

Abstract
Streptococcus mutans is a major aetiological agent of dental caries. Formation of biofilms is a key virulence factor of S. mutans. Drugs that inhibit S. mutans biofilms may have therapeutic potential. Dihydrofolate reductase (DHFR) plays a critical role in regulating the metabolism of folate. DHFR inhibitors are thus potent drugs and have been explored as anticancer and antimicrobial agents. In this study, a library of analogues based on a DHFR inhibitor, trimetrexate (TMQ), an FDA-approved drug, was screened and three new analogues that selectively inhibited S. mutans were identified. The most potent inhibitor had a 50% inhibitory concentration (IC50) of 454.0 ± 10.2 nM for the biofilm and 8.7 ± 1.9 nM for DHFR of S. mutans. In contrast, the IC50 of this compound for human DHFR was ca. 1000 nM, a >100-fold decrease in its potency, demonstrating the high selectivity of the analogue. An analogue that exhibited the least potency for the S. mutans biofilm also had the lowest activity towards inhibiting S. mutans DHFR, further indicating that inhibition of biofilms is related to reduced DHFR activity. These data, along with docking of the potent analogue to the modelled DHFR structure, suggested that the TMQ analogues indeed selectively inhibited S. mutans through targeting DHFR. These potent and selective small molecules are thus promising lead compounds to develop new effective therapeutics to prevent and treat dental caries.
Keywords: Streptococcus mutans, Biofilm, Dihydrofolate reductase, Trimetrexate analogues
1. Introduction
Dental caries is initiated by cariogenic bacteria under the carbohydrate-rich environment of the oral cavity. Streptococcus mutans is the principal causative agent for the development of dental caries. The ability of S. mutans to adhere to the tooth surface and to incorporate into a polymicrobial consortium is paramount for the development of the disease; thus, reducing the number of cariogenic bacteria in dental biofilms is the key to devising therapeutic and preventive strategies [1].
Various strategies aimed at preventing dental caries have been attempted on the basis of various criteria: increasing antimicrobial activity [2]; replacement of sucrose with other sweeteners [3]; inhibition of the key matrix-producing enzymeglucosyltransferases either by vaccine approaches [4] or enzymatic inhibitors [5]; and targeting another important surface protein antigen I/II using immunomodulatory monoclonal antibodies [6]. However, in vivo application of these promising approaches is uncertain.
Folate is essential for all organisms. It contributes to the production of cofactors required for the synthesis of DNA, RNA and amino acids. Dihydrofolate reductase (DHFR) is the key enzyme involved in the production of the cofactors, thereby playing a critical role in regulating folate metabolism [7].
The importance of DHFR in the folate cycle has been explored in order to develop drugs for tumour therapy as well as therapy against opportunistic infections of diverse origin [7–9]. Inhibition of DHFR leads to partial depletion of reduced intracellular folate, thereby limiting cell growth. Functionally, DHFR is highly conserved in all domains of life, however they are divergent in amino acid sequence, offering an opportunity to impart a high degree of selectivity for certain antifolate drugs against one organism versus another [10].
Antifolate drugs have been successful in the treatment of bacterial and parasitic infections and for cancer chemotherapy. A number of antifolate drugs have been developed to date [9,11–13]. Trimetrexate (TMQ) has been used clinically for treatment of the parasites Pneumocystis carinii and Toxoplasma gondii infections in acquired immunodeficiency syndrome (AIDS) patients [13]. To date, no DHFR inhibitor has been evaluated for its selectivity against the cariogenic bacterium S. mutans. Therefore, it is imperative to design new inhibitors that have high selectivity against S. mutans DHFR (SmDHFR). Considering that the primary sequence of SmDHFR shares only 28% identity with the human DHFR (hDHFR) sequence, it is conceivable to rationally design selective and potent inhibitors of S. mutans based on the structure of TMQ.
The goal of this study was to identify small-molecule inhibitors of DHFR that are capable of inhibiting S. mutans. A library of ca. 100 small-molecule compounds, which were designed based on TMQ, a US Food and Drug Administration (FDA)-approved drug [13], was screened and compounds that potently inhibit S. mutans were identified. More importantly, these compounds were selective against SmDHFR. Computer modelling and docking analysis supported the finding that the identified small molecule possesses much higher affinity for SmDHFR than for hDHFR. The study to design more potent and selective compounds inhibiting S. mutans is ongoing, which should facilitate the development of potent therapeutic drugs against S. mutans.
2. Materials and methods
2.1. Synthesis of small-molecule compounds and assembly of a screening library
Protocols used to synthesise small-molecule compounds are described in the Supplemental material. The small-molecule library was assembled by the Velu Research Group.
The synthetic processes and schemes are described in the main text and the Supplementary material (Fig. 1A; Supplementary Fig. S1). The chemical structures of TMQ and its analogues are depicted in the results section (Fig. 1B).
Fig. 1.

(A) Synthetic scheme of trimetrexate (TMQ) analogues and (B) molecular structures of TMQ, trimethoprim (TMP) and their analogues #66, #151, #153 and #154. Generally, the synthesis involved a reductive amination of 2-fluoro-5-formylbenzonitrile 4 with appropriate amines (RNH2) in the presence of NaCNBH3 and ZnCl2 in methanol to afford the aminated product 5 (59–100% yield). The aminated compound 5 was refluxed with guanidine carbonate in N,N-dimethyl acetamide to give the final products #66, #151, #153 and #154 (13–76% yield).
2.2. Bacterial strains and culture conditions
Streptococcus strains, including S. mutans UA159, Streptococcus sanguinis SK36 and Streptococcus gordonii DL1, were grown as previously described [14].
2.3. Biofilm formation and inhibition assays
Exponentially grown S. mutans and S. sanguinis bacteria were inoculated at 1:100 dilution with chemically-defined biofilm medium (CDBM) containing 1% sucrose for biofilm assays, whilst S. gordonii was inoculated at 1:50 dilution. Compounds at indicated concentrations were added to the inoculated bacterial cultures. The incubation time was 16 h for S. mutans and S. sanguinis and 12 h for S. gordonii to obtain reproducible and comparable biofilms. For control cultures, the corresponding volume of dimethyl sulphoxide (DMSO) was added. Crystal violet staining measured at an optical density of 562 nm was used to monitor biofilm formation described previously [14]. Minimum inhibitory concentrations (MICs) of the compounds were examined using a previously described method [14]. The concentration of potent compounds that inhibited S. mutans biofilm formation by 50% (IC50) was determined by serial dilution. Each assay was carried out with duplicate samples and was repeated three times.
2.4. Cloning, expression and purification of recombinant DHFR from Streptococcusmutans and human cells
The coding sequence for the S. mutans UA159 DHFR protein was amplified by PCR (GoTaq® DNA Polymerase; Promega) using genomic DNA as template and SmDHFR-specific primers (CGCGGATCCATGAACAATAAGCGAGAAAAG and GCCGCTCGAGTCATTCCTTTTTCTCAAGTAC). The resulting PCR product (513 bp) was digested with BamHI and XhoI and was ligated into the pET28-SUMO vector to construct pET28-SUMO-SmDHFR. The constructed plasmids were verified by DNA sequence analysis and were then transformed into Escherichia coli BL21. The pET28-SUMO-hDHFR was constructed using a similar experimental procedure with hDHFR-specific primers (CGCGGATCCATGGTTGGTTCGCTAAACTGC and CCTCTCGAGTTAATCATTCTT CTCATATACTTCAAATTTGTAC) and human cDNA.
Expression, production and purification of recombinant SmDHFR and hDHFR proteins were carried out using the experimental procedures described previously [15]. In brief, cell lysates prepared from induced recombinant strains were loaded into a Ni-NTA resin column (Novagen, USA) for initial purification and the 6×His-SUMO tag was removed from the fusion proteins with SUMO proteinase. The fractions were then subjected to size exclusion chromatography on a Superdex75 column (GE Life Science, USA). Concentrations of purified SmDHFR and hDHFR were determined by the Bradford method (Bio-Rad Protein Assay).
2.5. Assay of enzymatic activity of SmDHFR and hDHFR
The activity of purified SmDHFR and hDHFR was determined by monitoring the decrease in absorbance at 340 nm due to the oxidation of NADPH to NADP+ accompanying the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF) (coupled ε = 12.260 M−1cm−1) using a UV–visible spectrophotometer. Reaction velocities were measured at 25°C for 1 min. A DMSO concentration of ≤0.125% was used in the assays as the effect of the solvent at this concentration is negligible. Under optimised conditions for each enzyme, SmDHFR activity was measured in 50 mM Tris–HCl buffer (pH 7.0), whilst hDHFR activity was measured in 50 mM KH2PO4 (pH 7.3) containing 250 mM KCl and 5 mM β-mercaptoethanol [9]. The inhibitory activities of the compounds were determined by measuring reaction velocities at several fixed concentrations of DHF (15, 20, 25, 30 and 40 μM), NADPH (150 μM) and various concentrations of compounds (dissolved in 0.1% DMSO). The 200μL reaction was initiated with 0.2 μg of purified SmDHFR or 0.3 μg of hDHFR. Each measurement was performed in triplicate.
The double-reciprocal plots were used to calculate the reaction constant (Km) and inhibitory constant (Ki) for inhibition of SmDHFR and hDHFR by each compound. The concentration of each inhibitor required for inhibition of enzyme activity by 50% (IC50) was determined using the following equation: , where Vo and Vi are the initial velocities in the absence and presence of inhibitor, respectively, and Ci is the concentration of inhibitor. The selectivity index (SI) was calculated using the equation: . The SI of each analogue was compared with that of TMQ to assess the selectivity of each compound.
2.6. Sequence alignment, homology modelling and validation
The SmDHFR sequence was submitted to Phyre2 (Protein Homology and Analogy Recognition Engine v.2.0) (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) and Swiss-Model (http://www.swissmodel.expasy.org) for structural prediction and modelling. Streptococcus pneumoniae DHFR (SpDHFR) (PDB code, 3ix9; resolution, 1.95 Å) was selected for structural modelling, and the whole chain B of SpDHFR served as the template for the homology modelling of SmDHFR using Phyre2normal mode. The model was superposed with the SpDHFR template by VMD 1.9.1 to establish the agreed and disagreed regions [11]. The overall calculated root-mean-square deviation (RMSD) was taken from overlapping the backbone of all aligned residues, whilst the active-site RMSD was obtained from overlapping the homology model with the SpDHFR backbone containing residues 8–119. The SmDHFR model generated by Phyre2 was validated by PROCHECK and VERIFY3D provided by Structure Analysis and Verification Server (SAVES v.4.0) and was also assessed by Swiss-Model Assessment for QMEAN and QMEAN Z-Score.
2.7. Molecular docking and energy analysis
The computer system Ubuntu 10.04 LTS on Quad Core OptiPlex 780 was used to dock the lead compound #66 into the active sites of hDHFR (PDB code, 1pd8) and the modelled SmDHFR. The three-dimensional structure of compound #66 was obtained from VEGA ZZ program using TRIPOS and Gasteiger for force field and charge calculations, respectively [16].
Preliminary docking was carried out using FlexX. The top ten structures with the best scores from FlexX were redocked and their binding energy distribution was visualised by HYDE. LeadIT 2.0.1 package that contains FlexX and HYDE is commercially available via BioSolveIT. HYDE is an innovative redocking and lead optimisation program that determines protein–ligand binding energies based on two factors: hydrogen bonding and dehydration [17]. Here we exploited HYDE to understand binding of the lead compound to the homology model SmDHFR, since the HYDE scoring function is known to be suitable for substantially improving docking and reducing false positives [17]. The docking results were visualised and illustrated by Chimera 1.8.1 [18]. In PROCHECK, the Ramachandran plot contained 87% residues in favourable regions, whilst most of the remaining residues were in allowed regions and none were in disallowed regions. In addition, the overall G-factor measuring the stereochemical property was 0.17, significantly above the threshold of −0.5 [19]. Furthermore, VERIFY3D showed 98% of the residues with an averaged 3D-1D score >0.2 [20]. Likewise, the QMEAN of the model was 0.79 and the QMEAN Z-Score was 0.26. Thus the QMEAN score of the homology model SmDHFR differed within 1 standard deviation from the scores of high-resolution X-ray structures of similar size(Supplementary Table S1) [21]. Evaluation validated the model [20] since the scores for SmDHFR were similar or better than those of the SpDHFR X-ray crystal structure with the resolution of 1.95 Å.
2.8. Statistical analysis
Statistical tests were performed using SPSS v.16.0 software (SPSS Inc., Chicago, IL) and the results are presented as the mean ± standard deviation. Two-group comparisons were performed using Student’s t-test. A P-value of <0.05 was considered statistically significant.
3. Results
3.1. Synthesis of SmDHFR inhibitor library
To identify new derivatives that inhibit S. mutans biofilms, a new library of TMQ analogues was assembled and used in the screening. The general scheme employed for the synthesis of the new derivatives is represented by the synthesis of four TMQ analogues (#66, #151, #153 and #154) identified in this study (Fig. 1A), and the structure of each derivative is outlined (Fig. 1B).
3.2. Identification of small-molecule compounds that selectively inhibit biofilm formation of Streptococcus mutans
To identify desirable inhibitors for S. mutans biofilms, the library of 100 TMQ derivatives was screened by a biofilm assay described previously [14]. Three of the most potent small-molecule analogues (#66, #151 and #153) (Fig. 1B) were identified and were further evaluated in biofilm assays and were found to inhibit S. mutans biofilm formation as well as cell growth. The MICs of the three analogues were 0.781, 1.56 and 1.56 μM, respectively, whilst MICs for the control compound TMQ and the least active analogue #154 were 48.8 nM and 50μM, respectively. The follow-up dose-dependent studies determined the precise IC50 for each compound (Table 1).
Table 1.
Inhibitory activity against Streptococcus mutans growth, biofilm and S. mutans dihydrofolate reductase (SmDHFR)
| Compound | IC50 (nM)
|
||
|---|---|---|---|
| Growth | Biofilm | SmDHFR | |
| #66 | 604.1 ± 4.5 | 454.0 ± 10.2 * | 8.7 ± 1.9 |
| #151 | 1505.5 ± 28.5 | 1127.9 ± 13.1 * | 7.1 ± 0.9 |
| #153 | 1352.7 ± 57.5 | 942.1 ± 3.5 * | 8.2 ± 0.9 |
| #154 | 51 518.5 ± 260.2 | 71 026.3 ± 360.4 | 1074.8 ± 150.4 |
| TMQ | 42.48 ± 3.22 | 49.08 ± 4.1 | 2.2 ± 0.7 |
IC50, 50% inhibitory concentration; TMQ, trimetrexate.
Streptococcus mutans was treated with dimethyl sulphoxide (DMSO) (a negative control), TMQ (a positive control) or the library analogues. Cell growth and biofilm formation were determined. Lead compounds (TMQ and its analogues #66, #151, #153 and #154) were selected to examine their effects on SmDHFR enzymatic activity. The IC50 value of cell growth, biofilm formation and SmDHFR in the analogue-treated groups was calculated with a series of diluted analogue compounds. Values represent the mean ± standard deviation from three independent experiments.
Statistically significant difference observed from the various comparisons.
Analogue #66 inhibited S. mutans growth and biofilm at a low submicromolar range, whereas a higher dose of #151 and #153 was required to inhibit biofilm formation. TMQ inhibited bacterial growth and biofilm formation more effectively than its analogues. Among the new analogues, #66 exhibited the most potent activity with an inhibitory IC50 for biofilm at 454.0 ± 10.2 nM, whilst the IC50 for planktonic cells was much higher at 604.1 ± 4.5 nM (P < 0.05). Consistently, #151 and #153 also possessed similar modest selectivity toward inhibiting biofilm formation, whilst #154 and the control TMQ did not have any selectivity.
To determine whether the identified inhibitors had any selectivity against S. mutans, the effect of these TMQ analogues on cell growth and biofilm formation of commensal streptococci (S. sanguinis and S. gordonii) at the concentration that inhibited biofilm formation of S. mutans was evaluated. TMQ did not inhibit biofilm formation of S. sanguinis and S. gordonii (P > 0.05) at 1.56 μM. Similarly #66, #151 and #153 had no inhibitory effect on biofilm formation of S. sanguinis and S. gordonii (P > 0.05) at 6.25 μM. #153 failed to inhibit biofilm formation of S. sanguinis even at a higher concentration of 12.5μM (P > 0.05) (Fig. 2). These data suggest that the identified compounds did possess selectivity towards inhibiting S. mutans.
Fig. 2.

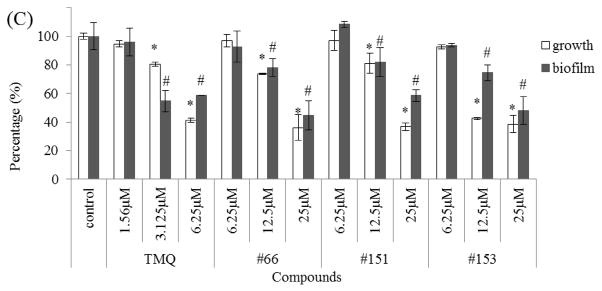
Effects of trimetrexate (TMQ) and its analogues (#66, #151 and #153) on cell growth and biofilm formation of commensal streptococci. (A) Streptococcus mutans, (B) Streptococcus sanguinis and (C) Streptococcus gordonii were treated with TMQ and its analogues at different concentrations and cell growth and biofilm formation of each strain were determined. The percentage cell growth or biofilm formation was normalised to the dimethyl sulphoxide (DMSO) control groups (100%). Values represent the mean ± standard deviation from three independent experiments. * Statistically significant difference observed in cell growth; # statistically significant difference observed in biofilm formation.
3.3. Characterisation of recombinant Streptococcus mutans and human DHFR
To determine whether the identified analogues inhibited biofilm formation of S. mutans via inhibiting the activity of DHFR as designed, and to evaluate the selectivity of the analogues against hDHFR, recombinant SmDHFR and hDHFR were first expressed and purified and the recombinant enzymes were then characterised. Both DHFRs exist as a monomer as observed from the gel filtration profile with an estimated molecular weight of 17 kDa and 18 kDa, respectively (data not shown). Enzymatic parameters were characterised as described previously [9].
First, the optimal concentration of SmDHFR to reach linear reaction velocity at the first 2 min was determined (0.001 μg/μL). This concentration was then used to characterise the enzyme. SmDHFR has its maximal activity at pH 7.0. The activity decreased by ca. 50% from pH 6.8 to pH 8.0 (Fig. 3). The hDHFR optimal reaction conditions were also determined in the same manner. The hDHFR enzyme reaction reached maximal velocity at pH 7.3 in 50 mM KH2PO4 buffer with 0.0015μg/μL of purified hDHFR. The purified hDHFR is stable and soluble at concentrations up to 25 mg/mL at 4°C and is catalytically active at room temperature. These parameters allowed us to determine the effects of the TMQ analogues on both DHFRs.
Fig. 3.
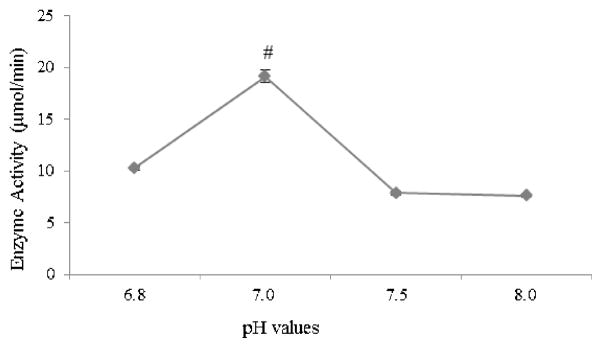
Effect of pH on Streptococcus mutans dihydrofolate reductase (SmDHFR) enzymatic activity. The assay was carried out in 30 μM dihydrofolate (DHF) and 0.001 μg/μL SmDHFR with varying pH values. # SmDHFR activity in the reaction at pH 7 was significantly different from the activities measured at other pH conditions.
3.4. Trimetrexate analogues selectively inhibited SmDHFR
Analogues identified for their ability to inhibit bacterial growth and biofilm formation were further assessed to determine whether the inhibition acted via inhibiting DHFR activity. The inhibitory activities of four analogues (#66, #151, #153 and #154) and TMQ, used as a positive control, were determined by kinetic assays established above.
Four inhibitors that were potent in vivo were also potent in vitro in the enzymatic kinetic assay (Tables 1 and 2). The biofilm IC50 values of potent analogues #66, #151 and #153 ranged from 0.45 μM to 1.1μM, whilst the SmDHFR IC50 was ca. 10 nM. The IC50 values of the least effective analogue #154 for the biofilm and SmDHFR were >100-fold higher than those of the analogue #66. Compared with the active analogues #66, #151 and #153, the analogue #154 had much higher IC50 for both SmDHFR and biofilm, suggesting that the biofilm IC50 correlated with the SmDHFR IC50. These data suggest that SmDHFR is the target for TMQ analogues in the inhibition of bacterialgrowth and biofilm formation.
Table 2.
Inhibitory activity against Streptococcus mutans dihydrofolate reductase (SmDHFR) and human DHFR (hDHFR)
| Compound | SmDHFR | hDHFR | SI c | |||
|---|---|---|---|---|---|---|
|
| ||||||
| IC50 (nM) a | Ki (nM) b | IC50 (nM) a | Ki (nM) b | IC50 | Ki | |
| #66 | 8.7 ± 1.9 | 3.4 ± 0.7 | 1027.0 ± 137.8 | 91.7 ± 4.6 | 117.8 | 26.7 |
| #151 | 7.1 ± 0.9 | 3.0 ± 0.1 | 52.8 ± 6.9 | 5.5 ± 1.0 | 7.5 | 1.8 |
| #153 | 8.2 ± 1.4 | 3.6 ± 0.1 | 53.5 ± 6.8 | 4.9 ± 1.5 | 6.5 | 1.4 |
| TMQ | 2.2 ± 0.7 | 1.3 ± 0.4 | 6.87 ± 0.52 | 1.88 | 3.1 | 1.4 |
IC50, 50% inhibitory concentration; Ki, inhibitory constant; SI, selectivity index.
IC50 = Ci/(Vo/Vi−1), where Vo and Vi are the initial velocities in the absence and presence of inhibitor, respectively, and Ci is the concentration of inhibitor.
Ki calculation uses the Michaelis–Menten equation for competitive inhibition.
SI is defined as IC50(hDHFR)/IC50(SmDHFR) or Ki(hDHFR)/Ki (SmDHFR).
The SIs of these inhibitors were determined by the IC50 ratio of hDHFR over SmDHFR. Although the IC50 value of SmDHFR for analogue #66 (8.7 ± 1.9 nM) was ca. 4-fold higher than that of TMQ (2.2 ± 0.7 nM), the selectivity of the analogue #66 (117.8) was much greater than that of TMQ (3.1) towards inhibiting S. mutans (Table 2). In addition, the DHFR selectivity by the analogue #66 was also greater than that of analogues #151 and #153 (117.8 vs. 7.5 and 6.5) (Table 2). Furthermore, the inhibitor constant Ki as an indicator for the potency of each analogue was also evaluated (Fig. 4; Supplementary Fig. S2; Table 2). The Ki of SmDHFR for analogue #66 was only ca. 2-fold higher than that of TMQ, but analogue #66 had the best SI(Ki) (26.7 vs. 1.4). The SI(Ki) of compound #66 (26.7) was much greater than that of the analogues #151(1.8) and #153 (1.4), further indicating that analogue #151 and #153 had lower selectivity against hDHFR. These data further demonstrated that compound #66 was the most potent and selective analogue towards inhibiting SmDHFR.
Fig. 4.

Inhibition of (A) Streptococcus mutans dihydrofolate reductase (SmDHFR) and (B) human DHFR (hDHFR) by analogue #66. The enzymatic activity of DHFR was measured at a series of fixed concentrations of dihydrofolate (DHF) (20–40 μM) with varying concentrations of analogue #66. The reaction rate was determined by monitoring the decrease in absorbance at 340 nm [oxidation of NADPH to NADP+ and reduction of DHF to tetrahydrofolate (THF)] (coupled ε = 12.260 M−1cm−1) using a UV–visible spectrophotometer. The double-reciprocal plots were used to calculate the reaction constant (Km) and the inhibitory constant (Ki) for the inhibition of SmDHFR and hDHFR by the analogue.
3.5. Analogue #66 bound tighter to SmDHFR than to hDHFR
To explain the biological activity and observed selectivity of TMQ analogues towards inhibiting SmDHFR versus hDHFR, homology modelling and docking analysis were employed since the crystal structure of SmDHFR is not available. A homology model of SmDHFR was generated based on an available X-ray crystal structure of SpDHFR (PDB code, 3ix9) [22]. SpDHFR shares 55% sequence identity with SmDHFR (Fig. 5). The overall predicated structure of SmDHFR is very similar to the X-ray crystal structure from SpDHFR. Superposition of the two enzymes showed that the RMSD difference only concentrated in one loop region (SmDHFR, 135 FPLRDFSS 142, and SpDHFR 130 FPAEFDLSL 138) (Fig. 6). It is known that loops have a tendency to change their conformation upon crystal contact, thus the model prediction is likely to be correct. In addition, the loop is far away from the active site, and the superposition of the active site residues exhibited significantly small RMSD.
Fig. 5.

Sequence alignment of Streptococcus pneumoniae dihydrofolate reductase (SpDHFR) and Streptococcus mutans DHFR (SmDHFR). SpDHFR shares 55% overall sequence identity and 79% active-site identity with SmDHFR. Stars highlight the identical active sites of SpDHFR and SmDHFR.
Fig. 6.

Superposition of the homology model of Streptococcus mutans dihydrofolate reductase (SmDHFR) and template Streptococcus pneumoniae DHFR (SpDHFR). Homology model SmDHFR (green) and 3D structure of SpDHFR (orange) are superposed. With an overall root-mean-square deviation (RMSD) value of 1.76 Å, the only difference between the model and the target is shown in the box. The location is far away from the ligand MTX (purple), a potential enzymatic active site of SpDHFR. The active site superposition of SpDHFR and SmDHFR has a root-mean0square deviation (RMSD) of 4.97 × 10−4 (range: SmDHFR amino acid residues 12–123, SpDHFR residues 8–119). The image and calculation were generated using VMD 1.9.1.
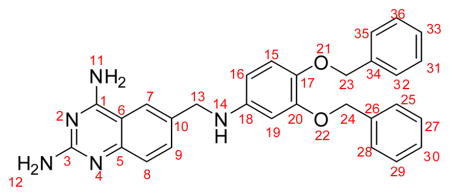
The structure of the analogue compound #66 generated by VEGA ZZ was docked into the active sites of hDHFR and SmDHFR using FlexX and HYDE (Fig. 7). A summary of the results (Table 3) shows the free energy contribution of each atom and fragment to the binding of SmDHFR and hDHFR to analogue #66. The free energy calculation indicated that analogue #66 bound to SmDHFR at a nanomolar range and to hDHFR at a micromolar range. To analyse the docking details, the contribution of each numbered atom to the ΔG values was assessed. The accumulated score for 2,4-diaminoquinazoline scaffold (atoms 1–12) was −19.6 kJ/mol for SmDHFR versus −12.5 kJ/mol for hDHFR, and the methyl amine linker (atoms 13–14) contributed −3.0 kJ/mol to SmDHFR versus 0.7 kJ/mol to hDHFR, whilst phenyl groups (atoms 15–36) exhibited almost the same binding energy.
Fig. 7.

Docking orientation of the analogue compound #66 into the active sites of (A) human dihydrofolate reductase (hDHFR) and (B) Streptococcus mutans DHFR (SmDHFR). Protein structural models are shown in cartoon diagrams. Ligand and active site residues are presented as stick models (colour code: orange/grey-C, red-O, blue-N). The analogue ligand #66 was docked with FlexX and was then redocked by HYDE. Images were generated using Chimera 1.8.1.
Table 3.
Analogue #66 with numbered atoms, and table with HYDE score for free energy binding of the analogue molecule #66 to Streptococcus mutans dihydrofolate reductase (SmDHFR) and human DHFR (hDHFR)
| ||
|---|---|---|
| Atom # | HYDE score
|
|
| Sm DHFR | hDHFR | |
| 1 | −2.2 | −1.5 |
| 2 | 1.6 | 3.7 |
| 3 | −0.9 | −1.1 |
| 4 | −3.8 | −5.8 |
| 5 | −0.2 | −0.3 |
| 6 | −0.5 | −0.2 |
| 7 | −2.9 | −0.2 |
| 8 | −1.2 | −1.3 |
| 9 | −2.7 | −4.0 |
| 10 | −0.1 | 0 |
| 11 | −2.5 | 5.6 |
| 12 | −4.2 | −7.4 |
| 13 | −3.9 | −0.5 |
| 14 | 0.9 | 1.2 |
| 15 | −0.2 | −2.7 |
| 16 | −2.2 | −1.4 |
| 17 | −0.2 | −0.8 |
| 18 | −0.7 | −1.9 |
| 19 | −1.5 | −1.1 |
| 20 | −0.4 | −1.0 |
| 21 | −0.6 | 0.6 |
| 22 | 0.2 | −2.7 |
| 23 | 0 | −2.2 |
| 24 | −6.6 | −1.7 |
| 25 | −2.6 | −3.4 |
| 26 | −0.3 | −0.1 |
| 27 | −2.3 | −1.2 |
| 28 | −2 | −0.4 |
| 29 | −0.3 | 0.8 |
| 30 | −0.1 | −0.4 |
| 31 | −0.1 | −0.1 |
| 32 | −0.1 | −1.3 |
| 33 | −0.1 | 0 |
| 34 | 0 | −0.9 |
| 35 | −0.7 | −0.7 |
| 36 | −1.1 | −0.2 |
| 1–12 | −19.6 | −12.5 |
| 13–14 | −3 | 0.7 |
| 15–36 | −21.9 | −22.8 |
| Total ΔG | −44.5 | −34.6 |
4. Discussion
A variety of natural products [23,24] and semisynthetic small molecules have been shown to inhibit biofilm formation of diverse bacteria [14]. However, the inhibitory mechanisms of these molecules are not well understood. In this study, we identified new small molecules that inhibited a well-defined target, DHFR. DHFR has been proven to be a viable target [8,10–13]. TMQ is a promising lipophilic antifolate drug [25]. Despite being an FDA-approved drug, TMQ has a very low selectivity against hDHFR and is thus not a desirable anti-S. mutans drug. The three analogues (#66, #151 and #153) identified retain the key structural components of TMQ, namely a 2,4-diaminoquinazoline scaffold, N-linker and a modified phenyl ring (Fig. 1B). As a result, all three compounds exhibit excellent inhibition against SmDHFR despite the activity not being as potent as that of TMQ. Importantly, analogue #66 is highly selective against SmDHFR versus hDHFR, being 38-fold greater than TMQ in its selectivity. In addition, it exhibits almost 26-fold higher potency toward inhibiting SmDHFR. Although the cytotoxicity of the analogue is unknown, it likely exhibits less adverse effects on human cells than TMQ owning to its high selectivity.
TMQ blocks the DHFR activity both of Trypanosoma cruzi and P. carinii [9,26]. However, the selectivity of this compound is very low [9]. Attempts to enhance the potency and selectivity of TMQ have achieved limited success until recently [27,28]. In contrast to TMQ, the SI of the analogue #66 against SmDHFR over hDHFR reached 117.8, indicating its potential as a leading compound to develop an anti-S. mutans drug.
Despite the low selectivity, TMQ is very potent against S. mutans. This is partly due to the lipophilic nature of TMQ, which allows it to readily permeate the cell envelop of S. mutans [29]. This feature can be harnessed to increase the potency of our lead compounds. Analogues #66, #151 and #153 did not inhibit cell growth or biofilm formation of the commensal streptococci at the IC50 value, whereas S. mutans was significantly inhibited, suggesting that the analogues possessed good selectivity. Another unique feature of the analogue #66 is its ability to selectively inhibit biofilm formation as well. It exhibited a greater selectivity in inhibiting S. mutans biofilm formation than TMQ. These features can be explored in the further design of more selective #66 derivatives.
To ascertain why the analogue compound #66 possessed high selectivity, the homology modelled SmDHFR and docking free energy were used to determine the binding affinity and selectivity of the analogue #66 by FlexX docking and HYDE redocking (Fig. 7). The docking predicts that the analogue compound #66 binds to SmDHFR in a nanomolar range and to hDHFR in a micromolar range, with an estimated 100-fold selectivity. Based on the free energy distribution, the discrimination largely stems from the 2,4-diaminoquinazoline scaffold and in part from the methyl amine linker. Perhaps binding of the 2,4-diaminoquinazoline scaffold and methyl amine linker are affected by the binding of bulky phenyl rings, since the other analogues #151 and #153 did not have significant selectivity despite their structural resemblance. Free-energy simulations and binding affinity determination based on homology modelling have been successfully used in the study of DHFR of Plasmodium falciparum [29]. The current model prediction is consistent with the experimental finding, demonstrating the validity of the approach. Recently structure-based design has led to the development of new DHFR inhibitors that are potent and selective against Staphylococcus aureus in vitro and in vivo [30], indicating the potential of developing more potent and selective antifolate drugs.
Two strategies can be employed to improve the activity of TMQ analogues against SmDHFR. One focuses on improving both potency and selectivity of the inhibitors by a structure-based design, whilst a complementary strategy will be to enhance solubility and permeability of the lead compounds. These efforts would be greatly facilitated by obtaining X-ray crystal structures of the DHFR–inhibitor complexes. Introduction of the bulky benzyl groups dramatically increases the selectivity of SmDHFR over hDHFR. Further structural modification may not only increase the selectivity but also enhance their potency in inhibiting bacterial biofilms
In conclusion, the TMQ analogues identified in this study selectively inhibited cell growth and biofilm formation of S. mutans biofilms. The 100-fold increase in the selectivity of this antifolate analogue against SmDHFR versus hDHFR is greater than those ever reported for DHFR inhibitors of a number of different bacteria [8,9,11,13]. Therefore, this analogue is a promising lead to develop effective antifolate therapeutic drugs for the prevention and treatment of dental caries.
Supplementary Material
Highlights.
Generated and screened a library of dihydrofolate reductase (DHFR) inhibitor analogues based on an FDA-approved drug.
Small molecules inhibited DHFR activity in vitro and blocked biofilm formation in vivo.
The most potent analogue selectively inhibited the cariogenic bacterium Streptococcus mutans.
Computer modelling reveal why the analogue has the observed selectivity.
The potent analogue has the potential to be developed into an anticaries drug.
Acknowledgments
The authors would like to thank Dr Christian Lemmen for advice related to LeadIT.
Funding: This study was funded by the National Institute of Dental and Craniofacial Research, National Institutes of Health (NIDCR/NIH) [R01 DE022350] and an International Association for Dental Research/GlaxoSmithKline (IADR/GSK) Innovation in Oral Care Award.
Footnotes
Competing interests: None declared.
Ethical approval: Not required.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Loesche WJ. Role of Streptococcus mutans in human dental decay. Microbiol Rev. 1986;50:353–80. doi: 10.1128/mr.50.4.353-380.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hada LS, Kakiuchi N, Hattori M, Namba T. Identification of antibacterial principles against Streptococcus mutans and inhibitory principles against glucosyltransferase from the seed of Areca catechu L. Phytother Res. 1989;3:140–4. [Google Scholar]
- 3.Ooshima T, Izumitani A, Minami T, Yoshida T, Sobue S, Fujiwara T, et al. Noncariogenicity of maltitol in specific pathogen-free rats infected with mutans streptococci. Caries Res. 1992;26:33–7. doi: 10.1159/000261424. [DOI] [PubMed] [Google Scholar]
- 4.Hamada S, Horikoshi T, Minami T, Kawabata S, Hiraoka J, Fujiwara T, et al. Oral passive immunization against dental caries in rats by use of hen egg yolk antibodies specific for cell-associated glucosyltransferase of Streptococcus mutans. Infect Immun. 1991;59:4161–7. doi: 10.1128/iai.59.11.4161-4167.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kakiuchi N, Hattori M, Nishizawa M, Yamagishi T, Okuda T, Namba T. Studies on dental caries prevention by traditional medicines. VIII. Inhibitory effect of various tannins on glucan synthesis by glucosyltransferase from Streptococcus mutans. Chem Pharm Bull (Tokyo) 1986;34:720–5. doi: 10.1248/cpb.34.720. [DOI] [PubMed] [Google Scholar]
- 6.Robinette RA, Heim KP, Oli MW, Crowley PJ, McArthur WP, Brady LJ. Alterations in immunodominance of Streptococcus mutans AgI/II: lessons learned from immunomodulatory antibodies. Vaccine. 2014;32:375–82. doi: 10.1016/j.vaccine.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schnell JR, Dyson HJ, Wright PE. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu Rev Biophys Biomol Struct. 2004;33:119–40. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- 8.Gonen N, Assaraf YG. Antifolates in cancer therapy: structure, activity and mechanisms of drug resistance. Drug Resist Updat. 2012;15:183–210. doi: 10.1016/j.drup.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Senkovich O, Bhatia V, Garg N, Chattopadhyay D. Lipophilic antifolate trimetrexate is a potent inhibitor of Trypanosoma cruzi: prospect for chemotherapy of Chagas’ disease. Antimicrob Agents Chemother. 2005;49:3234–8. doi: 10.1128/AAC.49.8.3234-3238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roth B. Design of dihydrofolate reductase inhibitors from X-ray crystal structures. Fed Proc. 1986;45:2765–72. [PubMed] [Google Scholar]
- 11.Suling W, Reynolds R, Barrow E, Wilson L, Piper J, Barrow W. Susceptibilities of Mycobacterium tuberculosis and Mycobacterium avium complex to lipophilic deazapteridine derivatives, inhibitors of dihydrofolate reductase. J Antimicrob Chemother. 1998;42:811–5. doi: 10.1093/jac/42.6.811. [DOI] [PubMed] [Google Scholar]
- 12.Rosowsky A, Mota CE, Wright JE, Freisheim JH, Heusner JJ, McCormack JJ, et al. 2,4-Diaminothieno[2,3-d]pyrimidine analogues of trimetrexate and piritrexim as potential inhibitors of Pneumocystis carinii and Toxoplasma gondii dihydrofolate reductase. J Med Chem. 1993;36:3103–12. doi: 10.1021/jm00073a009. [DOI] [PubMed] [Google Scholar]
- 13.Fischi MA, Dickinson GM, La Vole L. Safety and efficacy of sulfamethoxazole and trimethoprim chemoprophylaxis for Pneumocystis carinii pneumonia in AIDS. JAMA. 1988;259:1185–9. doi: 10.1001/jama.259.8.1185. [DOI] [PubMed] [Google Scholar]
- 14.Liu C, Worthington RJ, Melander C, Wu H. A new small molecule specifically inhibits the cariogenic bacterium Streptococcus mutans in multispecies biofilms. Antimicrob Agents Chemother. 2011;55:2679–87. doi: 10.1128/AAC.01496-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou M, Zhu F, Li Y, Zhang H, Wu H. Gap1 functions as a molecular chaperone to stabilize its interactive partner Gap3 during biogenesis of serine-rich repeat bacterial adhesin. Mol Microbiol. 2012;83:866–78. doi: 10.1111/j.1365-2958.2012.07970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedretti A, Villa L, Vistoli G. VEGA: a versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J Mol Graph Model. 2002;21:47–9. doi: 10.1016/s1093-3263(02)00123-7. [DOI] [PubMed] [Google Scholar]
- 17.Schneider N, Hindle S, Lange G, Klein R, Albrecht J, Briem H, et al. Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J Comput Aided Mol Des. 2012;26:701–23. doi: 10.1007/s10822-011-9531-0. [DOI] [PubMed] [Google Scholar]
- 18.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 19.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 20.Eisenberg D, Lüthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 21.Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–50. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee J, Yennawar NH, Gam J, Benkovic SJ. Kinetic and structural characterization of dihydrofolate reductase from Streptococcus pneumoniae. Biochemistry. 2010;49:195–206. doi: 10.1021/bi901614m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duarte S, Gregoire S, Singh AP, Vorsa N, Schaich K, Bowen WH, et al. Inhibitory effects of cranberry polyphenols on formation and acidogenicity of Streptococcus mutans biofilms. FEMS Microbiol Lett. 2006;257:50–6. doi: 10.1111/j.1574-6968.2006.00147.x. [DOI] [PubMed] [Google Scholar]
- 24.Percival RS, Devine DA, Duggal MS, Chartron S, Marsh PD. The effect of cocoa polyphenols on the growth, metabolism, and biofilm formation by Streptococcus mutans and Streptococcus sanguinis. Eur J Oral Sci. 2006;114:343–8. doi: 10.1111/j.1600-0722.2006.00386.x. [DOI] [PubMed] [Google Scholar]
- 25.Blanke CD. A double-blind placebo-controlled randomized phase III trial of 5-fluorouracil and leucovorin, plus or minus trimetrexate, in previously untreated patients with advanced colorectal cancer. Ann Oncol. 2002;13:87–91. doi: 10.1093/annonc/mdf043. [DOI] [PubMed] [Google Scholar]
- 26.Kovacs JA, Allegra CJ, Swan JC, Drake JC, Parrillo JE, Chabner BA, et al. Potent antipneumocystis and antitoxoplasma activities of piritrexim, a lipid-soluble antifolate. Antimicrob Agents Chemother. 1988;32:430–3. doi: 10.1128/aac.32.4.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schormann N, Velu SE, Murugesan S, Senkovich O, Walker K, Chenna BC, et al. Synthesis and characterization of potent inhibitors of Trypanosoma cruzi dihydrofolate reductase. Bioorg Med Chem. 2010;18:4056–66. doi: 10.1016/j.bmc.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 28.White EL, Ross LJ, Cunningham A, Escuyer V. Cloning, expression, and characterization of Mycobacterium tuberculosis dihydrofolate reductase. FEMS Microbiol Lett. 2004;232:101–5. doi: 10.1016/S0378-1097(04)00038-2. [DOI] [PubMed] [Google Scholar]
- 29.Gangjee A, Vidwans AP, Vasudevan A, Queener SF, Kisliuk RL, Cody V, et al. Structure-based design and synthesis of lipophilic 2,4-diamino-6-substituted quinazolines and their evaluation as inhibitors of dihydrofolate reductases and potential antitumor agents. J Med Chem. 1998;41:3426–34. doi: 10.1021/jm980081y. [DOI] [PubMed] [Google Scholar]
- 30.Lam T, Hilgers M, Cunningham ML, Kwan BP, Nelson KJ, Brown-Driver V, et al. Structure-based design of new dihydrofolate reductase antibacterial agents: 7-(benzimidazol-1-yl)-2,4-diaminoquinazolines. J Med Chem. 2014;57:651–68. doi: 10.1021/jm401204g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.