

Abstract
Purpose of review
Iron homeostasis and erythropoiesis regulate each other to ensure optimal delivery of oxygen and iron to cells and tissues. Defining the mechanisms of this crosstalk is important for understanding the pathogenesis of common conditions associated with disordered iron metabolism and erythropoiesis.
Recent findings
Stress erythropoiesis causes suppression of hepcidin to increase iron availability for hemoglobin synthesis. The erythroid hormone erythroferrone (ERFE) was identified as the mediator of this process. ERFE and additional candidates (TWSG1 and GDF15) may also mediate hepcidin suppression in ineffective erythropoiesis. Several mechanisms by which iron regulates erythropoiesis were also recently identified. Iron deficiency suppresses erythropoietin production via the IRP1–HIF2α axis to prevent excessive iron usage by erythropoiesis during systemic iron restriction. Iron restriction also directly impairs erythroid maturation by inhibiting aconitase, and this can be reversed by the administration of the aconitase product isocitrate. Another novel target is GDF11, which is thought to autoinhibit erythroid maturation. GDF11 traps show promising pharmacologic activity in models of both ineffective erythropoiesis and iron-restricted anemia.
Summary
This review summarizes exciting advances in understanding the mechanisms of iron and erythropoietic regulation, and development of novel therapeutic tools for disorders resulting from dysregulation of iron metabolism or erythropoiesis.
Keywords: erythroferrone, erythropoiesis, GDF11, hepcidin, iron
INTRODUCTION
Iron is an essential trace element for nearly all organisms. At the systemic level, iron is required as a component of heme, including in hemoglobin and myoglobin, which are vital for the delivery and storage of oxygen. Iron is also required for cell viability as a constituent of iron-containing proteins that are involved in DNA synthesis and repair, energy metabolism and cell proliferation [1]. Conversely, iron overload can lead to cellular toxicity via iron-generated oxyradicals and peroxidation of lipid membranes [2]. To maintain the appropriate balance, organisms have developed complex cellular and systemic iron-regulatory mechanisms.
Basal systemic iron homeostasis is maintained primarily by the recycling of iron from senescent erythrocytes with a small contribution from dietary iron absorption. During times of stress erythropoiesis, iron consumption by the bone marrow can increase up to 10-fold [3]. Thus, rapidly acting compensatory mechanisms have evolved to increase dietary iron absorption (up to 20-fold [4]) and mobilization of iron from stores, to increase iron availability for hemoglobin synthesis. Recent years have yielded exciting glimpses into the complex regulatory mechanisms linking iron and erythropoiesis, and these advances will be important for clinical applications in the near future.
HEPCIDIN AND IRON REGULATION
The primary regulator of systemic iron homeostasis, and thus iron availability for erythropoiesis, is hepcidin, a 25-amino-acid peptide hormone that is produced by hepatocytes [5]. Hepcidin acts by binding to ferroportin, the sole known cellular iron exporter, and causing its internalization and degradation within lysosomes [6,7]. As ferroportin is expressed on duodenal enterocytes, macrophages and hepatocytes, hepcidin controls the flow of iron from gut absorption, recycling of senescent erythrocytes, and cellular iron stores. Injecting a single dose of synthetic hepcidin into mice caused a rapid and dramatic drop in serum iron [8]. Prolonged hepcidin overexpression in transgenic mice resulted in ironrestricted erythropoiesis and iron deficiency [9]. Conversely, inactivation of the hepcidin gene in mice caused severe iron overload [10,11], and mutations in the hepcidin gene in humans are associated with juvenile hemochromatosis, a particularly severe form of genetic iron overload [12].
Hepcidin production is homeostatically regulated by iron concentrations and erythropoiesis. When plasma iron is increased, Fe-Tf concentrations are sensed by the TfR1/HFE complex and by TfR2. The exact mechanism by which HFE and TfR2 stimulate hepcidin production is still unclear, but involves the potentiation of bone morphogenetic protein (BMP)/SMAD signaling [13]. Increase in intracellular iron concentrations in the liver also stimulates hepcidin production. The cell types and signaling pathways involved are not well understood, although current evidence suggests that they ultimately converge on the BMP receptor–Smad pathway [13,14].
Hepcidin production is suppressed during times of intense iron utilization and reparative or ineffective erythropoiesis [15,16], resulting in increased iron delivery to the erythron. The pathways involved in hepcidin regulation by erythropoiesis are only now beginning to be characterized and are discussed later in the review.
Inflammation is an important pathological regulator of hepcidin. Interleukin-6 potently stimulates hepcidin transcription [17,18], with synergistic effects from the BMP pathway [19,20]. Increased hepcidin in inflammatory conditions leads to hypoferremia and iron-restricted erythropoiesis. Hepcidin is also regulated by various endocrine stimuli, including growth hormones [21], steroid hormones [22] and gluconeogenic signals [23], but the pathophysiological relevance of these regulators has not yet been delineated. Better understanding of the intricate cross-talk among hepcidin, iron and erythropoiesis is the key to developing effective therapeutics for common iron and erythropoietic disorders.
CLINICAL IMPLICATIONS OF HEPCIDIN DYSREGULATION
Both inappropriately increased and inappropriately suppressed production of hepcidin result in clinical syndromes with disordered iron and erythropoietic homeostasis. Increased hepcidin production contributes to the pathogenesis of various iron-restricted anemias. Anemia of inflammation, also known as anemia of chronic disease, is a complicating factor of a broad spectrum of inflammatory disorders, includingchronicrenalfailure,autoimmunediseases, infections and certain cancers [24]. Hepcidin is elevated in these conditions because of the increased production of cytokines (e.g. interleukin-6, BMP2) [25,26], although in chronic kidney disease, decreased kidney clearance of hepcidin may also contribute [27]. In a mouse model of anemia of inflammation, ablation of hepcidin ameliorated anemia and allowed faster hemoglobin recovery [28■,29■], suggesting that therapeutic lowering of hepcidin may be useful for the treatment of iron-restricted anemias associated with increased hepcidin.
Increased production of hepcidin can occur even in the absence of inflammation. In iron-refractory iron-deficiency anemia, loss-of-function mutations in the negative hepcidin regulator, TMPRSS6, lead to increased hepcidin production and inadequate iron supply for erythropoiesis [30].
Conversely, in anemias with ineffective erythropoiesis such as β-thalassemia, hepcidin production is suppressed [31,32], causing hyperabsorption of dietary iron by enterocytes and systemic iron overload even in the absence of transfusions. β-thalassemia is characterized by chronic anemia with compensatory proliferation of the immature erythroblast pool and apoptosis at the polychromatophilic stage [33■■]. Increase in nontransferrin bound iron as a consequence of inappropriately low hepcidin further contributes to ineffective erythropoiesis by stimulating free radical damage of erythroid precursors and promoting α-globin precipitation [34,35]. Moderate overexpression of hepcidin in mouse models of β-thalassemia, which results in moderate iron restriction, was shown to ameliorate ineffective erythropoiesis by decreasing α-globin precipitates and improving RBC survival. Improvements in hematological parameters in thalassemic mice were observed with transgenic overexpression of hepcidin [35], administration of minihepcidins [36], Tmprss6 ablation [37] or administration of Tmprss6 siRNAs [38■] or antisense oligos [39■]. A subset of myelodysplastic syndromes (MDS) is also characterized by ineffective erythropoiesis and iron overload toxicities, which can be attenuated with the use of chelating therapies [40]. It remains to be seen to what extent hepcidin suppression contributes to the iron overload and ineffective erythropoiesis in MDS.
‘ERYTHROID REGULATORS’ OF HEPCIDIN EXPRESSION
Although increased erythropoietic activity has long been known to suppress hepcidin expression, the specific mechanisms and ‘erythroid regulators’ involved are still under investigation. Treating mice with the myeloablative drug carboplatin prevented hepcidin suppression after erythropoietin (EPO) injections, ruling out EPO as a direct suppressor of hepcidin [16]. Since those initial studies, several candidates have been proposed as erythroid regulators of hepcidin expression.
Erythroferrone
A newly described regulator of hepcidin expression is the hormone erythroferrone (ERFE), a member of the C1q-tumor necrosis factor-related family of proteins [41■■] (Fig. 1). ERFE is an EPO-responsive gene. Mice injected with EPO rapidly (within 4 h) increased ERFE mRNA expression by erythroid precursors in the bone marrow and spleen through the stress erythropoiesis-related JAK2-STAT5 signaling pathway [41■■]. In contrast, ERFE mRNA expression in mice was not regulated by chronic iron dextran injections or by acute inflammation (4 h lipopolysaccharide treatment). Treatment of erythroid precursors by dimethyloxaloylglycine in vitro also had no effect on ERFE mRNA expression [41■■], indicating that hypoxia-inducible factor (HIF) transcription factors do not directly control ERFE transcription.
FIGURE 1.
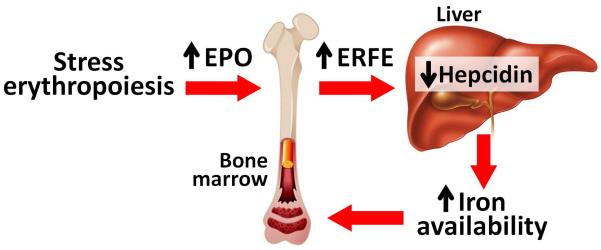
A novel pathway by which erythropoietic activity regulates iron homeostasis. During stress erythropoiesis (e.g. after blood loss), increased erythropoietin (EPO) levels cause increased production of the hormone erythroferrone (ERFE) by erythroid progenitors in the bone marrow (as well as the spleen in mice). ERFE induction by EPO is dependent on the JAK2/STAT5 signaling. ERFE then targets hepatocytes in the liver via an unidentified pathway to suppress hepcidin expression. As a result, iron availability for hemoglobin synthesis in erythroid precurosors is increased.
ERFE plays an important role in ensuring iron supply during stress erythropoiesis in vivo. In contrast to their wild-type counterparts, ERFE knockout mice failed to suppress hepcidin acutely in response to phlebotomy or EPO injections, indicating that ERFE is necessary for rapid hepcidin suppression in the setting of increased erythroid activity [41■■]. Without hepcidin suppression, ERFE knockout mice showed delayed recovery of hemoglobin after hemorrhage [41■■]. ERFE ablation in a mouse model of anemia of inflammation using heat-killed Brucella abortus resulted in a more severe anemia with inappropriately elevated hepcidin levels, indicating that ERFE may play an important role in the recovery from anemia that occurs with inflammation [42■■]. ERFE may also contribute to the pathological hepcidin suppression in conditions of ineffective erythropoiesis. β-thalassemia intermedia mice (Hbbth3/+) had dramatically increased ERFE mRNA levels in bone marrow and spleen [41■■], as would be expected for a condition characterized by high EPO levels and increased number of erythroid precursors. In comparison with decreased hepcidin mRNA in th3/+ mice, ablation of ERFE in thalassemic mice restored liver hepcidin expression to the levels observed in wild-type mice. This was associated with a moderate decrease in liver iron concentrations, without any changes in hematological parameters [41■■]. Further studies are necessary to elucidate the specific roles of ERFE in the various human anemia syndromes, including hemoglobinopathies, anemia of inflammation and chronic kidney disease, and iron deficiency.
Growth differentiation factor 15
Growth differentiation factor 15 (GDF15), a member of the transforming growth factor-β superfamily, is produced by the late-stage erythroid precursors [43]. GDF15 has been proposed to suppress hepcidin expression during ineffective erythropoiesis. β-thalassemia patients had extremely elevated GDF15 serum levels that correlated positively with levels of soluble transferrin receptor, EPO, and ferritin. Suppression of hepcidin mRNA in primary human hepatocytes by serum from β-thalassemia patients was at least partially dependent on GDF15 [43]. Another study examining the serum of congenital dyserythropoietic anemia patients found elevated levels of GDF15 that inversely correlated with hepcidin levels [44]. Unlike the human disease, however, mouse models of β-thalassemia do not show greatly increased GDF15 levels [45], and this has hampered mechanistic studies on the role of GDF15 in hepcidin regulation in ineffective erythropoiesis.
Recent translational studies found no significant contribution of GDF15 levels to hepcidin suppression in iron deficiency [46,47], anemia of chronic disease [47] or chronic myeloproliferative diseases [48]. Phlebotomy of wild-type and Gdf15−/− mice showed similar suppression of hepatic hepcidin mRNA in both groups of mice [49■]. Based on these studies, GDF15 does not appear to be necessary for physiological suppression of hepcidin. Further studies are necessary to determine its pathological role in human disease.
Twisted gastrulation protein homolog 1
Twisted gastrulation protein homolog 1 (TWSG1) is a BMP-binding protein produced at an earlier stage of erythroblast differentiation than GDF15 [43]. Twsg1 mRNA levels were increased in Hbbth3/+ and Hbbth3/th3 mouse spleen, bone marrow and liver [50]. However, TWSG1 has not been evaluated in human patients, and there is no in-vivo evidence that TWSG1 plays a regulatory role in pathological hepcidin suppression. A phenylhydrazine mouse model of stimulated erythropoiesis with iron loading showed no increase in Twsg1 mRNA expression despite reduced hepcidin levels [45]. Similarly, neither Twsg1 nor Gdf15 mRNA was increased in the bone marrow of mice within 24 h after hemorrhage, despite strong hepcidin suppression by 12–15 h [41■■]. Thus, like GDF15, TWSG1 does not appear to be a physiological suppressor of hepcidin and its role in ineffective erythropoiesis remains to be demonstrated.
ERYTHROPOIETIC REGULATION BY IRON STATUS
The hormone EPO has long been recognized as the primary driver of erythropoiesis. By binding to EPO receptors, EPO stimulates the proliferation and terminal differentiation of erythroid precursors while inhibiting RBC precursor apoptosis [51]. Recent studies have described a novel mechanism by which iron status modulates production of EPO through the involvement of the iron regulatory protein 1 (IRP1). IRPs posttranscriptionally regulate the stability or translation of transcripts containing iron-responsive elements (IREs), including TfR1, ferroportin, HIF2α and ferritin [52]. The activity of IRP1 is modulated by intracellular iron concentrations. In times of cellular iron repletion, synthesis of the iron sulfur cluster of IRP1 is increased, and IRP1 converts from the apoprotein form to cytosolic aconitase, the iron-dependent enzyme that catalyzes the conversion of citrate to isocitrate [53]. Conversely, iron depletion promotes accumulation of the RNA-binding form of IRP1. Recently, IRP1 was shown to repress HIF2α expression in the kidney and liver tissues [54■■-56■■], presumably by binding to the 5′IRE of HIF2α transcript. Irp1−/− mice, particularly after being challenged with iron deficiency [55■■] or iron demands of rapid growth [54■■,56■■], increased HIF2α expression in the kidney, which stimulated EPO production and caused severe polycythemia [54■■-56■■]. This suggested a new role of IRP1 in modulating erythropoiesis (Fig. 2a): during iron deficiency, IRP1 in the kidneys tempers the production of EPO by repressing HIF2α, decreasing the production of red blood cells and thereby diminishing iron drain from the systemic iron pool at the expense of iron needs of other tissues.
FIGURE 2.
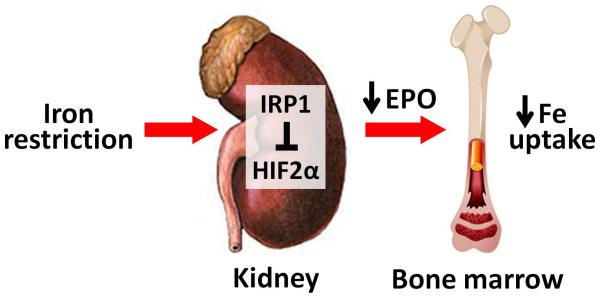
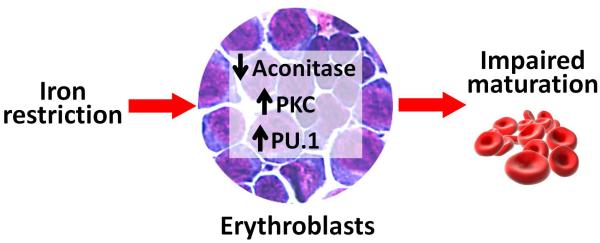
Novel pathways by which iron restriction regulates erythropoietic activity. (a) In iron-restricted conditions, a decrease in intracellular iron in renal interstitial fibroblasts results in iron regulatory protein 1 (IRP1) binding to the 5′ iron-responsive element in hypoxia-inducible factor 2α (HIF2α) mRNA and suppression of HIF2α translation. Lower HIF2α levels lead to decreased production of erythropoietin (EPO) and eventually lesser utilization of iron by developing RBC. This is thought to be a ‘safeguard’ mechanism to balance the usage of iron by erythron vs. other tissues during the times of iron deficiency. (b) Iron restriction directly affects the maturation of erythroblasts by causing inactivation of aconitase, hyperactivation of protein kinase C (PKC) and induction of the erythroid-inhibitory transcription factor PU.1. These effects of iron restriction can be reversed by the treatment with isocitrate, mimicking the enzymatic activity of aconitase.
Another novel regulatory mechanism was recently described that explains how iron restriction suppresses erythroid development. Erythroid aconitase (likely the mitochondrial form [57]) and its product isocitrate have recently been postulated to mediate the effect of iron restriction on erythroid development (Fig. 2b). Iron restriction was shown to inhibit aconitase activity, leading to protein kinase C α/β hyperactivation and overexpression of PU.1, a transcription factor that inhibits erythroid development [58■■]. The effect on PU.1 was particularly prominent in the presence of inflammation. Exogenous supplementation of isocitrate to iron-deprived erythroid cultures was able to reverse abnormalities in the GPA–CD41 ratio, cellular structure, hemoglobinization, globin expression and IRP1 expression [59]. Exogenous isocitrate treatment also prevented the inhibitory effect of interferon-γ on in-vitro proliferation and differentiation of iron-restricted erythroid progenitors [58■■]. Accordingly, isocitrate injections in vivo in a rat model of anemia of inflammation improved hemoglobin and marrow erythropoiesis, indicating that isocitrate or compounds mimicking its activity may be useful as treatment for anemias associated with inflammation and iron restriction.
NOVEL THERAPIES FOR ERYTHROPOIETIC DYSREGULATION
Much attention has been focused on the development of therapeutics for anemias resulting from dysfunctional iron homeostasis. Several therapies are under development that target increased hepcidin levels in anemia of inflammation, chronic kidney disease and cancer [60■], and clinical trials are already under way. The discovery of ERFE as a potent suppressor of hepcidin expression makes it another viable candidate for therapeutic development for iron-restricted anemias. Other therapies based on hepcidin mimics or TMPRSS6 antigene approaches are under development for iron-loading anemias [60■]. Apart from hepcidin, GDF11 was recently described as a promising target for novel therapeutics for both iron-loading and iron-restricted anemias.
The role of GDF11 in erythropoiesis was recognized through the clinical use of sotatercept (ACE-011), a ligand trap consisting of the extracellular domain of activing receptor IIA (ActRIIA) linked to the human IgG1 Fc domain. Sotatercept, which binds GDF11, was developed to treat bone loss, but studies revealed the unexpected side-effect of increased hemoglobin levels [61]. Recently, GDF11 was found to be increased in splenic erythroblasts and sera from thalassemic mice and cultured erythroblasts and sera from β-thalassemic patients [33■■]. GDF11 is thought to act as an autocrine inhibitor of late-stage erythroid differentiation while maintaining the survival of immature erythroid progenitors [33■■,62■■]. GDF11 inactivation using a murine version of sotatercept (RAP-011) improved anemia, iron overload and ineffective erythropoiesis in a mouse model of β-thalassemia intermedia (Hbbth1/th1). Another study similarly found increased GDF11 mRNA expression in bone marrow and spleen in a mouse model of MDS that is characterized by ineffective erythropoiesis. Administration of a different ActRIIA ligand trap (RAP-536) improved anemia and ineffective erythropoiesis in this murine MDS model [62■■].
ActRIIA ligand traps may also be useful for treatment of iron-restricted anemias associated with elevated hepcidin. RAP-011 effectively increased hemoglobin concentration in hepcidin transgenic mice [63], and RAP-536 treatment increased RBC count and hemoglobin in a mouse model of chronic kidney disease [62■■]. Outside of its potential role in erythropoiesis, GDF11 has also been implicated in regulation of neurogenesis [64], retinal development [65], axial skeleton patterning [66], kidney development [66] and cardiomyocyte aging [67]. It remains to be determined whether ActRIIA ligand traps are sufficiently selective to be used as erythropoiesis-stimulating agents.
CONCLUSION
Remarkable scientific advances in recent years have widely expanded our understanding of iron and erythropoietic regulation, as well as its relevance to human disease. Future studies will realize the potential of these advances in clinical therapeutic applications.
KEY POINTS.
ERFE is an erythroid hormone that mediates the suppression of hepcidin during stress erythropoiesis to increase iron availability for hemoglobin synthesis.
Iron deficiency suppresses erythropoiesis by stimulating the IRP–HIF2α axis to reduce erythropoietin production, and by inhibiting aconitase-induced isocitrate activity to suppress early erythropoiesis.
Novel therapies for the treatment of ineffective or iron-restricted erythropoiesis include isocitrate supplementation and GDF11 inhibition by a ligand trap protein.
Acknowledgements
The authors thank Dr Tomas Ganz for insightful discussions.
Financial support and sponsorship
This work was supported by the NIH grant DK090554.
Footnotes
Conflicts of interest
E.N. is a stockholder of Intrinsic LifeSciences, Merganser Biotech and Silarus Therapeutics. A.K. has no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Ganz T, Nemeth E. Regulation of iron acquisition and iron distribution in mammals. Biochim Biophys Acta. 2006;1763:690–699. doi: 10.1016/j.bbamcr.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 2.Ramm GA, Ruddell RG. Hepatotoxicity of iron overload: mechanisms of iron-induced hepatic fibrogenesis. Semin Liver Dis. 2005;25:433–449. doi: 10.1055/s-2005-923315. [DOI] [PubMed] [Google Scholar]
- 3.Finch C. Regulators of iron balance in humans. Blood. 1994;84:1697–1702. [PubMed] [Google Scholar]
- 4.Ganz T, Nemeth E. Iron metabolism: interactions with normal and disordered erythropoiesis. Cold Spring Harb Perspect Med. 2012;2:a011668. doi: 10.1101/cshperspect.a011668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 6.Ganz T, Nemeth E. The hepcidin–ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematology Am Soc Hematol Educ Program. 2011;2011:538–542. doi: 10.1182/asheducation-2011.1.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 8.Rivera S, Liu L, Nemeth E, et al. Hepcidin excess induces the sequestration of iron and exacerbates tumor-associated anemia. Blood. 2005;105:1797–1802. doi: 10.1182/blood-2004-08-3375. [DOI] [PubMed] [Google Scholar]
- 9.Roy CN, Mak HH, Akpan I, et al. Hepcidin antimicrobial peptide transgenic mice exhibit features of the anemia of inflammation. Blood. 2007;109:4038–4044. doi: 10.1182/blood-2006-10-051755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Viatte L, Lesbordes-Brion JC, Lou DQ, et al. Deregulation of proteins involved in iron metabolism in hepcidin-deficient mice. Blood. 2005;105:4861–4864. doi: 10.1182/blood-2004-12-4608. [DOI] [PubMed] [Google Scholar]
- 12.Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33:21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 13.Corradini E, Rozier M, Meynard D, et al. Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe. Gastroenterology. 2011;141:1907–1914. doi: 10.1053/j.gastro.2011.06.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramos E, Kautz L, Rodriguez R, et al. Evidence for distinct pathways of hepcidin regulation by acute and chronic iron loading in mice. Hepatology. 2011;53:1333–1341. doi: 10.1002/hep.24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganz T, Nemeth E. Hepcidin and disorders of iron metabolism. Annu Rev Med. 2011;62:347–360. doi: 10.1146/annurev-med-050109-142444. [DOI] [PubMed] [Google Scholar]
- 16.Pak M, Lopez MA, Gabayan V, et al. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108:3730–3735. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–1276. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez R, Jung CL, Gabayan V, et al. Hepcidin induction by pathogens and pathogen-derived molecules is strongly dependent on interleukin-6. Infect Immun. 2014;82:745–752. doi: 10.1128/IAI.00983-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armitage AE, Eddowes LA, Gileadi U, et al. Hepcidin regulation by innate immune and infectious stimuli. Blood. 2011;118:4129–4139. doi: 10.1182/blood-2011-04-351957. [DOI] [PubMed] [Google Scholar]
- 20.Mayeur C, Lohmeyer LK, Leyton P, et al. The type I BMP receptor Alk3 is required for the induction of hepatic hepcidin gene expression by interleukin-6. Blood. 2014;123:2261–2268. doi: 10.1182/blood-2013-02-480095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodnough JB, Ramos E, Nemeth E, Ganz T. Inhibition of hepcidin transcription by growth factors. Hepatology. 2012;56:291–299. doi: 10.1002/hep.25615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Latour C, Kautz L, Besson-Fournier C, et al. Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology. 2014;59:683–694. doi: 10.1002/hep.26648. [DOI] [PubMed] [Google Scholar]
- 23.Vecchi C, Montosi G, Garuti C, et al. Gluconeogenic signals regulate iron homeostasis via hepcidin in mice. Gastroenterology. 2014;146:1060–1069. doi: 10.1053/j.gastro.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005;352:1011–1023. doi: 10.1056/NEJMra041809. [DOI] [PubMed] [Google Scholar]
- 25.Theurl I, Aigner E, Theurl M, et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: diagnostic and therapeutic implications. Blood. 2009;113:5277–5286. doi: 10.1182/blood-2008-12-195651. [DOI] [PubMed] [Google Scholar]
- 26.Maes K, Nemeth E, Roodman GD, et al. In anemia of multiple myeloma, hepcidin is induced by increased bone morphogenetic protein 2. Blood. 2010;116:3635–3644. doi: 10.1182/blood-2010-03-274571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young B, Zaritsky J. Hepcidin for clinicians. Clin J Am Soc Nephrol. 2009;4:1384–1387. doi: 10.2215/CJN.02190309. [DOI] [PubMed] [Google Scholar]
- 28■.Gardenghi S, Renaud TM, Meloni A, et al. Distinct roles for hepcidin and interleukin-6 in the recovery from anemia in mice injected with heat-killed Brucella abortus. Blood. 2014;123:1137–1145. doi: 10.1182/blood-2013-08-521625. This study used a mouse model of inflammation in wild-type, hepcidin KO, and interleukin-6 knock-out mice to quantify the contributions of interleukin-6 and hepcidin to acute and severe anemia of inflammation.
- 29■.Kim A, Fung E, Parikh SG, et al. A mouse model of anemia of inflammation: complex pathogenesis with partial dependence on hepcidin. Blood. 2014;123:1129–1136. doi: 10.1182/blood-2013-08-521419. The study provided a comprehensive characterization of the features of a mouse model of anemia of inflammation, including suppressed erythropoiesis, shortened erythrocyte lifespan and the contribution of hepcidin
- 30.Heeney MM, Finberg KE. Iron-refractory iron deficiency anemia (IRIDA) Hematol Oncol Clin North Am. 2014;28:637–652. doi: 10.1016/j.hoc.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Pasricha SR, Frazer DM, Bowden DK, Anderson GJ. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with beta-thalassemia major: a longitudinal study. Blood. 2013;122:124–133. doi: 10.1182/blood-2012-12-471441. [DOI] [PubMed] [Google Scholar]
- 32.Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92:583–588. doi: 10.3324/haematol.10842. [DOI] [PubMed] [Google Scholar]
- 33■■.Dussiot M, Maciel TT, Fricot A, et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat Med. 2014;20:398–407. doi: 10.1038/nm.3468. Using a mouse model, this study demonstrated that ActRIIA ligand traps may suppress the deleterious effects of GDF11 on erythroid maturation and improve ineffective erythropoiesis in β-thalassemia.
- 34.Li H, Rybicki AC, Suzuka SM, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16:177–182. doi: 10.1038/nm.2073. [DOI] [PubMed] [Google Scholar]
- 35.Gardenghi S, Ramos P, Marongiu MF, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest. 2010;120:4466–4477. doi: 10.1172/JCI41717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casu CGS, Nemeth E, Ganz T, et al. Low dose minihepcidin peptide causes improvement of anemia by increased erythropoietic efficiency in a mouse model of thalassemia intermedia. Am J Hematol. 2013;88:E43. [Google Scholar]
- 37.Nai A, Pagani A, Mandelli G, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood. 2012;119:5021–5029. doi: 10.1182/blood-2012-01-401885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38■.Schmidt PJ, Toudjarska I, Sendamarai AK, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(−/−) mice and ameliorates anemia iron overload in murine beta-thalassemia intermedia. Blood. 2013;121:1200–1208. doi: 10.1182/blood-2012-09-453977. Using mouse models, this study demonstrated the potential of Tmprss6 siRNA to treat hereditary hemochromatosis and thalassemia by increasing hepcidin, reducing iron overload and improving ineffective erythropoiesis
- 39■.Guo S, Casu C, Gardenghi S, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest. 2013;123:1531–1541. doi: 10.1172/JCI66969. This study showed that mouse models of β-thalassemia and hereditary hemochromatosis could be treated with TMPRSS6 antisense oligonucleotides to improve iron overload and anemia.
- 40.Shenoy N, Vallumsetla N, Rachmilewitz E, et al. Impact of iron overload and potential benefit from iron chelation in low-risk myelodysplastic syndrome. Blood. 2014;124:873–881. doi: 10.1182/blood-2014-03-563221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41■■.Kautz L, Jung G, Valore EV, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46:678–684. doi: 10.1038/ng.2996. This study identified ERFE as a key erythroid regulator mediating erythropioesis induced hepcidin suppression.
- 42■■.Kautz L, Jung G, Nemeth E, Ganz T. Erythroferrone contributes to recovery from anemia of inflammation. Blood. 2014;124:2569–2574. doi: 10.1182/blood-2014-06-584607. This study used ERFE knock-out mice to demonstrate that ERFE contributes to recovery from anemia of inflammation.
- 43.Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096–1101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 44.Tamary H, Shalev H, Perez-Avraham G, et al. Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I. Blood. 2008;112:5241–5244. doi: 10.1182/blood-2008-06-165738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frazer DM, Wilkins SJ, Darshan D, et al. Stimulated erythropoiesis with secondary iron loading leads to a decrease in hepcidin despite an increase in bone morphogenetic protein 6 expression. Br J Haematol. 2012;157:615–626. doi: 10.1111/j.1365-2141.2012.09104.x. [DOI] [PubMed] [Google Scholar]
- 46.Tanno T, Rabel A, Lee YT, et al. Expression of growth differentiation factor 15 is not elevated in individuals with iron deficiency secondary to volunteer blood donation. Transfusion. 2010;50:1532–1535. doi: 10.1111/j.1537-2995.2010.02601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Theurl I, Finkenstedt A, Schroll A, et al. Growth differentiation factor 15 in anaemia of chronic disease, iron deficiency anaemia and mixed type anaemia. Br J Haematol. 2010;148:449–455. doi: 10.1111/j.1365-2141.2009.07961.x. [DOI] [PubMed] [Google Scholar]
- 48.Tarkun P, Mehtap O, Atesoglu EB, et al. Serum hepcidin and growth differentiation factor-15 (GDF-15) levels in polycythemia vera and essential thrombocythemia. Eur J Haematol. 2013;91:228–235. doi: 10.1111/ejh.12150. [DOI] [PubMed] [Google Scholar]
- 49■.Casanovas G, Vujic Spasic M, Casu C, et al. The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematologica. 2013;98:444–447. doi: 10.3324/haematol.2012.069807. Using Gdf15 knockout mice, this study directly demonstrated that Gdf15 does not function as an erythroid suppressor of hepcidin during recovery from hemorrhage.
- 50.Tanno T, Porayette P, Sripichai O, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood. 2009;114:181–186. doi: 10.1182/blood-2008-12-195503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elliott S, Pham E, Macdougall IC. Erythropoietins: a common mechanism of action. Exp Hematol. 2008;36:1573–1584. doi: 10.1016/j.exphem.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 52.Zhang DL, Ghosh MC, Rouault TA. The physiological functions of iron regulatory proteins in iron homeostasis – an update. Front Pharmacol. 2014;5:124. doi: 10.3389/fphar.2014.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rouault TA. Cell biology. An ancient gauge for iron. Science. 2009;326:676–677. doi: 10.1126/science.1181938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54■■.Anderson SA, Nizzi CP, Chang YI, et al. The IRP1-HIF-2alpha axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013;17:282–290. doi: 10.1016/j.cmet.2013.01.007. This article described the distinct physiological roles of IRP1 and IRP2, and identified IRP1 as a key modulator of HIF-2α activity and EPO production.
- 55■■.Ghosh MC, Zhang DL, Jeong SY, et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2α. Cell Metab. 2013;17:271–281. doi: 10.1016/j.cmet.2012.12.016. The study identified IRP1 as a translational repressor of HIF2α and demonstrated that ablation of IRP1 together with low-iron diet induced severe polycythemia and pulmonary hypertension.
- 56■■.Wilkinson N, Pantopoulos K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2alpha mRNA translation. Blood. 2013;122:1658–1668. doi: 10.1182/blood-2013-03-492454. The study demonstrated that IRP1 and not IRP2 is the principal regulator of HIF2α production. Ablation of IRP1 in mice resulted in translational derepression of HIF2α, increased production of EPO and extramedullary hematopoiesis.
- 57.Talbot AL, Bullock GC, Delehanty LL, et al. Aconitase regulation of erythropoiesis correlates with a novel licensing function in erythropoietin-induced ERK signaling. PLoS One. 2011;6:e23850. doi: 10.1371/journal.pone.0023850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58■■.Richardson CL, Delehanty LL, Bullock GC, et al. Isocitrate ameliorates anemia by suppressing the erythroid iron restriction response. J Clin Invest. 2013;123:3614–3623. doi: 10.1172/JCI68487. This is the first comprehensive study examining the hematologic effects of exogeneous isocitrate in a mouse model of chronic inflammation, including the mechanism of action involving PKCs and PU.1.
- 59.Bullock GC, Delehanty LL, Talbot AL, et al. Iron control of erythroid development by a novel aconitase-associated regulatory pathway. Blood. 2010;116:97–108. doi: 10.1182/blood-2009-10-251496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60■.Fung E, Nemeth E. Manipulation of the hepcidin pathway for therapeutic purposes. Haematologica. 2013;98:1667–1676. doi: 10.3324/haematol.2013.084624. This is a detailed review of the pharmacological strategies under development for modulating hepcidin activity for the treatment of various hematologic disorders.
- 61.Ruckle J, Jacobs M, Kramer W, et al. Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J Bone Miner Res. 2009;24:744–752. doi: 10.1359/jbmr.081208. [DOI] [PubMed] [Google Scholar]
- 62■■.Suragani RN, Cadena SM, Cawley SM, et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20:408–414. doi: 10.1038/nm.3512. This is a compelling study of the therapeutic effects of an ActRIIB ligand trap in treating the anemia and ineffective erythropoiesis of MDS.
- 63.Langdon JM, Barkataki S, Berger AE, et al. RAP-011, an activin receptor ligand trap, increases hemoglobin concentration in hepcidin transgenic mice. Am J Hematol. 2015;90:8–14. doi: 10.1002/ajh.23856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu HH, Ivkovic S, Murray RC, et al. Autoregulation of neurogenesis by GDF11. Neuron. 2003;37:197–207. doi: 10.1016/s0896-6273(02)01172-8. [DOI] [PubMed] [Google Scholar]
- 65.Kim J, Wu HH, Lander AD, et al. GDF11 controls the timing of progenitor cell competence in developing retina. Science. 2005;308:1927–1930. doi: 10.1126/science.1110175. [DOI] [PubMed] [Google Scholar]
- 66.McPherron AC, Lawler AM, Lee SJ. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat Genet. 1999;22:260–264. doi: 10.1038/10320. [DOI] [PubMed] [Google Scholar]
- 67.Loffredo FS, Steinhauser ML, Jay SM, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153:828–839. doi: 10.1016/j.cell.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]