

Abstract
To evaluate the growth-inhibitory properties of the potent multi-kinase antagonist Regorafenib (Fluoro-Sorafenib), which was synthesized as a more potent Sorafenib, a Raf inhibitor and to determine whether similar mechanisms were involved, human hepatoma cell lines were grown in the presence or absence of Regorafanib and examined for growth inhibition. Western blots were performed for Raf targets, for apoptosis and autophagy. Regorafenib inhibited growth of human Hep3B, PLC/PRF/5 and HepG2 cells in a concentration- and time-dependent manner. Multiple signaling pathways were altered, including MAP kinases phospho-ERK and phospho-JNK and its target phospho-c-Jun. There was evidence for apoptosis by FACS, cleavage of caspases and increased Bax levels; as well as induction of autophagy, as judged by increased Beclin-1 and LC3 (II) levels. Prolonged drug exposure resulted in cell quiescence. Full growth recovery occurred after drug removal, unlike with doxorubicin chemotherapy. Regorafenib is a potent inhibitor of cell growth. Cells surviving Regorafenib treatment remain viable, but quiescent and capable of regrowth following drug removal. The reversibility of tumor cell growth suppression after drug removal may have clinical implications.
Keywords: growth, apoptosis, reversibility, hepatoma, Regorafenib
INTRODUCTION
Sorafenib is a multi-kinase growth inhibitor, which was designed as a Raf antagonist (Dumas, 2000) and inhibits cell growth of many tumor types (Wilhelm et al., 2004, 2008; Adnane et al., 2006; Liu et al., 2006). It is the first FDA-approved oral therapy for hepatoma or HCC (hepatocellular carcinoma), following a phase III international, multi-center, 602 patient randomized, placebo-controlled trial (Llovet et al., 2006). The Sorafenib treatment arm median overall survival in this trial was 10.7 months and showed a 10 weeks advantage over placebo treated control patients, and the tumor objective partial response rate was 2%. The results from an Asian trial showed lower median overall survival of 6.5 months (Cheng et al., 2009). It has now become a standard of care for non-surgically treatable HCC patients. More effective therapy is still needed to increase responses and enhance survival. There are many new kinase-inhibitory and cell cycle modulating agents now in clinical trials for HCC (Huynh et al., 2010; Wörns and Galle, 2010) including Brivanib, Linifanib, Bevacizumab, Erlotinib, Sirolimus, Bortezomib (www.clinicaltrials.org), as well as a Sorafenib analog, Fluoro-Sorafenib (Regorafenib) (Wilhelm et al., 2011). We report here that Regorafenib inhibits hepatoma cell growth and induces cell quiescence. Unlike chemotherapy actions, these effects are reversible after drug removal from the cell cultures.
MATERIALS and METHODS
Drugs and cells
Regorafenib was a gift of Bayer Corp (West Haven, CT USA), Doxorubicin (Dox) was purchased from Pfizer (Rome, Italy). Regorafenib was dissolved in dimethylsulfoxide (DMSO) at a stock concentration of 10 mM, and then diluted to appropriate concentrations with Dulbecco’s Modified Eagle’s Medium (DMEM) and DMSO at 0.1% (v/v). The cells untreated (DMSO at 0.1% in DMEM) were used as controls. Dox was dissolved at stock concentration of 3 mM in 0.9% of NaCl and then dissolved to appropriate concentrations. The cells treated with saline solvent in DMEM were used as controls.
Hep3B, HepG2 and PLC/PRF/5 human hepatoma cells were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). Though these cell lines are most widely used as in vitro model for pharmacological studies, each has specific genetic and biochemical features. HepG2 cells show a wild-type p53 gene, PLC cells a mutant p53 and Hep3B are a p53-null cells (McClendon et al., 2011) and each cell line shows also a different expression of drug-metabolizing enzymes (Guo et al., 2011).
We have focused our attention on Hep3B cells in order: (i) to compare with prior papers on Sorafenib and hepatoma cells; and (ii) to study the quiescence status induced by Regorafenib in a p53-independent metabolic system.
Cell culture
Cells were cultured in DMEM in monolayer culture, and supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100μg/ml streptomycin, and incubated at 37°C in a humidified atmosphere containing 5% CO2 in air. At confluence, the growing cells were harvested by means of trypsinization and serially sub-cultured with a 1:4 split ratio.
All cell culture components were purchased from Sigma- Aldrich (Milan, Italy).
Regorafenib treatment
Each cell line was seeded at 0.3×105 cells/2ml of DMEM containing 10% FBS in 35 mm tissue culture dishes (Corning Costar Co., Milan, Italy). The cells were incubated for 24 h to allow attachment, and then the medium was replaced by fresh culture medium containing Regorafenib at increasing concentrations (1 μM, 2.5 μM, 5 μM, 7.5 μM and 10 μM). In these experimental conditions, the cells were allowed to grow for 72 or 96 h.
Time-course experiments on Hep3B cells were performed with 7.5 μM of Regorafenib at short (15, 60, 180 min.), middle (24, 48, 72 and 96 h) or long times (up to seven days). When the cells were treated for long times the drug was replaced with a fresh one. Each experiment included a control with the equivalent concentration of DMSO (solvent control) as the one used for adding Regorafenib. Each experiment was performed in triplicate and repeated 3 times. Subsequent analyses were performed at specific Regorafenib concentrations and incubation times.
Assessment of cell proliferation
After cells had been cultured for 72 h with different drug concentrations, the proliferative response was estimated by colorimetric 3-(4,5 di-methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test and 5-bromo-2’-deoxy-uridine Labeling and Detection (BrdU) kit (Roche Diagnostics GmbH, Mannheim, Germany). To determine cell growth by the colorimetric test, MTT stock solution (5mg/ml in medium) was added to each dish at a volume of one-tenth the original culture volume and incubated for 2 h at 37°C in humidified CO2. At the end of incubation period, the medium was removed, and the blue formazan crystals were solubilized with acidic isopropanol (0.1 N HCl in absolute isopropanol). MTT conversion to formazan by metabolically viable cells was monitored using a spectrophotometer at an optical density of 570 nm. The rate of DNA synthesis after drug treatment was evaluated by measuring BrdU incorporation into newly synthesized DNA, following the supplier’s instructions, using BrdU kit. The trypan blue exclusion assay was used to evaluate cell viability. Cell viability was expressed as the percentage of cell survival compared with that of control (DMSO alone).
Each experiment was performed in triplicate and repeated 3 times.
Recovery/Reversibility
To study the recovery in cell proliferation after drug withdrawal, Hep3B cells were treated with Regorafenib 5 or 7.5 μM for 3-7 days, then the medium was removed and replaced with fresh medium without drug. The rate of cell recovery was evaluated by MTT test at different subsequent time points.
Doxorubicin treatment at 0.01or 0.05 or 0.1 μM was used as positive control to study the apoptotic process.
FACS analysis for apoptosis
The FITC-annexin V kit (Immunotech SAS, Beckman Coulter Company, Marseille Cedex, France) was used to detect apoptosis as specified by the supplier. Briefly, 1×106 cells treated with various Regorafenib concentrations for 48 h were harvested and washed with PBS. Cells were resuspended in binding buffer and then incubated for 5 min at room temperature in the dark after 5 μl AnnexinV-FITC and 10μl 7-amino actinomycin D (7AAD) intercalates into DNA. Intact cells were discriminated from apoptotic cells. Data was acquired on BD FACSort flow cytometer using CellQuest software (BD, San Jose, CA) and analyzed by Kaluza software (Beckman Coulter, Brea, CA).
Western blotting analysis
To evaluate Regorafenib effects at different time after cell treatment, cellular MAPK, apoptosis and autophagy pathways were analyzed by Western blotting analysis. After harvest, the cells were washed twice with cold phosphate-buffered saline (PBS) and then lysed in RIPA buffer (Sigma-Aldrich, Milan; Italy). The total soluble proteins were subjected to Western blotting analysis. Protein concentration was determined using Micro BCA Protein Assay kit (Thermo Scientific, Rockford, IL USA) as described in manufacture manual. Equal amounts of protein (20 μg) were resolved on SDS–PAGE and transferred to polyvinyldifluoride (PVDF) filters (BioRad, Milan; Italy). The filters were blocked 5% (w/v) nonfat dry milk for 2 h at room temperature and then probed with primary antibody overnight at 4°C. The primary antibodies were directed against the following proteins: ERK and phospho-ERK (42-44 kDa), JNK and phospho-JNK (46-54 kDa), c-Jun and phospho-c-Jun (39 kDa), and β-actin (42 kDa) (Cell Signaling, Beverly, MA, USA); cleaved caspase-3 (11 kDa), -7 (10 kDa), -8 (10 kDa), -9 (10 kDa), MAP LC3-II (14-16 kDa), Bcl-XL (30 kDa), phospho-survivin (16 kDa), Bcl-2 (26 kDa), Bax (23 kDa) and Beclin-1 (60 kDa) (Santa Cruz Biotechnology, Santa Cruz, CA; USA). After three washes, incubation was followed by reaction with horseradish peroxidase-conjugated specific secondary antibody for 1 h at room temperature and the bands were visualized by enhanced chemiluminescence detection system (Bio-Rad). The primary antibodies against proteins with molecular weight similar to β-actin (42 kDa), used as internal control, were removed by stripping buffer (Thermo Fisher Scientific, MA, USA) and filter was re-probed with the antibody against β-actin.
Densitometric values were normalized using β-actin and analyzed with ChemiDoc XRS apparatus and software (BioRad).
For each time point, the variations in protein phosphorylation levels were analyzed by comparing the single target phosphorylated or not phosphorylated protein with the respective control (protein in untreated cells), arbitrarily considered as unit.
Statistical analysis
All experiments were done in triplicate. Data are presented as mean ± standard deviation (S.D.) and evaluated by one-way ANOVA followed by Dunnett’s post test used for multiple comparisons. P values of <0.05 were considered statistically significant.
RESULTS
Hepatoma cell growth inhibition
Hep3B, PLC/PRF/5 and HepG2 human hepatoma cell lines were treated in log phase growth with various concentrations of Regorafenib or with solvent alone. Regorafenib caused a concentration-dependent decrease in Hep3B cell growth, having an IC50 of 5 μM when compared to untreated controls (Fig. 1A). PLC/PRF/5 cells were similar in their responses to Hep3B cells, but HepG2 cells were more sensitive and had approximate IC50 of 1 μM. Further experiments were done with Hep3B cells using 7.5 μM Regorafenib that should read 50% in cell viability. This is an optimal condition for studying the cell quiescence status and reversibility. In order to examine the minimum time of exposure of Hep3B cells to drug for the growth-inhibition, cells were treated with drug for increasing time windows, with all cells harvested after 96 h in culture. After only 24 h, the drug exposure showed minimal effect on subsequent growth (Fig. 1B). However, major inhibition of cell growth was seen after 48 h of cell exposure to the drug.
Fig. 1. Hepatoma cell growth inhibition.
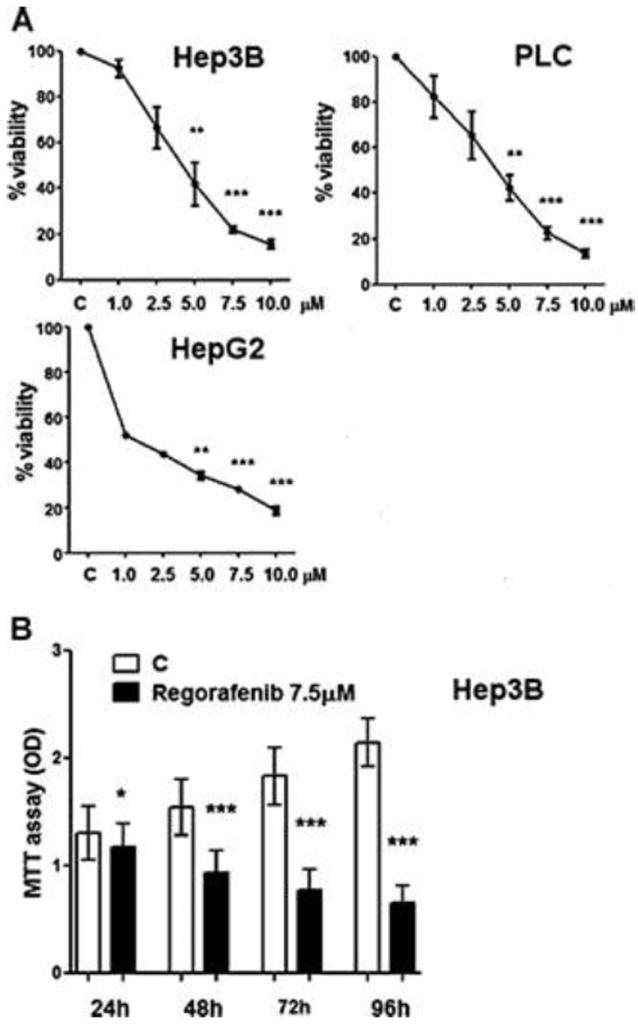
A. Hep3B, PLC/PRF/5, HepG2 human hepatoma cell lines were cultured in presence of Regorafenib at various concentrations or in solvent alone. At 72 h, cells were harvested for MTT assay. The relative cell viability (%) was calculated as a percentage relative to the untreated control cells.: OD of treated cells / OD of control cells × 100 %.
B. Hep3B cells were cultured in presence of 7.5 μM Regorafenib for either 0-24, 0-48 or 0-72 h and the medium was then changed to drug-free medium or for 0-96 h. All cells were harvested at 96 h and the viability was evaluated by the MTT assay.
ERK signaling effects
Hep3B cells, after few hours of drug exposure, showed a rapid and profound decrease in phospho-ERK, but not in total ERK (Fig. 2A). The decrease in phospho-ERK levels was transient and the levels later returned to baseline levels, despite continued growth arrest and continued presence in the culture of Regorafenib (Figs. 2A and 2B).
Fig. 2. Regorafenib-mediated cell signaling.
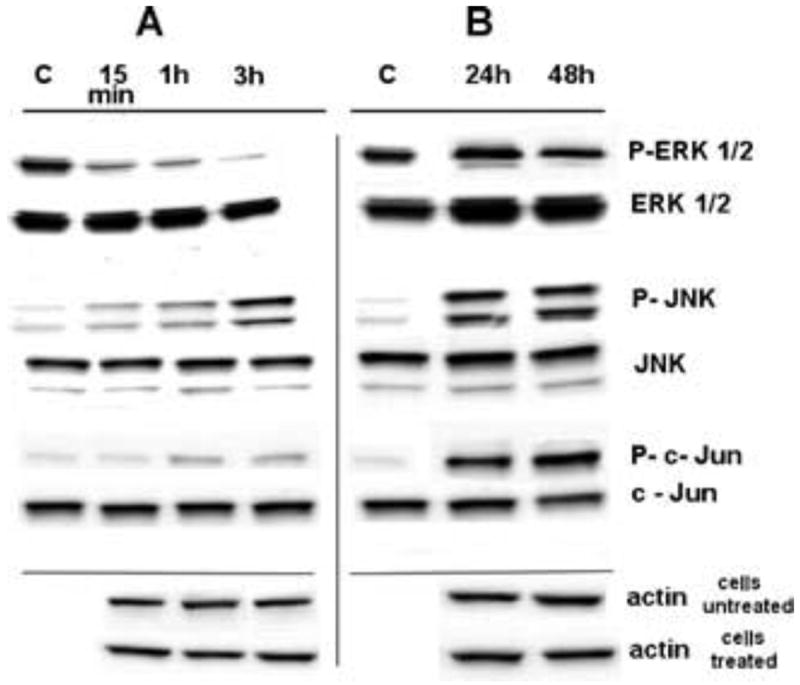
A. Regorafenib-mediated early cell signaling. A representative time-course of Regorafenib-induced phosphorylation of phospho-ERK, phospho-JNK and phospho-c-Jun in Hep3B cells treated continuously in culture with 7.5 μM Regorafenib for 15, 60 or 180 min. C, controls (solvent alone, drug-untreated cells).
B. Same treatments as Fig. 2A, but cells were treated for and then harvested at 24 or 48 h.
The β-actin, used as loading control, in treated and untreated cells show similar relative optical density at each time point. This reflects the accurate amount of loaded target proteins.
Apoptosis and autophagy
Exposure of Hep3B cells to increasing concentrations of Regorafenib resulted in apoptosis, as judged by FACS analysis, with up to 66% of cells becoming apoptotic at the highest drug concentrations which were used (Fig. 3A). However, at the IC50 concentration of 5 μM, only 13% of the cells were found to be apoptotic. Evidence to support the presence of apoptosis included the cleavage of caspase 3 and its activating caspases 8 and 9, with an induction of pro-apoptotic Bax and a decrease in anti-apoptotic Bcl-2 but not in Bcl-xL (Figs. 3B, 3C and Tab. 1 for quantitation), and a decrease in anti-apoptotic survivin levels. There was also an induction of phospho-JNK, an apoptotic mediator, but not total JNK. In addition, Regorafenib subsequently increased the levels of phospho-c-Jun, a JNK target, but not total c-Jun. The autophagy investigation showed that Beclin-1 was induced by Regorafenib action (Fig. 3C), with subsequent increase in the levels of LC3 (II) and change in the LC3 (I/II) ratio (Fig. 3C). Since it is known that Bcl-2, anti-apoptotic proteins, inhibit Beclin 1-dependent autophagy, here, we show that Bcl-2 levels decreased as Beclin-1 levels increased.
Fig. 3. Apoptosis and autophagy.
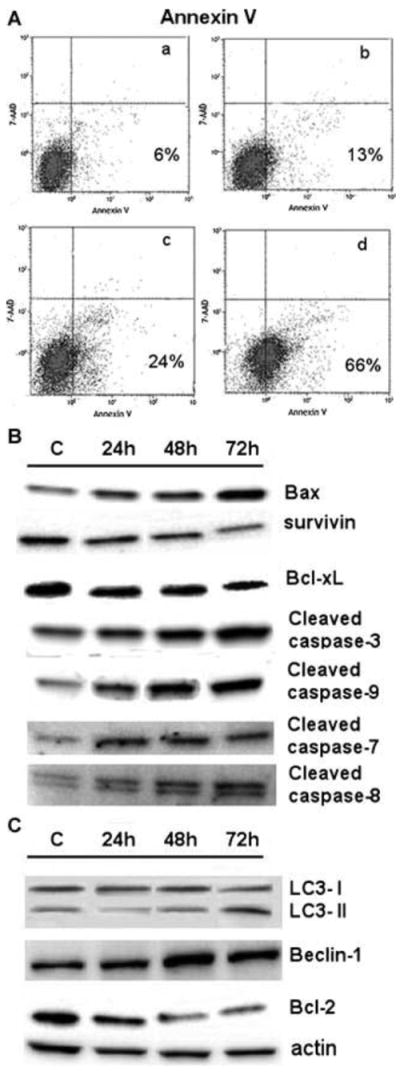
A. FACS measurement for apoptosis after culture of Hep3B cells for 48 h with 2.5 (a), 5 (b), 7.5 (c) or 10 (d) μM Regorafenib.
B and C. Western blotting of lysates from Hep3B cells treated with 7.5 μM Regorafenib for 24, 48 or 72 h and probed for apoptosis (B) and autophagy (C) markers.
C, controls (solvent alone, drug-untreated cells). The β-actin protein was used as loading control.
Table 1.
| MAPK pathway | 15 min | 1h | 3h | 24h | 48h |
|---|---|---|---|---|---|
| P-ERK 1/2 | 0.30 ± 0.01 *** | 0.22 ± 0.01 *** | 0.05 ± 0.01 *** | 1.36 ± 0.06 ** | 0.94 ± 0.04 |
| P-JNK | 2.58 ± 0.67 * | 4.57 ± 1.13 ** | 10.38 ± 0.85 *** | 13.40 ± 0.93 *** | 14.38 ± 0.45 *** |
| P-c-Jun | 0.71 ± 0.14 * | 2.37 ± 0.68 * | 2.56 ± 0.81 * | 12.71 ± 0.93 *** | 16.63 ± 1.83 *** |
| Apoptosis | 24h | 48h | 72h |
|---|---|---|---|
| Bax | 1.75 ± 0.17 ** | 2.15 ± 0.30 ** | 3.33 ± 0.48 ** |
| Bcl-2 | 0.71 ± 0.07 * | 0.34 ± 0.02 *** | 0.27 ± 0.01 *** |
| Bcl-XL | 0.81 ± 0.10 | 0.73 ± 0.06 * | 0.51 ± 0.02 ** |
| Survivin | 0.85 ± 0.03 | 0.69 ± 0.02 ** | 0.39 ± 0.04 *** |
| Cleaved caspase-3 | 2.03 ± 0.03 | 2.48 ± 0.10 ** | 3.61 ± 0.05 *** |
| Cleaved caspase-7 | 3.30 ± 0.15 *** | 3.00 ± 0.23 *** | 3.05 ± 0.16 *** |
| Cleaved caspase-8 | 1.34 ± 0.06 ** | 1.59 ± 0.09 ** | 2.04 ± 0.06 *** |
| Cleaved caspase-9 | 1.95 ± 0.05 *** | 3.04 ± 0.04 *** | 3.11 ± 0.05 *** |
| Autophagy | |||
| Beclin-1 | 1.77 ± 0.02 * | 2.67 ± 0.07 *** | 2.10 ± 0.02 * |
| LC 3 - II | 0.46 ± 0.06 ** | 0.97 ± 0.07 | 2.36 ± 0.03 *** |
The values represent the fold change of each phosphorylated protein from Regorafenib-treated Hep3B cells, compared to respective protein controls (solvent alone, drug-untreated cells). Controls were given an arbitrary value of unity.
All experiments were performed in triplicate and the results are expressed as mean ± SD. The statistical analysis was performed with one-way analysis of variance (ANOVA) and Dunnett’s post test. The asterisks indicate statistically different differences:
for P < 0.05;
for P < 0.001;
for P < 0.0001.
Quiescence and recovery
We tried and failed to make Regorafanib-resistant Hep3B cells by sub-culturing our cells in Regorafenib at increasing concentrations over several weeks. However, we noted that after many days of incubation with Regorafenib 5 μM, the cells remained healthy, did not grow, die nor detach. We considered the possibility that we might be observing a drug-induced cell quiescence. We examined cell growth and viability (MTT), DNA synthesis (BrdU), total cell protein amounts and cell viability (trypan blue dye exclusion test) over several days of drug exposure, but found little loss in cell number or viability (Tab. 2). The MTT results were almost unchanged between 48 and 72 h of drug exposure and cell viability loss as judged by trypan blue dye exclusion test was minimally different between treated cells and controls at 72 h. BrdU measurements were greater in control cells than in cells treated with drug at 72 h, as was total cellular protein, consistent with continued growth of control cells. Late signaling processes were examined at 24 and 48 h of continuous Regorafenib exposure and we found that phospho-ERK had increased back to baseline levels as noted above, unlike phospho-JNK levels which stayed altered (Fig. 2B), despite cell quiescence and the continued presence of Regorafenib.
Table 2.
Hep 3B quiescence.
| C | Regorafenib | |||
|---|---|---|---|---|
| 5.0 μM | 7.5 μM | |||
| MTT | 24h | 0.43 ± 0.10 | 0.34 ± 0.08 | 0.33 ± 0.07 ** |
| 48h | 0.87 ± 0.20 | 0.65 ± 0.19 * | 0.46 ± 0.11 *** | |
| 72h | 1.13 ± 0.06 | 0.64 ± 0.03 * | 0.45 ± 0.10 *** | |
|
| ||||
| Trypan blue | 24h | 0 | 3.10 ± 2.52 | 4.66 ± 2.83 |
| 48h | 1.50 ± 1.10 | 3.83 ± 2.80 | 2.23 ± 0.85 | |
| 72h | 1.63 ± 0.91 | 2.90 ± 2.69 | 1.76 ± 0.65 | |
|
| ||||
| Total proteins | 24h | 3.28 ± 0.12 | 3.24 ± 0.20 | 3.17 ± 1.18 |
| 48h | 6.20 ± 0.44 | 3.44 ± 0.22 | 4.15 ± 0.57 | |
| 72h | 9.05 ± 1.14 | 4.83 ± 2.79 | 4.98 ± 0.60 | |
|
| ||||
| BrdU | 24h | 1 | 1.10 ± 0.05 | 0.70 ± 0.03 |
| 48h | 1 | 0.80 ± 0.09 * | 0.60 ± 0.08 * | |
| 72h | 1 | 0.70 ± 0.004 * | 0.40 ± 0.005 *** | |
The values (mean ± SD) are expressed as: OD unit for MTT; percentage of dead cells for trypan blue staining; μg/μl for total proteins; treated/control cell ratio of OD units for BrdU. C = control
The statistical analysis was performed with one-way analysis of variance (ANOVA) and Dunnett’s post test for each time considered (24, 48 and 72h). The asterisks indicate statistically different differences:
for P < 0.05;
for P < 0.001;
for P < 0.0001.
Multi-kinase antagonists were designed to inhibit cell growth, rather than causing cell death and necrosis, unlike chemotherapy. We therefore investigated whether the cell growth inhibition was reversible. Cells were treated with Regorafenib till quiescence and the medium was then re-placed with drug-free medium (t0 or time zero). Over the subsequent days, normal cell growth resumed (Figs. 4A). However, parallel dishes of cells that were treated with Doxorubicin (Fig. 4B) that caused cell growth inhibition, but not resumption of growth or reversibility after drug removal, when used under identical conditions as Regorafenib.
Fig. 4. Reversibility of drug effects on cell growth.
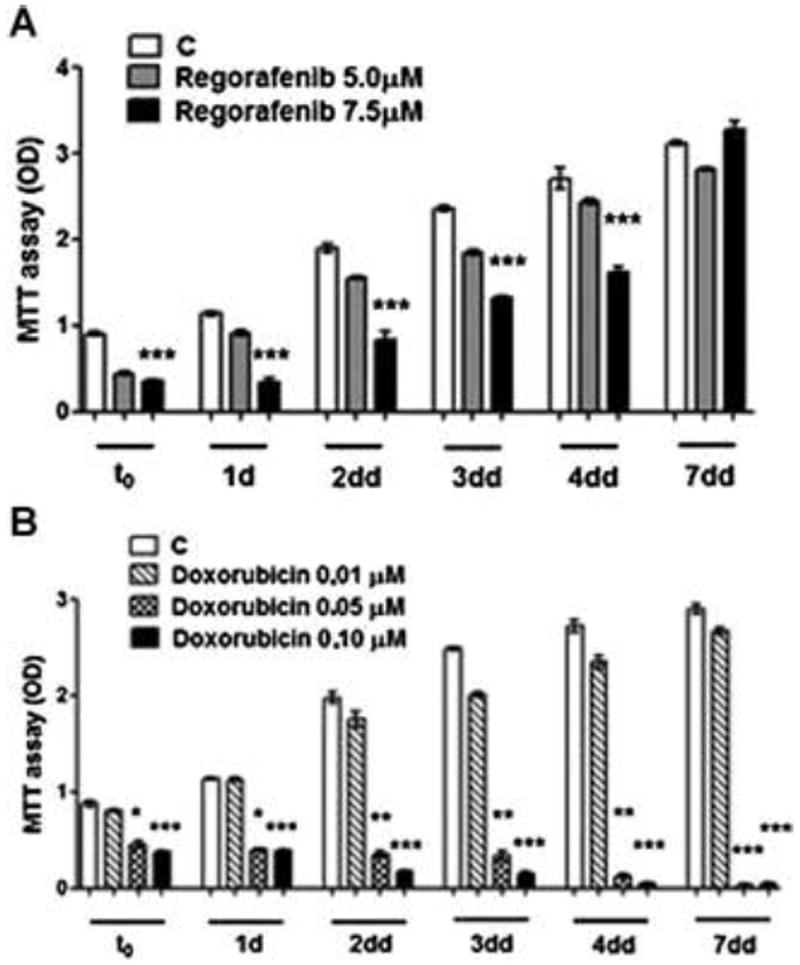
Hep3B cells were treated either with Regorafenib (4A) or Doxorubicin (4B) at different concentrations for 72 h in culture and the medium was then removed (t0). The cells were washed with fresh medium and then cultured for the indicated further days in drug-free medium. The cells were harvested and the viability has been evaluated by the MTT assay.
DISCUSSION
Sorafenib has become a first line of therapy for non-surgically treatable clinical hepatoma. It prolongs survival by about 10 weeks and can be taken orally. It thus represents a promising step for building upon. Many clinical trials are now in progress to assess the possibility of combining it either with other cell cycle inhibitors that target other pathways, or with chemotherapy. The observation in the phase III SHARP trial that it could extend survival with minimal tumor shrinkage (Llovet et al., 2008), suggests the possibility that survival can be enhanced without the tumor necrosis and cell death that is associated with cytotoxic chemotherapy, and may thus have significance for the development of pre-clinical models and endpoints for the evaluation of new anti-cancer compounds. Regorafenib has been recently introduced into clinical trials, as a potentially more potent analog for hepatoma treatment, and has a slightly different spectrum of kinase inhibitory profile than Sorafenib (Wilhelm et al., 2011). We found that Regorafenib inhibited growth of three hepatoma cell lines in the low μM range. We evaluated the time of exposure of cells to drug for growth inhibition, and found that more than 24 h was necessary (Fig.1), suggestive of the involvement of other actions for growth inhibition.
Although this class of agent was designed to inhibit the Raf/MEK/ERK pathway, these changes in early signaling events would seem to be insufficient to fully explain its growth-inhibitory actions, given the length of exposure time of cells to drug that was needed. We also found an increase in phospho-JNK, an apoptotic mediator, but not total JNK, which is not characteristic of published Sorafenib effects in hepatomas (Wei et al., 2010). Unlike ERK, the JNK changes only started around 3 h and were pronounced at 24 and 48 h. Increased levels of phospho-c-Jun, a target of phospho-JNK, were also seen at late time points. Given that JNK is involved in both apoptosis and autophagy, we looked for evidence of them both.
FACS analysis showed an induction in the percentage of apoptotic cells, in a drug concentration-dependent manner, supported by Western blot changes of decreased phospho-survivin and enhanced caspase cleavage, increased Bax and decreased anti-apoptotic Bcl-xL and Bcl-2 (Fig. 3B and Tab. 1 for quantitation). Since the level of apoptosis did not seem to be a sufficient explanation for the profound growth inhibition, given that at the IC50, only 13% of cells were apoptotic, we also looked for evidence for autophagy, which is increasingly recognized as being important in drug mediated cancer cell growth inhibition (Kimmelman, 2011) with several mechanisms (Codogno et al., 2011). We found an increase in Beclin-1 and a decrease in Beclin-1 controlling Bcl-2 and a later increase in LC3-II levels (Fig. 3C), supportive of autophagy. Bcl-2 has a controlling function for both apoptosis and autophagy and inhibits Beclin-1 (Pattingre et al., 2005; Guo et al., 2002).
We endeavored to make Regorafenib-resistant cells. However, once drug was removed, the cells re-grew similarly to controls. This was tested by inhibiting cell growth and then removing the drug, replacing it with drug-free medium and measuring subsequent growth (Fig. 4). The cells appeared healthy after several days of incubation with drug, with little loss of viability (Tab. 2). The cells recovered and grew as they did prior to drug exposure. This was different from cells treated in an identical fashion with doxorubicin, a cytotoxic chemotherapic commonly used to treat hepatoma patients (Gish et al., 2007; Yeo et al., 2005), and then released from drug exposure, which did not recover. This is consistent with the action of the Regorafenib class of compound as a cell signaling antagonist, and not a cytotoxin, although cells treated at 10 μM concentration eventually died. If these results have a reflection in patients, they might suggest the need for long-term treatment, with concern for patient ‘drug holidays’ that often result from clinical toxicities and the need to temporarily withhold drug. The apparent health of the cells after several days in stable, viable, non-growing conditions in culture (Tab. 2), is different from our experience with chemotherapy. We found that phospho-ERK signaling at late time points had increased to baseline levels, despite continued cellular quiescence. Selective MEK inhibition by Sorafenib was reported to decrease phospho-ERK in hepatoma cells, yet increased phospho-MEK, suggesting likely feedback loops and multiple interacting factors in this pathway (Liu et al., 2006). Presumably additional changes are keeping the cells quiescent, that were not measured in these experiments, likely involving other effectors, such as p38 (Sosa, 2011) or matrix (Barkan et al., 2010).
The dormancy is thought to be a major clinical problem and may explain late cancer recurrence after definitive primary therapies in patients. Given the clinical significance of tumor dormancy and the limited experimental data on its mechanisms (Sosa et al., 2011; Goss and Chambers, 2010), quiescence induced by Regorafenib might provide a useful, if simple model for its study. Furthermore, Raf-independent mechanisms have been recently found for Sorafenib-mediated apoptosis, including changes in Mcl-1 (Matsuda and Fukumoto, 2011), indicating that both Sorafenib and Regorafenib likely alter multiple growth-regulatory pathways.
Abbreviations
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun NH2-terminal kinase
- FACS
fluorescence activated cell sorting
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- BrdU
5-bromo-2’-deoxy-uridine
Footnotes
Conflict of Interest Statement: None declared.
Contributor Information
Brian I. Carr, Email: brianicarr@hotmail.com.
Aldo Cavallini, Email: cavaldo@libero.it.
Catia Lippolis, Email: catia.lippolis20@libero.it.
Rosalba D’Alessandro, Email: dalessandro.rosalba@libero.it.
Caterina Messa, Email: caterina.messa@irccsdebellis.it.
Maria Grazia Refolo, Email: mgrefolo@libero.it.
Angela Tafaro, Email: angelatafaro@libero.it.
References
- Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597–612. doi: 10.1016/S0076-6879(05)07047-3. [DOI] [PubMed] [Google Scholar]
- Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer. 2010;46:1181–1188. doi: 10.1016/j.ejca.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, Liu J, Wang J, Tak WY, Pan H, Burock K, Zou J, Voliotis D, Guan Z. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2011;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- Dumas J, Sibley R, Riedl B, Monahan MK, Lee W, Lowinger TB, Redman AM, Johnson JS, Kingery-Wood J, Scott WJ, Smith RA, Bobko M, Schoenleber R, Ranges GE, Housley TJ, Bhargava A, Wilhelm SM, Shrikhande A. Discovery of a new class of p38 kinase inhibitors. Bioorg Med Chem Lett. 2000;10:2047–2050. doi: 10.1016/s0960-894x(00)00270-5. [DOI] [PubMed] [Google Scholar]
- Gish RG, Porta C, Lazar L, Ruff P, Feld R, Croitoru A, Feun L, Jeziorski K, Leighton J, Gallo J, Kennealey GT. Phase III randomized controlled trial comparing the survival of patients with unresectable hepatocellular carcinoma treated with nolatrexed or doxorubicin. J Clin Oncol. 2007;25:3069–3075. doi: 10.1200/JCO.2006.08.4046. [DOI] [PubMed] [Google Scholar]
- Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat Rev Cancer. 2010;10:871–877. doi: 10.1038/nrc2933. [DOI] [PubMed] [Google Scholar]
- Guo L, Dial S, Shi L, Branham W, Liu J, Fang JL, Green B, Deng H, Kaput J, Ning B. Similarities and differences in the expression of drug-metabolizing enzymes between human hepatic cell lines and primary human hepatocytes. Drug Metab Dispos. 2011;39:528–533. doi: 10.1124/dmd.110.035873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo XZ, Shao XD, Liu MP, Xu JH, Ren LN, Zhao JJ, Li HY, Wang D. Effect of bax, bcl-2 and bcl-xL on regulating apoptosis in tissues of normal liver and hepatocellular carcinoma. World J Gastroenterol. 2002;8:1059–1062. doi: 10.3748/wjg.v8.i6.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh H. Molecularly targeted therapy in hepatocellular carcinoma. Biochem Pharmacol. 2010;80:550–560. doi: 10.1016/j.bcp.2010.03.034. [DOI] [PubMed] [Google Scholar]
- Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2010;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Häussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Fukumoto M. Sorafenib: complexities of Raf-dependent and Raf-independent signaling are now unveiled. Med Mol Morphol. 2011;44:183–189. doi: 10.1007/s00795-011-0558-z. [DOI] [PubMed] [Google Scholar]
- McClendon AK, Dean JL, ERTEL A, Fu Z, Rivadeneira DB, Reed CA, Bourgo RJ, Witkiewicz A, Addya S, Mayhew CN, Grimes HL, Fortina P, Knudsen ES. RB and p53 cooperate to prevent liver tumorigenesis in response to tissue damage. Gastroenterology. 2011;141:1439–1450. doi: 10.1053/j.gastro.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Sosa MS, Avivar-Valderas A, Bragado P, Wen HC, Aguirre-Ghiso JA. ERK1/2 and p38α/β signaling in tumor cell quiescence: opportunities to control dormant residual disease. Clin Cancer Res. 2011;17:5850–5857. doi: 10.1158/1078-0432.CCR-10-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G, Wang M, Hyslop T, Wang Z, Carr BI. Vitamin K enhancement of sorafenib-mediated HCC cell growth inhibition in vitro and in vivo. Int J Cancer. 2010;127:2949–2958. doi: 10.1002/ijc.25498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129–3140. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, Thierauch KH, Zopf D. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129:245–255. doi: 10.1002/ijc.25864. [DOI] [PubMed] [Google Scholar]
- Wörns MA, Galle PR. Novel inhibitors in development for hepatocellular carcinoma. Expert Opin Investig Drugs. 2010;19:615–629. doi: 10.1517/13543781003767418. [DOI] [PubMed] [Google Scholar]
- Yeo W, Mok TS, Zee B, Leung TW, Lai PB, Lau WY, Koh J, Mo FK, Yu SC, Chan AT, Hui P, Ma B, Lam KC, Ho WM, Wong HT, Tang A, Johnson PJ. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97:1532–1538. doi: 10.1093/jnci/dji315. [DOI] [PubMed] [Google Scholar]