259 phosphorylation result in increased ERK activity and are found in a number of “RASopathy”111, 112 conditions including Noonan’s and LEOPARD syndromes113 characterized by presence of arterio-venous malformations. In addition, increased ERK signaling has been implicated in aneurysm development.114, 115
In agreement with the concept of signaling cross-talk discussed above, a number of studies have demonstrated that inhibition of PI3K can be used to activate ERK signaling. Thus, treatment of zebrafish embryos with a PI3K chemical inhibitor resulted in increased ERK activation that lead to excessive formation of arterial vasculature including duplication of the dorsal aorta107. Other demonstrated that inhibition of PI3K activity can reverse decreased VEGF-dependent ERK activation in a number of settings including reduction of VEGFR2 Y1175 phosphorylation by excessive contact with PTP1b in synectin null, myosin-VI and Nrp1cyto knock-in67, 68 and decreased VEGFR2 activation by VEGF in hypercholesterolemic conditions116. This restoration of ERK activation, in turn, resulted in increased arteriogenesis and functional blood flow improvement.
Arteriogenesis drivers
The critical importance of VEGFR2-dependent ERK activation for arterial fate specification and arteriogenesis raises a critical question of the stimulus (or stimuli) inducing this activity. While it is tacitly assumed that VEGF-A is that signal, the direct experimental evidence to that effect is fragmentary. VEGF knockout in mice induces a complete failure of endothelial cell formation that, by itself, cannot be used to deduce VEGF role in arterial fate specification. A decrease in VEGF levels (mouse VEGF hypomorphs, low-dose morpholino knockdown in zebrafish) leads to a partial (regional) loss of arterial marker expression117.
A new level of complexity has arisen with the discovery of anti-angiogenic form of VEGF-A, termed VEGF-A165b, likely generated by translational readthrough118. The isoform has been detected in patients with peripheral artery disease and is capable of reducing blood flow recovery in the hindlimb ischemia model in mice119.
On the other hand, experiments with constitutively active Raf1 (Raf1S259A) demonstrate that it is sufficient to activate ERK without any growth factor input. This form of Raf1 is resistant to phosphorylation on the Ser259 site that renders it inactive. The excessive activation of ERK induced by the introduction of Raf1S259SA leads to increased arteriogenesis and increased arterial lumen diameter (Fig 3C,D)78. These data also suggest that the absence of ERK activation in endothelial cells required for arteriogenesis may well be the consequence of the presence of an inhibitory input (phosphorylation-dependent inactivation of Raf1) rather than the absence of a stimulatory (growth factor) input.
The situation is equally unclear in the case of adult arteriogenesis. Many growth factors, including VEGF-A, FGF2 and HGF among others have been suggested as possible drivers42, 120. In addition, physical forces such as shear stress play an important role4, 79. Shear stress is able to induce VEGFR2 activation in a ligand-independent manner121. Shear stress signal transduction in the endothelium involves the VE-cadherin-VEGFR2-PECAM complex122. Mice with a homozygous disruption of global PECAM expression demonstrate dramatically reduced arterial remodeling and arteriogenesis.59 However, whether this is primarily due to the loss of endothelial PECAM vs. PECAM in other cell types has not been established.
Shear stress has a number of other effects on the endothelium including induction of endothelial VEGF expression123 and activation of NFκB signaling124. One outcome is increased expression of adhesion proteins including ICAM-1 and VCAM125 leading to increased monocyte adhesion to the flow-activated endothelium126. This factor appears critical to shear stress-induced accumulation of monocytes at the sites of arteriogenesis as suppression of NFκB activation in the endothelium leads to a profound reduction in monocytes/macrophages accumulation60.
One of the consequences of shear stress signaling is activation of endothelial NFkB leading to expression of various adhesions molecules such as ICAM-1 and VCAM-160, 127, 128. This, in turn, facilitates accumulation of inflammatory cells, including blood-derived macrophages (Fig 4). Another factor driving macrophage accumulation is angiopoietin-1 that is also produced by activated endothelial cells129. The presence of macrophage at sites of arteriogenesis has been long appreciated4, 93, 94. They are the primary source of VEGF and are also capable of producing other “angiogenic” growth factors including FGF2 and PlGF among others130, 131.
Figure 4. Regulation of arteriogenesis.
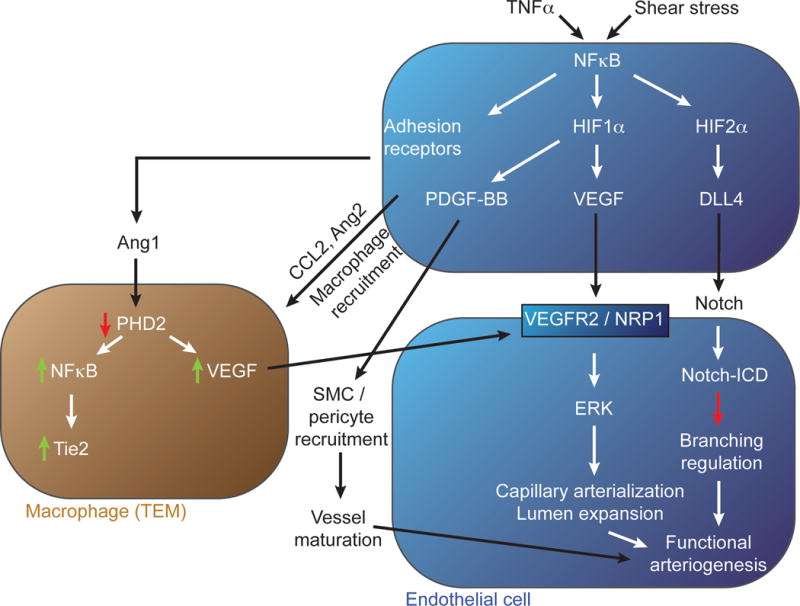
Inflammatory (e.g. TNFα) and mechanical (e.g. shear stress) stimuli initiate arteriogenic signaling in a resting endothelial cell (top, blue). Activation of NFκB signaling by these stimuli leads to increase HIF1α and HIF2α levels, expression of adhesion receptors and production of PDGF-BB, Ang1 and Ang2. Ang2 in turn induces accumulation of a specific macrophage population that, under control of Ang1, reduce their PHD2 levels thereby increasing VEGF production. The macrophage-produced VEGF (and to a lesser extent endothelial-derived VEGF) activate arteriogenic signaling via VEGFR2/Nrp1 complex. HIF2α-induced expression of Dll4 activates Notch signaling in neighboring endothelial cells thereby controlling branching extent. PDGF-BB plays an important role in recruitment of mural cells and maturation of the new arterial network.
Regulation of the extent of arteriogenesis and arterial branching
On par with induction of arterial fate specification and growth of the arterial tree, regulation of the extent of this growth is another key to effective arteriogenesis. Remarkably, virtually nothing is known regarding the signals that control the extent of vascular tree formation. Endothelial Notch activation induced by Dll1 is involved in regulation of arteriogenesis132 while Dll4 binding is considered the principle mechanism controlling the extent of branching133–135. Loss of a single Dll4 allele, or inhibition of Notch signaling, significantly augments the extent of arterial branching and artery-to-artery connections61, 135. Yet, despite the overall increase in arterial density and the number of collateral connections, tissue perfusion is not improved at baseline and is distinctly reduced in adult mice following a major arterial trunk ligation61.
Despite an important role played by Dll4, regulation of its expression remains poorly understood. Among the known regulators are Sox7 and/or Sox18 transcription factors136 and Wnt137 that may act via Sox172. More directly linked to arteriogenesis is the recently described regulation of Dll4 expression by NFκB60. Expression of a dominant-negative IκBα construct in endothelial cells results in a nearly complete suppression of inflammation- or shear-stress-induced NFκB activation thus eliminating signaling input of the major arteriogenesis triggers. This leads to reduction of expression of key molecules involved in arteriogenic response: Dll4, PDGF-BB and endothelial adhesion molecules such as ICAM-1 and VCAM (Fig 4). Reduction in Dll4 levels reduced Notch signaling and hence increased branching, while decreased PDGF-BB levels likely account for reduced maturation of neovasculature due to impaired mural cell recruitment and differentiation. Finally, reduced adhesion molecule expression leads to a profound decrease in recruitment of blood-derived monocytes/macrophages thereby reducing local VEGF concentration60. The resultant phenotype is characterized by vastly excessive, hyperbranched and immature arterial vasculature and a dramatic reduction in tissue perfusion, similarly to the phenotype observed in Dll4 heterozygous mice60, 61.
NFκB directly regulates VEGF-A and PDGF-BB expression via HIF1α138, 139 and Dll4 via HIF2α60. The latter conclusion is supported by the observation of increased arterial branching and decreased tissue perfusion in mice with endothelial HIF2α deletion that resulted in decreased Dll4 expression62.
Summary and practical implications of the new knowledge
The emerging data firmly identifies endothelial ERK signaling as the key driver of arterial fate specification during development as well as developmental and adult arteriogenesis. The regulation of this signaling cascade (Fig 5) is complex and is still not fully understood.
Figure 5. Key arteriogenic events.
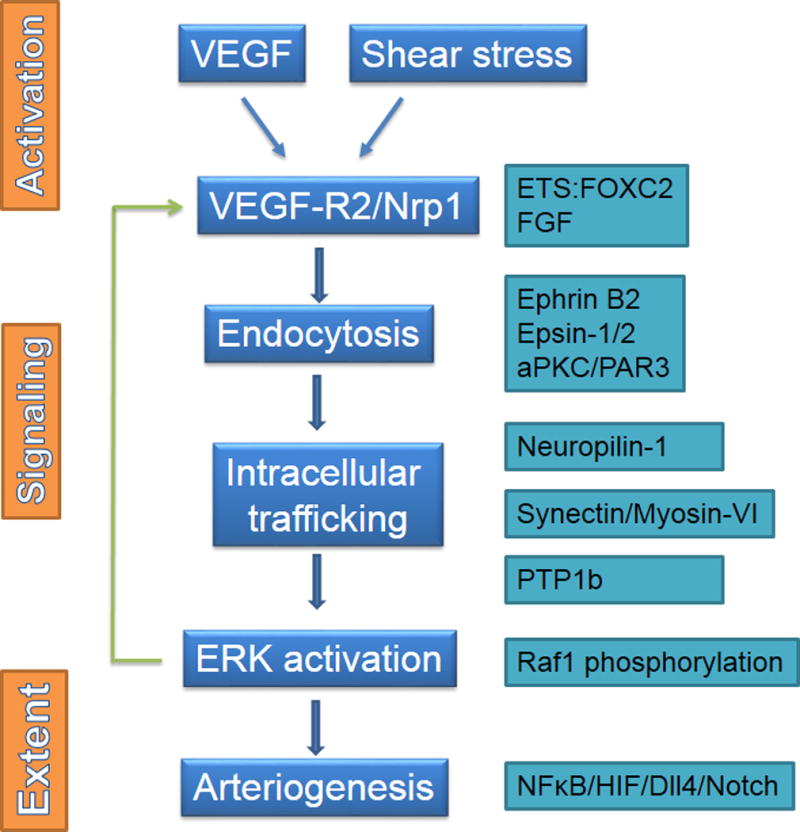
Key arteriogenic events including activation, regulation of signal transduction and arteriogenesis extent. See text for details.
At the level of an endothelial cell, expression of VEGFR2 and Nrp1 is required for arteriogenic signaling. Activation of the VEGFR2/Nrp1 complex, either by a ligand (e.g. VEGF-A) or in a non-ligand-dependent fashion (shear stress and perhaps other physical factors), leads to its endocytosis, a step required for activation of ERK signaling. Initial steps involved in VEGF/BVEGFR2/Nrp1 complex entrance into the cell are poorly understood but involve ephrin B2100, epsins 1 and 2140, 141, and polarity proteins aPKC and PAR3142 among others.
Once internalized, VEGFR2-containing endosomes undergo intracellular trafficking away from PTP1b-rich areas of the cytoplasm, allowing for a full phosphorylation of Y1175 site critical to ERK activation via the PLCγ/Raf1 cascade65, 67, 68, 75, 87, 143, 144.
The state of Raf1 phosphorylation is another critical control point as phosphorylation of its Ser259 site leads to suppression of MEK-dependent ERK activation. Activation ERK signaling either by suppression of Raf1Ser259 phosphorylation or by introduction of constitutive-active MEK/ERK constructs promotes arteriogenesis77, 78, 107.
This scheme suggests the presence of several critical checkpoints (Fig 5). One is the availability of VEGF-A. The other is the ability of the shear stress or another physical stimulus (radial wall stress, for example) to activate VEGFR2 in a ligand-independent manner. Importantly, the relative contributions of these two factors to VEGFR2 activation are not understood.
During embryonic arteriogenesis nerves are likely the key source of VEGF10, 11 while in the case of adult arteriogenesis, it is largely dependent on the presence of blood-derived monocytes/macrophages4, 92. In the case of adult arteriogenesis in disease settings it is doubtful that reduced VEGF levels are ever they key factors in impaired neovascular response. Therefore therapies aimed at providing exogenous VEGF in order to stimulate arteriogenesis are unlikely to be effective. At the same time, non-ligand-dependent activation of VEGFR2 signaling in pathological conditions has not been fully explored.
Regardless of the stimulus, VEGFR2 becomes the central molecule driving all subsequent events. A decline in VEGFR2 levels seen in disease states such as diabetes70 and hypercholesterolemia52, 116 may account for poor arteriogenesis in these settings. In addition, maintenance of endothelial VEGFR2 expression is an active process that requires a continuous FGF signaling input145 acting via ETS and Forkhead transcription factors146, 147.
Impaired VEGFR2 trafficking leads to a decline in its activity due to PTP1b-dependent dephosphorylation of the Y1175 site and inhibition of PTP1b activity appears effective in restoration of ERK activation in certain circumstances68, 75. An equally effective strategy may involve suppression of Akt77, 107 or another kinases capable of Raf1 Ser259 phosphorylation, thereby also leading to ERK activation.
Finally, the effective size of the arterial tree is controlled via NFκB-dependent regulation of Dll4 expression and subsequent Notch signaling activation60–62. Remarkably, in the absence of sufficient Notch activation the increased number of arterial conduits and collaterals leads to ineffective circulation suggesting that an optimal tree size and full maturation of the newly formed vasculature are required for effective tissue perfusion.
In a larger context, endothelial cells have emerged as central regulators of arteriogenesis87. The presence of inflammatory stimuli such as TNFα or shear stress leads to activation of NFκB. This, in turn, leads to three key events: 1) induction of HIF2α expression that then stimulates Dll4 expression and activates Notch signaling; 2) induction of HIF1α expression that leads to increased PDGF-BB and VEGF-A production and 3) expression of adhesion molecules such as VCAM and ICAM-1 that facilitate blood monocyte recruitment that subsequently become the key source of VEGF-A fully activating this cascade (Fig 5).
This emerging paradigm has direct implications with regard to potential therapies, as endothelial ERK signaling is clearly an appealing target. In the past efforts and therapeutic stimulation of neovascularization focused on growth factor (predominantly VEGF-A) therapy. While new data clearly supports the biological importance of this molecule in adult arteriogenesis, clinical trials experience to date equally clearly points to its futility42, 148, 149. The principle cause of failures is likely unresponsiveness of the diseased endothelium to VEGF stimulation that is due to either a reduction in VEGFR2 levels or a decrease in its cellular uptake upon stimulation.
Given this, the ability to stimulate arteriogenesis by interfering with endogenous regulators of VEGFR2 signaling such as PTP1b to enhance signaling of partially endocytosed VEGF/VEGFR2/Nrp1 complexes or to directly activate ERK activation via suppression of Raf1 Ser259 phosphorylation or kinases that phosphorylate this site may prove practically viable.
Acknowledgments
SOURCES OF FUNDING: Supported in part by NIH grants HL053793, HL062289, P01 HL107205 (MS) and R01 HL 111504 (AE)
Nonstandard Abbreviations and Acronyms
- ALK1
activin receptor-like kinase 1
- aPKC
atypical PKC
- AVM
arterio-venous malformations
- CADASIL
cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- CCM
cerebral cavernous malformation
- Dll4
delta-like 4
- EEA1
early endosome antigen 1
- ERK
extracellular receptor kinase
- FGF
fibroblast growth factor
- GDNF
glial-derived neurotrophic factor
- HIF
hypoxia-inducible factor
- HGF
hepatocyte growth factor
- ICAM-1
intercellular adhesion molecule-1
- LATS1
large tumor suppressor kinase 1
- LEOPARD
lentigenes, EKG, ocular hyperteleorism, pulmonary stenosis, abnormal genitalia, retardation of growth, deafness
- MAPK
mitogen-activated protein kinase
- MCP1
monocyte chemoattractant protein 1
- MEK
mitogen extracellular kinase
- MRTF-A
myocardin-related transcription factor-A
- NFkB
nuclear factor kappa B
- NGF
nerve growth factor
- Nrp1
neuropilin-1
- PECAM
platelet-endothelial cell adhesion molecule
- PHD2
prolyl hydroxylase 2
- PI3K
phosphoinositol-3-kinase
- PLCγ
phospholipase gamma
- PlGF
platelet growth factor
- PDGF-BB
platelet-derived growth factor BB
- PTP1b
phosphotyrosine phosphatase 1b
- Raf1
rapidly accelerating fibrosarcoma
- RGS5
regulator of G-protein signaling 5
- Shh
sonic hedgehog
- Sox
Sry-related HMG box
- SMC
smooth muscle cells
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- VCAM-1
vascular cell adhesion protein 1
Footnotes
DISCLOSURES
None.
References
- 1.Faber JE, Chilian WM, Deindl E, van Royen N, Simons M. A brief etymology of the collateral circulation. Arterioscler Thromb Vasc Biol. 2014;34:1854–1859. doi: 10.1161/ATVBAHA.114.303929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morini MF, Dejana E. Transcriptional regulation of arterial differentiation via wnt, sox and notch. Current opinion in hematology. 2014 doi: 10.1097/MOH.0000000000000043. [DOI] [PubMed] [Google Scholar]
- 3.Marcelo KL, Goldie LC, Hirschi KK. Regulation of endothelial cell differentiation and specification. Circulation research. 2013;112:1272–1287. doi: 10.1161/CIRCRESAHA.113.300506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schaper W. Collateral circulation: Past and present. Basic research in cardiology. 2009;104:5–21. doi: 10.1007/s00395-008-0760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng X, Xu C, Smith AO, Stratman AN, Zou Z, Kleaveland B, Yuan L, Didiku C, Sen A, Liu X, Skuli N, Zaslavsky A, Chen M, Cheng L, Davis GE, Kahn ML. Dynamic regulation of the cerebral cavernous malformation pathway controls vascular stability and growth. Developmental cell. 2012;23:342–355. doi: 10.1016/j.devcel.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dejana E, Orsenigo F. Endothelial adherens junctions at a glance. Journal of cell science. 2013;126:2545–2549. doi: 10.1242/jcs.124529. [DOI] [PubMed] [Google Scholar]
- 7.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single vegf allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 8.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the vegf gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 9.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 10.Mukouyama YS, Gerber HP, Ferrara N, Gu C, Anderson DJ. Peripheral nerve-derived vegf promotes arterial differentiation via neuropilin 1-mediated positive feedback. Development. 2005;132:941–952. doi: 10.1242/dev.01675. [DOI] [PubMed] [Google Scholar]
- 11.Mukouyama YS, Shin D, Britsch S, Taniguchi M, Anderson DJ. Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell. 2002;109:693–705. doi: 10.1016/s0092-8674(02)00757-2. [DOI] [PubMed] [Google Scholar]
- 12.Lawson ND, Vogel AM, Weinstein BM. Sonic hedgehog and vascular endothelial growth factor act upstream of the notch pathway during arterial endothelial differentiation. Developmental cell. 2002;3:127–136. doi: 10.1016/s1534-5807(02)00198-3. [DOI] [PubMed] [Google Scholar]
- 13.Kohli V, Schumacher JA, Desai SP, Rehn K, Sumanas S. Arterial and venous progenitors of the major axial vessels originate at distinct locations. Developmental cell. 2013;25:196–206. doi: 10.1016/j.devcel.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seki T, Yun J, Oh SP. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circulation research. 2003;93:682–689. doi: 10.1161/01.RES.0000095246.40391.3B. [DOI] [PubMed] [Google Scholar]
- 15.Shovlin CL. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood reviews. 2010;24:203–219. doi: 10.1016/j.blre.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Somekawa S, Imagawa K, Hayashi H, Sakabe M, Ioka T, Sato GE, Inada K, Iwamoto T, Mori T, Uemura S, Nakagawa O, Saito Y. Tmem100, an alk1 receptor signaling-dependent gene essential for arterial endothelium differentiation and vascular morphogenesis. Proc Natl Acad Sci U S A. 2012;109:12064–12069. doi: 10.1073/pnas.1207210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tual-Chalot S, Mahmoud M, Allinson KR, Redgrave RE, Zhai Z, Oh SP, Fruttiger M, Arthur HM. Endothelial depletion of acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PloS one. 2014;9:e98646. doi: 10.1371/journal.pone.0098646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.David L, Mallet C, Keramidas M, Lamande N, Gasc JM, Dupuis-Girod S, Plauchu H, Feige JJ, Bailly S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circulation research. 2008;102:914–922. doi: 10.1161/CIRCRESAHA.107.165530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of bmp9 and bmp10 as functional activators of the orphan activin receptor-like kinase 1 (alk1) in endothelial cells. Blood. 2007;109:1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- 21.Scharpfenecker M, van Dinther M, Liu Z, van Bezooijen RL, Zhao Q, Pukac L, Lowik CW, ten Dijke P. Bmp-9 signals via alk1 and inhibits bfgf-induced endothelial cell proliferation and vegf-stimulated angiogenesis. Journal of cell science. 2007;120:964–972. doi: 10.1242/jcs.002949. [DOI] [PubMed] [Google Scholar]
- 22.Kim JH, Peacock MR, George SC, Hughes CC. Bmp9 induces ephrinb2 expression in endothelial cells through an alk1-bmprii/actrii-id1/id3-dependent pathway: Implications for hereditary hemorrhagic telangiectasia type ii. Angiogenesis. 2012;15:497–509. doi: 10.1007/s10456-012-9277-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larrivee B, Prahst C, Gordon E, del Toro R, Mathivet T, Duarte A, Simons M, Eichmann A. Alk1 signaling inhibits angiogenesis by cooperating with the notch pathway. Developmental cell. 2012;22:489–500. doi: 10.1016/j.devcel.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindskog H, Kim YH, Jelin EB, Kong Y, Guevara-Gallardo S, Kim TN, Wang RA. Molecular identification of venous progenitors in the dorsal aorta reveals an aortic origin for the cardinal vein in mammals. Development. 2014;141:1120–1128. doi: 10.1242/dev.101808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quillien A, Moore JC, Shin M, Siekmann AF, Smith T, Pan L, Moens CB, Parsons MJ, Lawson ND. Distinct notch signaling outputs pattern the developing arterial system. Development. 2014;141:1544–1552. doi: 10.1242/dev.099986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moyon D, Pardanaud L, Yuan L, Breant C, Eichmann A. Plasticity of endothelial cells during arterial-venous differentiation in the avian embryo. Development. 2001;128:3359–3370. doi: 10.1242/dev.128.17.3359. [DOI] [PubMed] [Google Scholar]
- 27.Herbert SP, Huisken J, Kim TN, Feldman ME, Houseman BT, Wang RA, Shokat KM, Stainier DY. Arterial-venous segregation by selective cell sprouting: An alternative mode of blood vessel formation. Science. 2009;326:294–298. doi: 10.1126/science.1178577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buschmann I, Pries A, Styp-Rekowska B, Hillmeister P, Loufrani L, Henrion D, Shi Y, Duelsner A, Hoefer I, Gatzke N, Wang H, Lehmann K, Ulm L, Ritter Z, Hauff P, Hlushchuk R, Djonov V, van Veen T, le Noble F. Pulsatile shear and gja5 modulate arterial identity and remodeling events during flow-driven arteriogenesis. Development. 2010;137:2187–2196. doi: 10.1242/dev.045351. [DOI] [PubMed] [Google Scholar]
- 29.le Noble F, Moyon D, Pardanaud L, Yuan L, Djonov V, Matthijsen R, Breant C, Fleury V, Eichmann A. Flow regulates arterial-venous differentiation in the chick embryo yolk sac. Development. 2004;131:361–375. doi: 10.1242/dev.00929. [DOI] [PubMed] [Google Scholar]
- 30.Kudo FA, Muto A, Maloney SP, Pimiento JM, Bergaya S, Fitzgerald TN, Westvik TS, Frattini JC, Breuer CK, Cha CH, Nishibe T, Tellides G, Sessa WC, Dardik A. Venous identity is lost but arterial identity is not gained during vein graft adaptation. Arterioscler Thromb Vasc Biol. 2007;27:1562–1571. doi: 10.1161/ATVBAHA.107.143032. [DOI] [PubMed] [Google Scholar]
- 31.Muto A, Yi T, Harrison KD, Davalos A, Fancher TT, Ziegler KR, Feigel A, Kondo Y, Nishibe T, Sessa WC, Dardik A. Eph-b4 prevents venous adaptive remodeling in the adult arterial environment. The Journal of experimental medicine. 2011;208:561–575. doi: 10.1084/jem.20101854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mack CP. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol. 2011;31:1495–1505. doi: 10.1161/ATVBAHA.110.221135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majesky MW, Dong XR, Regan JN, Hoglund VJ. Vascular smooth muscle progenitor cells: Building and repairing blood vessels. Circulation research. 2011;108:365–377. doi: 10.1161/CIRCRESAHA.110.223800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 35.Greif DM, Kumar M, Lighthouse JK, Hum J, An A, Ding L, Red-Horse K, Espinoza FH, Olson L, Offermanns S, Krasnow MA. Radial construction of an arterial wall. Developmental cell. 2012;23:482–493. doi: 10.1016/j.devcel.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fouillade C, Monet-Lepretre M, Baron-Menguy C, Joutel A. Notch signalling in smooth muscle cells during development and disease. Cardiovascular research. 2012;95:138–146. doi: 10.1093/cvr/cvs019. [DOI] [PubMed] [Google Scholar]
- 37.Tang Y, Urs S, Boucher J, Bernaiche T, Venkatesh D, Spicer DB, Vary CP, Liaw L. Notch and transforming growth factor-beta (tgfbeta) signaling pathways cooperatively regulate vascular smooth muscle cell differentiation. The Journal of biological chemistry. 2010;285:17556–17563. doi: 10.1074/jbc.M109.076414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basu S, Srinivasan DK, Yang K, Raina H, Banerjee S, Zhang R, Fisher SA, Proweller A. Notch transcriptional control of vascular smooth muscle regulatory gene expression and function. The Journal of biological chemistry. 2013;288:11191–11202. doi: 10.1074/jbc.M112.442996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Honma Y, Araki T, Gianino S, Bruce A, Heuckeroth R, Johnson E, Milbrandt J. Artemin is a vascular-derived neurotropic factor for developing sympathetic neurons. Neuron. 2002;35:267–282. doi: 10.1016/s0896-6273(02)00774-2. [DOI] [PubMed] [Google Scholar]
- 40.Kuruvilla R, Zweifel LS, Glebova NO, Lonze BE, Valdez G, Ye H, Ginty DD. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of trka trafficking and retrograde signaling. Cell. 2004;118:243–255. doi: 10.1016/j.cell.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 41.Brunet I, Gordon E, Han J, Cristofaro B, Broqueres-You D, Liu C, Bouvree K, Zhang J, del Toro R, Mathivet T, Larrivee B, Jagu J, Pibouin-Fragner L, Pardanaud L, Machado MJ, Kennedy TE, Zhuang Z, Simons M, Levy BI, Tessier-Lavigne M, Grenz A, Eltzschig H, Eichmann A. Netrin-1 controls sympathetic arterial innervation. The Journal of clinical investigation. 2014;124:3230–3240. doi: 10.1172/JCI75181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simons M. Angiogenesis: Where do we stand now? Circulation. 2005;111:1556–1566. doi: 10.1161/01.CIR.0000159345.00591.8F. [DOI] [PubMed] [Google Scholar]
- 43.Limbourg A, Korff T, Napp LC, Schaper W, Drexler H, Limbourg FP. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nature protocols. 2009;4:1737–1746. doi: 10.1038/nprot.2009.185. [DOI] [PubMed] [Google Scholar]
- 44.Helisch A, Wagner S, Khan N, Drinane M, Wolfram S, Heil M, Ziegelhoeffer T, Brandt U, Pearlman JD, Swartz HM, Schaper W. Impact of mouse strain differences in innate hindlimb collateral vasculature. Arterioscler Thromb Vasc Biol. 2006;26:520–526. doi: 10.1161/01.ATV.0000202677.55012.a0. [DOI] [PubMed] [Google Scholar]
- 45.Chalothorn D, Clayton JA, Zhang H, Pomp D, Faber JE. Collateral density, remodeling, and vegf-a expression differ widely between mouse strains. Physiol Genomics. 2007;30:179–191. doi: 10.1152/physiolgenomics.00047.2007. [DOI] [PubMed] [Google Scholar]
- 46.Chalothorn D, Faber JE. Strain-dependent variation in collateral circulatory function in mouse hindlimb. Physiol Genomics. 2010;42:469–479. doi: 10.1152/physiolgenomics.00070.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pohl T, Seiler C, Billinger M, Herren E, Wustmann K, Mehta H, Windecker S, Eberli FR, Meier B. Frequency distribution of collateral flow and factors influencing collateral channel development. Functional collateral channel measurement in 450 patients with coronary artery disease. Journal of the American College of Cardiology. 2001;38:1872–1878. doi: 10.1016/s0735-1097(01)01675-8. [DOI] [PubMed] [Google Scholar]
- 48.Sealock R, Zhang H, Lucitti JL, Moore SM, Faber JE. Congenic fine-mapping identifies a major causal locus for variation in the native collateral circulation and ischemic injury in brain and lower extremity. Circulation research. 2014;114:660–671. doi: 10.1161/CIRCRESAHA.114.302931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Epstein SE, Lassance-Soares RM, Faber JE, Burnett MS. Effects of aging on the collateral circulation, and therapeutic implications. Circulation. 2012;125:3211–3219. doi: 10.1161/CIRCULATIONAHA.111.079038. [DOI] [PubMed] [Google Scholar]
- 50.Ruiter MS, van Golde JM, Schaper NC, Stehouwer CD, Huijberts MS. Diabetes impairs arteriogenesis in the peripheral circulation: Review of molecular mechanisms. Clin Sci (Lond) 2010;119:225–238. doi: 10.1042/CS20100082. [DOI] [PubMed] [Google Scholar]
- 51.Hazarika S, Dokun AO, Li Y, Popel AS, Kontos CD, Annex BH. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: Differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circulation research. 2007;101:948–956. doi: 10.1161/CIRCRESAHA.107.160630. [DOI] [PubMed] [Google Scholar]
- 52.Tirziu D, Moodie KL, Zhuang ZW, Singer K, Helisch A, Dunn JF, Li W, Singh J, Simons M. Delayed arteriogenesis in hypercholesterolemic mice. Circulation. 2005;112:2501–2509. doi: 10.1161/CIRCULATIONAHA.105.542829. [DOI] [PubMed] [Google Scholar]
- 53.Heil M, Ziegelhoeffer T, Wagner S, Fernandez B, Helisch A, Martin S, Tribulova S, Kuziel WA, Bachmann G, Schaper W. Collateral artery growth (arteriogenesis) after experimental arterial occlusion is impaired in mice lacking cc-chemokine receptor-2. Circulation research. 2004;94:671–677. doi: 10.1161/01.RES.0000122041.73808.B5. [DOI] [PubMed] [Google Scholar]
- 54.Sheridan KM, Ferguson MJ, Distasi MR, Witzmann FA, Dalsing MC, Miller SJ, Unthank JL. Impact of genetic background and aging on mesenteric collateral growth capacity in fischer 344, brown norway, and fischer 344 × brown norway hybrid rats. American journal of physiology. Heart and circulatory physiology. 2007;293:H3498–3505. doi: 10.1152/ajpheart.00040.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meier P, Seiler C. The coronary collateral circulation-past, present and future. Current cardiology reviews. 2013 doi: 10.2174/1573403X113099990004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Royen N, Voskuil M, Hoefer I, Jost M, de Graaf S, Hedwig F, Andert JP, Wormhoudt TA, Hua J, Hartmann S, Bode C, Buschmann I, Schaper W, van der Neut R, Piek JJ, Pals ST. Cd44 regulates arteriogenesis in mice and is differentially expressed in patients with poor and good collateralization. Circulation. 2004;109:1647–1652. doi: 10.1161/01.CIR.0000124066.35200.18. [DOI] [PubMed] [Google Scholar]
- 57.Chalothorn D, Zhang H, Smith JE, Edwards JC, Faber JE. Chloride intracellular channel-4 is a determinant of native collateral formation in skeletal muscle and brain. Circulation research. 2009;105:89–98. doi: 10.1161/CIRCRESAHA.109.197145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fang JS, Angelov SN, Simon AM, Burt JM. Cx37 deletion enhances vascular growth and facilitates ischemic limb recovery. American journal of physiology. Heart and circulatory physiology. 2011;301:H1872–1881. doi: 10.1152/ajpheart.00683.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen Z, Rubin J, Tzima E. Role of pecam-1 in arteriogenesis and specification of preexisting collaterals. Circulation research. 2010;107:1355–1363. doi: 10.1161/CIRCRESAHA.110.229955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tirziu D, Jaba IM, Yu P, Larrivee B, Coon BG, Cristofaro B, Zhuang ZW, Lanahan AA, Schwartz MA, Eichmann A, Simons M. Endothelial nuclear factor-kappab-dependent regulation of arteriogenesis and branching. Circulation. 2012;126:2589–2600. doi: 10.1161/CIRCULATIONAHA.112.119321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cristofaro B, Shi Y, Faria M, Suchting S, Leroyer AS, Trindade A, Duarte A, Zovein AC, Iruela-Arispe ML, Nih LR, Kubis N, Henrion D, Loufrani L, Todiras M, Schleifenbaum J, Gollasch M, Zhuang ZW, Simons M, Eichmann A, le Noble F. Dll4-notch signaling determines the formation of native arterial collateral networks and arterial function in mouse ischemia models. Development. 2013;140:1720–1729. doi: 10.1242/dev.092304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skuli N, Majmundar AJ, Krock BL, Mesquita RC, Mathew LK, Quinn ZL, Runge A, Liu L, Kim MN, Liang J, Schenkel S, Yodh AG, Keith B, Simon MC. Endothelial hif-2alpha regulates murine pathological angiogenesis and revascularization processes. The Journal of clinical investigation. 2012;122:1427–1443. doi: 10.1172/JCI57322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arnold C, Feldner A, Pfisterer L, Hodebeck M, Troidl K, Genove G, Wieland T, Hecker M, Korff T. Rgs5 promotes arterial growth during arteriogenesis. EMBO molecular medicine. 2014;6:1075–1089. doi: 10.15252/emmm.201403864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toyota E, Warltier DC, Brock T, Ritman E, Kolz C, O’Malley P, Rocic P, Focardi M, Chilian WM. Vascular endothelial growth factor is required for coronary collateral growth in the rat. Circulation. 2005;112:2108–2113. doi: 10.1161/CIRCULATIONAHA.104.526954. [DOI] [PubMed] [Google Scholar]
- 65.Chittenden TW, Claes F, Lanahan AA, Autiero M, Palac RT, Tkachenko EV, Elfenbein A, Ruiz de Almodovar C, Dedkov E, Tomanek R, Li W, Westmore M, Singh JP, Horowitz A, Mulligan-Kehoe MJ, Moodie KL, Zhuang ZW, Carmeliet P, Simons M. Selective regulation of arterial branching morphogenesis by synectin. Developmental cell. 2006;10:783–795. doi: 10.1016/j.devcel.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 66.Dedkov EI, Thomas MT, Sonka M, Yang F, Chittenden TW, Rhodes JM, Simons M, Ritman EL, Tomanek RJ. Synectin/syndecan-4 regulate coronary arteriolar growth during development. Dev Dyn. 2007;236:2004–2010. doi: 10.1002/dvdy.21201. [DOI] [PubMed] [Google Scholar]
- 67.Lanahan A, Zhang X, Fantin A, Zhuang Z, Rivera-Molina F, Speichinger K, Prahst C, Zhang J, Wang Y, Davis G, Toomre D, Ruhrberg C, Simons M. The neuropilin 1 cytoplasmic domain is required for vegf-a-dependent arteriogenesis. Developmental cell. 2013;25:156–168. doi: 10.1016/j.devcel.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, Carmeliet P, Simons M. Vegf receptor 2 endocytic trafficking regulates arterial morphogenesis. Developmental cell. 2010;18:713–724. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Waltenberger J. Vegf resistance as a molecular basis to explain the angiogenesis paradox in diabetes mellitus. Biochem Soc Trans. 2009;37:1167–1170. doi: 10.1042/BST0371167. [DOI] [PubMed] [Google Scholar]
- 70.Sasso FC, Torella D, Carbonara O, Ellison GM, Torella M, Scardone M, Marra C, Nasti R, Marfella R, Cozzolino D, Indolfi C, Cotrufo M, Torella R, Salvatore T. Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signaling in the myocardium of type 2 diabetic patients with chronic coronary heart disease. Journal of the American College of Cardiology. 2005;46:827–834. doi: 10.1016/j.jacc.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 71.Warren CM, Ziyad S, Briot A, Der A, Iruela-Arispe ML. A ligand-independent vegfr2 signaling pathway limits angiogenic responses in diabetes. Science signaling. 2014;7:ra1. doi: 10.1126/scisignal.2004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437:169–183. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- 73.Shibuya M. Vegfr and type-v rtk activation and signaling. Cold Spring Harbor perspectives in biology. 2013;5:a009092. doi: 10.1101/cshperspect.a009092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M. Essential role of flk-1 (vegf receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci U S A. 2005;102:1076–1081. doi: 10.1073/pnas.0404984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lanahan AA, Lech D, Dubrac A, Zhang J, Zhuang ZW, Eichmann A, Simons M. Ptp1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation. 2014;130:902–909. doi: 10.1161/CIRCULATIONAHA.114.009683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hong CC, Kume T, Peterson RT. Role of crosstalk between phosphatidylinositol 3-kinase and extracellular signal-regulated kinase/mitogen-activated protein kinase pathways in artery-vein specification. Circulation research. 2008;103:573–579. doi: 10.1161/CIRCRESAHA.108.180745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ren B, Deng Y, Mukhopadhyay A, Lanahan AA, Zhuang ZW, Moodie KL, Mulligan-Kehoe MJ, Byzova TV, Peterson RT, Simons M. Erk1/2-akt1 crosstalk regulates arteriogenesis in mice and zebrafish. The Journal of clinical investigation. 2010;120:1217–1228. doi: 10.1172/JCI39837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deng Y, Larrivee B, Zhuang ZW, Atri D, Moraes F, Prahst C, Eichmann A, Simons M. Endothelial raf1/erk activation regulates arterial morphogenesis. Blood. 2013;121:3988–3996. doi: 10.1182/blood-2012-12-474601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pipp F, Boehm S, Cai WJ, Adili F, Ziegler B, Karanovic G, Ritter R, Balzer J, Scheler C, Schaper W, Schmitz-Rixen T. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler Thromb Vasc Biol. 2004;24:1664–1668. doi: 10.1161/01.ATV.0000138028.14390.e4. [DOI] [PubMed] [Google Scholar]
- 80.Heil M, Eitenmuller I, Schmitz-Rixen T, Schaper W. Arteriogenesis versus angiogenesis: Similarities and differences. Journal of cellular and molecular medicine. 2006;10:45–55. doi: 10.1111/j.1582-4934.2006.tb00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hoefer IE, den Adel B, Daemen MJ. Biomechanical factors as triggers of vascular growth. Cardiovascular research. 2013;99:276–283. doi: 10.1093/cvr/cvt089. [DOI] [PubMed] [Google Scholar]
- 82.Chilian WM, Penn MS, Pung YF, Dong F, Mayorga M, Ohanyan V, Logan S, Yin L. Coronary collateral growth–back to the future. Journal of molecular and cellular cardiology. 2012;52:905–911. doi: 10.1016/j.yjmcc.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu J, deMuinck ED, Zhuang Z, Drinane M, Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B, Sessa WC. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci U S A. 2005;102:10999–11004. doi: 10.1073/pnas.0501444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dai X, Faber JE. Endothelial nitric oxide synthase deficiency causes collateral vessel rarefaction and impairs activation of a cell cycle gene network during arteriogenesis. Circulation research. 2010;106:1870–1881. doi: 10.1161/CIRCRESAHA.109.212746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ahn GO, Seita J, Hong BJ, Kim YE, Bok S, Lee CJ, Kim KS, Lee JC, Leeper NJ, Cooke JP, Kim HJ, Kim IH, Weissman IL, Brown JM. Transcriptional activation of hypoxia-inducible factor-1 (hif-1) in myeloid cells promotes angiogenesis through vegf and s100a8. Proc Natl Acad Sci U S A. 2014;111:2698–2703. doi: 10.1073/pnas.1320243111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mac Gabhann F, Peirce SM. Collateral capillary arterialization following arteriolar ligation in murine skeletal muscle. Microcirculation. 2010;17:333–347. doi: 10.1111/j.1549-8719.2010.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moraes F, Paye J, Mac Gabhann F, Zhuang ZW, Zhang J, Lanahan AA, Simons M. Endothelial cell-dependent regulation of arteriogenesis. Circulation research. 2013;113:1076–1086. doi: 10.1161/CIRCRESAHA.113.301340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hinkel R, Trenkwalder T, Petersen B, Husada W, Gesenhues F, Lee S, Hannappel E, Bock-Marquette I, Theisen D, Leitner L, Boekstegers P, Cierniewski C, Muller OJ, le Noble F, Adams RH, Weinl C, Nordheim A, Reichart B, Weber C, Olson E, Posern G, Deindl E, Niemann H, Kupatt C. Mrtf-a controls vessel growth and maturation by increasing the expression of ccn1 and ccn2. Nature communications. 2014;5:3970. doi: 10.1038/ncomms4970. [DOI] [PubMed] [Google Scholar]
- 89.Deindl E, Hoefer IE, Fernandez B, Barancik M, Heil M, Strniskova M, Schaper W. Involvement of the fibroblast growth factor system in adaptive and chemokine-induced arteriogenesis. Circulation research. 2003;92:561–568. doi: 10.1161/01.RES.0000061181.80065.7D. [DOI] [PubMed] [Google Scholar]
- 90.Murakami M, Simons M. Fibroblast growth factor regulation of neovascularization. Current opinion in hematology. 2008;15:215–220. doi: 10.1097/MOH.0b013e3282f97d98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morrison AR, Yarovinsky TO, Young BD, Moraes F, Ross TD, Generi N, Zhang J, Zhuang ZW, Sinusas AJ, Pardi R, Schwartz MA, Simons M, Bender JR. Chemokine-coupled β2 integrin-induced macrophage rac2-myosin iia interaction regulates vegf-a mrna stability and arteriogenesis. The Journal of experimental medicine. 2014 doi: 10.1084/jem.20132130. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML, Finisguerra V, Deschoemaeker S, Bruyere F, Wenes M, Hamm A, Serneels J, Magat J, Bhattacharyya T, Anisimov A, Jordan BF, Alitalo K, Maxwell P, Gallez B, Zhuang ZW, Saito Y, Simons M, De Palma M, Mazzone M. Macrophage skewing by phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature. 2011;479:122–126. doi: 10.1038/nature10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Heil M, Ziegelhoeffer T, Pipp F, Kostin S, Martin S, Clauss M, Schaper W. Blood monocyte concentration is critical for enhancement of collateral artery growth. American journal of physiology. Heart and circulatory physiology. 2002;283:H2411–2419. doi: 10.1152/ajpheart.01098.2001. [DOI] [PubMed] [Google Scholar]
- 94.van Royen N, Hoefer I, Buschmann I, Kostin S, Voskuil M, Bode C, Schaper W, Piek JJ. Effects of local mcp-1 protein therapy on the development of the collateral circulation and atherosclerosis in watanabe hyperlipidemic rabbits. Cardiovascular research. 2003;57:178–185. doi: 10.1016/s0008-6363(02)00615-6. [DOI] [PubMed] [Google Scholar]
- 95.Voskuil M, van Royen N, Hoefer IE, Seidler R, Guth BD, Bode C, Schaper W, Piek JJ, Buschmann IR. Modulation of collateral artery growth in a porcine hindlimb ligation model using mcp-1. American journal of physiology. Heart and circulatory physiology. 2003;284:H1422–1428. doi: 10.1152/ajpheart.00506.2002. [DOI] [PubMed] [Google Scholar]
- 96.Fisher CE, Michael L, Barnett MW, Davies JA. Erk map kinase regulates branching morphogenesis in the developing mouse kidney. Development. 2001;128:4329–4338. doi: 10.1242/dev.128.21.4329. [DOI] [PubMed] [Google Scholar]
- 97.Horowitz A, Simons M. Branching morphogenesis. Circulation research. 2008;103:784–795. doi: 10.1161/CIRCRESAHA.108.181818. [DOI] [PubMed] [Google Scholar]
- 98.Rosario M, Birchmeier W. How to make tubes: Signaling by the met receptor tyrosine kinase. Trends in cell biology. 2003;13:328–335. doi: 10.1016/s0962-8924(03)00104-1. [DOI] [PubMed] [Google Scholar]
- 99.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls vegfr-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174:593–604. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, Acker-Palmer A. Ephrin-b2 regulates vegfr2 function in developmental and tumour angiogenesis. Nature. 2010;465:487–491. doi: 10.1038/nature08995. [DOI] [PubMed] [Google Scholar]
- 101.Simons M. An inside view: Vegf receptor trafficking and signaling. Physiology (Bethesda) 2012;27:213–222. doi: 10.1152/physiol.00016.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Osborne JK, Zaganjor E, Cobb MH. Signal control through raf: In sickness and in health. Cell Res. 2012;22:14–22. doi: 10.1038/cr.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A, Kolch W. Raf family kinases: Old dogs have learned new tricks. Genes & cancer. 2011;2:232–260. doi: 10.1177/1947601911407323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Romano D, Nguyen LK, Matallanas D, Halasz M, Doherty C, Kholodenko BN, Kolch W. Protein interaction switches coordinate raf-1 and mst2/hippo signalling. Nat Cell Biol. 2014;16:673–684. doi: 10.1038/ncb2986. [DOI] [PubMed] [Google Scholar]
- 105.Fey D, Croucher DR, Kolch W, Kholodenko BN. Crosstalk and signaling switches in mitogen-activated protein kinase cascades. Frontiers in physiology. 2012;3:355. doi: 10.3389/fphys.2012.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.O’Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase mst2 in suppression of apoptosis by the proto-oncogene product raf-1. Science. 2004;306:2267–2270. doi: 10.1126/science.1103233. [DOI] [PubMed] [Google Scholar]
- 107.Hong CC, Peterson QP, Hong JY, Peterson RT. Artery/vein specification is governed by opposing phosphatidylinositol-3 kinase and map kinase/erk signaling. Curr Biol. 2006;16:1366–1372. doi: 10.1016/j.cub.2006.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nejigane S, Takahashi S, Haramoto Y, Michiue T, Asashima M. Hippo signaling components, mst1 and mst2, act as a switch between self-renewal and differentiation in xenopus hematopoietic and endothelial progenitors. The International journal of developmental biology. 2013;57:407–414. doi: 10.1387/ijdb.130010st. [DOI] [PubMed] [Google Scholar]
- 109.Dai X, She P, Chi F, Feng Y, Liu H, Jin D, Zhao Y, Guo X, Jiang D, Guan KL, Zhong TP, Zhao B. Phosphorylation of angiomotin by lats1/2 kinases inhibits f-actin binding, cell migration, and angiogenesis. The Journal of biological chemistry. 2013;288:34041–34051. doi: 10.1074/jbc.M113.518019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou J. An emerging role for hippo-yap signaling in cardiovascular development. Journal of biomedical research. 2014;28:251–254. doi: 10.7555/JBR.28.20140020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tidyman WE, Rauen KA. The rasopathies: Developmental syndromes of ras/mapk pathway dysregulation. Current opinion in genetics & development. 2009;19:230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Atri D, Larrivee B, Eichmann A, Simons M. Endothelial signaling and the molecular basis of arteriovenous malformation. Cellular and molecular life sciences : CMLS. 2013 doi: 10.1007/s00018-013-1475-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kobayashi T, Aoki Y, Niihori T, Cave H, Verloes A, Okamoto N, Kawame H, Fujiwara I, Takada F, Ohata T, Sakazume S, Ando T, Nakagawa N, Lapunzina P, Meneses AG, Gillessen-Kaesbach G, Wieczorek D, Kurosawa K, Mizuno S, Ohashi H, David A, Philip N, Guliyeva A, Narumi Y, Kure S, Tsuchiya S, Matsubara Y. Molecular and clinical analysis of raf1 in noonan syndrome and related disorders: Dephosphorylation of serine 259 as the essential mechanism for mutant activation. Human mutation. 2010;31:284–294. doi: 10.1002/humu.21187. [DOI] [PubMed] [Google Scholar]
- 114.Ghosh A, DiMusto PD, Ehrlichman LK, Sadiq O, McEvoy B, Futchko JS, Henke PK, Eliason JL, Upchurch GR., Jr The role of extracellular signal-related kinase during abdominal aortic aneurysm formation. Journal of the American College of Surgeons. 2012;215:668–680. doi: 10.1016/j.jamcollsurg.2012.06.414. e661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin ii type 2 receptor signaling attenuates aortic aneurysm in mice through erk antagonism. Science. 2011;332:361–365. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jin F, Hagemann N, Brockmeier U, Schafer ST, Zechariah A, Hermann DM. Ldl attenuates vegf-induced angiogenesis via mechanisms involving vegfr2 internalization and degradation following endosome-trans-golgi network trafficking. Angiogenesis. 2013;16:625–637. doi: 10.1007/s10456-013-9340-2. [DOI] [PubMed] [Google Scholar]
- 117.Swift MR, Weinstein BM. Arterial-venous specification during development. Circulation research. 2009;104:576–588. doi: 10.1161/CIRCRESAHA.108.188805. [DOI] [PubMed] [Google Scholar]
- 118.Eswarappa SM, Potdar AA, Koch WJ, Fan Y, Vasu K, Lindner D, Willard B, Graham LM, DiCorleto PE, Fox PL. Programmed translational readthrough generates antiangiogenic vegf-ax. Cell. 2014;157:1605–1618. doi: 10.1016/j.cell.2014.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kikuchi R, Nakamura K, MacLauchlan S, Ngo DT, Shimizu I, Fuster JJ, Katanasaka Y, Yoshida S, Qiu Y, Yamaguchi TP, Matsushita T, Murohara T, Gokce N, Bates DO, Hamburg NM, Walsh K. An antiangiogenic isoform of vegf-a contributes to impaired vascularization in peripheral artery disease. Nat Med. 2014;20:1464–1471. doi: 10.1038/nm.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 121.Chiu JJ, Lee PL, Chen CN, Lee CI, Chang SF, Chen LJ, Lien SC, Ko YC, Usami S, Chien S. Shear stress increases icam-1 and decreases vcam-1 and e-selectin expressions induced by tumor necrosis factor-[alpha] in endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:73–79. doi: 10.1161/01.ATV.0000106321.63667.24. [DOI] [PubMed] [Google Scholar]
- 122.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 123.dela Paz NG, Walshe TE, Leach LL, Saint-Geniez M, D’Amore PA. Role of shear-stress-induced vegf expression in endothelial cell survival. Journal of cell science. 2012;125:831–843. doi: 10.1242/jcs.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ganguli A, Persson L, Palmer IR, Evans I, Yang L, Smallwood R, Black R, Qwarnstrom EE. Distinct nf-kappab regulation by shear stress through ras-dependent ikappabalpha oscillations: Real-time analysis of flow-mediated activation in live cells. Circulation research. 2005;96:626–634. doi: 10.1161/01.RES.0000160435.83210.95. [DOI] [PubMed] [Google Scholar]
- 125.Hay DC, Beers C, Cameron V, Thomson L, Flitney FW, Hay RT. Activation of nf-kappab nuclear transcription factor by flow in human endothelial cells. Biochimica et biophysica acta. 2003;1642:33–44. doi: 10.1016/s0167-4889(03)00084-3. [DOI] [PubMed] [Google Scholar]
- 126.Weber KS, Draude G, Erl W, de Martin R, Weber C. Monocyte arrest and transmigration on inflamed endothelium in shear flow is inhibited by adenovirus-mediated gene transfer of ikappab-alpha. Blood. 1999;93:3685–3693. [PubMed] [Google Scholar]
- 127.Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10:53–62. doi: 10.1038/nrm2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Conway DE, Schwartz MA. Flow-dependent cellular mechanotransduction in atherosclerosis. Journal of cell science. 2013;126:5101–5109. doi: 10.1242/jcs.138313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hamm A, Veschini L, Takeda Y, Costa S, Delamarre E, Squadrito ML, Henze AT, Wenes M, Serneels J, Pucci F, Roncal C, Anisimov A, Alitalo K, De Palma M, Mazzone M. Phd2 regulates arteriogenic macrophages through tie2 signalling. EMBO molecular medicine. 2013;5:843–857. doi: 10.1002/emmm.201302695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pipp F, Heil M, Issbrucker K, Ziegelhoeffer T, Martin S, van den Heuvel J, Weich H, Fernandez B, Golomb G, Carmeliet P, Schaper W, Clauss M. Vegfr-1-selective vegf homologue plgf is arteriogenic: Evidence for a monocyte-mediated mechanism. Circulation research. 2003;92:378–385. doi: 10.1161/01.RES.0000057997.77714.72. [DOI] [PubMed] [Google Scholar]
- 131.Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory m2, but not pro-inflammatory m1 macrophages promote angiogenesis in vivo. Angiogenesis. 2014;17:109–118. doi: 10.1007/s10456-013-9381-6. [DOI] [PubMed] [Google Scholar]
- 132.Sorensen I, Adams RH, Gossler A. Dll1-mediated notch activation regulates endothelial identity in mouse fetal arteries. Blood. 2009;113:5680–5688. doi: 10.1182/blood-2008-08-174508. [DOI] [PubMed] [Google Scholar]
- 133.Suchting S, Freitas C, le Noble F, Benedito R, Breant C, Duarte A, Eichmann A. The notch ligand delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci U S A. 2007;104:3225–3230. doi: 10.1073/pnas.0611177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Roca C, Adams RH. Regulation of vascular morphogenesis by notch signaling. Genes Dev. 2007;21:2511–2524. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- 135.Scehnet JS, Jiang W, Kumar SR, Krasnoperov V, Trindade A, Benedito R, Djokovic D, Borges C, Ley EJ, Duarte A, Gill PS. Inhibition of dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood. 2007;109:4753–4760. doi: 10.1182/blood-2006-12-063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sacilotto N, Monteiro R, Fritzsche M, Becker PW, Sanchez-Del-Campo L, Liu K, Pinheiro P, Ratnayaka I, Davies B, Goding CR, Patient R, Bou-Gharios G, De Val S. Analysis of dll4 regulation reveals a combinatorial role for sox and notch in arterial development. Proc Natl Acad Sci U S A. 2013;110:11893–11898. doi: 10.1073/pnas.1300805110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Corada M, Nyqvist D, Orsenigo F, Caprini A, Giampietro C, Taketo MM, Iruela-Arispe ML, Adams RH, Dejana E. The wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating dll4/notch signaling. Developmental cell. 2010;18:938–949. doi: 10.1016/j.devcel.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Taylor CT, Cummins EP. The role of nf-kappab in hypoxia-induced gene expression. Annals of the New York Academy of Sciences. 2009;1177:178–184. doi: 10.1111/j.1749-6632.2009.05024.x. [DOI] [PubMed] [Google Scholar]
- 139.van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by nf-kappab. Biochem J. 2008;412:477–484. doi: 10.1042/BJ20080476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pasula S, Cai X, Dong Y, Messa M, McManus J, Chang B, Liu X, Zhu H, Mansat RS, Yoon SJ, Hahn S, Keeling J, Saunders D, Ko G, Knight J, Newton G, Luscinskas F, Sun X, Towner R, Lupu F, Xia L, Cremona O, De Camilli P, Min W, Chen H. Endothelial epsin deficiency decreases tumor growth by enhancing vegf signaling. The Journal of clinical investigation. 2012;122:4424–4438. doi: 10.1172/JCI64537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tessneer KL, Pasula S, Cai X, Dong Y, McManus J, Liu X, Yu L, Hahn S, Chang B, Chen Y, Griffin C, Xia L, Adams RH, Chen H. Genetic reduction of vascular endothelial growth factor receptor 2 rescues aberrant angiogenesis caused by epsin deficiency. Arterioscler Thromb Vasc Biol. 2014;34:331–337. doi: 10.1161/ATVBAHA.113.302586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nakayama A, Nakayama M, Turner CJ, Hoing S, Lepore JJ, Adams RH. Ephrin-b2 controls pdgfrbeta internalization and signaling. Genes Dev. 2013;27:2576–2589. doi: 10.1101/gad.224089.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Prahst C, Heroult M, Lanahan AA, Uziel N, Kessler O, Shraga-Heled N, Simons M, Neufeld G, Augustin HG. Neuropilin-1-vegfr-2 complexing requires the pdz-binding domain of neuropilin-1. The Journal of biological chemistry. 2008;283:25110–25114. doi: 10.1074/jbc.C800137200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gelfand MV, Hagan N, Tata A, Oh WJ, Lacoste B, Kang KT, Kopycinska J, Bischoff J, Wang JH, Gu C. Neuropilin-1 functions as a vegfr2 co-receptor to guide developmental angiogenesis independent of ligand binding. eLife. 2014:3. doi: 10.7554/eLife.03720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Murakami M, Nguyen LT, Hatanaka K, Schachterle W, Chen PY, Zhuang ZW, Black BL, Simons M. Fgf-dependent regulation of vegf receptor 2 expression in mice. The Journal of clinical investigation. 2011;121:2668–2678. doi: 10.1172/JCI44762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.De Val S, Chi NC, Meadows SM, Minovitsky S, Anderson JP, Harris IS, Ehlers ML, Agarwal P, Visel A, Xu SM, Pennacchio LA, Dubchak I, Krieg PA, Stainier DY, Black BL. Combinatorial regulation of endothelial gene expression by ets and forkhead transcription factors. Cell. 2008;135:1053–1064. doi: 10.1016/j.cell.2008.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wythe JD, Dang LT, Devine WP, Boudreau E, Artap ST, He D, Schachterle W, Stainier DY, Oettgen P, Black BL, Bruneau BG, Fish JE. Ets factors regulate vegf-dependent arterial specification. Developmental cell. 2013;26:45–58. doi: 10.1016/j.devcel.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Silvestre JS, Smadja DM, Levy BI. Postischemic revascularization: From cellular and molecular mechanisms to clinical applications. Physiological reviews. 2013;93:1743–1802. doi: 10.1152/physrev.00006.2013. [DOI] [PubMed] [Google Scholar]
- 149.Ouma GO, Zafrir B, Mohler ER, 3rd, Flugelman MY. Therapeutic angiogenesis in critical limb ischemia. Angiology. 2013;64:466–480. doi: 10.1177/0003319712464514. [DOI] [PMC free article] [PubMed] [Google Scholar]