

Abstract
The adoptive transfer of genetically engineered T cells with cancer-targeting receptors has shown tremendous promise for eradicating tumors in clinical trials. This form of cellular immunotherapy presents a unique opportunity to incorporate advanced systems and synthetic biology approaches to create cancer therapeutics with novel functions. Here, we first review the development of synthetic receptors, switches, and circuits to control the location, duration, and strength of T cell activity against tumors. In addition, we discuss the cellular engineering and genome editing of host cells (or the chassis) to improve the efficacy of cell-based cancer therapeutics, and to reduce the time and cost of manufacturing.
Keywords: Synthetic biology, genetic circuits, chimeric antigen receptors, immunotherapy, cancer
Emergence of cellular immunotherapy
The intricate relationship between tumors and the immune system has been the subject of intense research, providing both insight into cancer progression [1, 2] and an arena for therapeutic intervention [3]. The immune system can directly attack tumors, and harnessing this power to eradicate tumors is a major goal in immunotherapy. The involvement of the immune cells in combating tumors was demonstrated when a lower rate of relapse was observed in cancer patients who underwent a hematopoietic stem cell transplant (HSCT) (see Glossary) to replace their bone marrow after chemotherapy [4–6]. This effect has been attributed to fresh T cells from the transplant engaging and killing the tumor in a graft-versus-tumor (GVT) response. However, this response is also correlated to graft-versus-host disease (GVHD), wherein the donor T cells begin to attack the host’s own tissue. This potential autoimmune response has limited the use of stem cell transplants for cancer treatment as a universal solution. Instead, aiding a patient’s own immune system to fight cancer may provide more viable, widespread therapies. However, cancer cells have also evolved strategies to oppose immune action [2, 3, 7]. As such, a major goal of cancer immunotherapy is to overcome these immunosuppressive mechanisms, including the use of cytokines to promote T cell proliferation [8–10] or antibody checkpoint blockers to prevent the signaling of inhibitory or apoptotic pathways of a T cell [11–13]. Cytokines and checkpoint blockers have shown great promise in therapy [14, 15], and several high profile drugs have been approved in recent years. These forms of immunotherapy aid the body’s response against cancer, but immune cells can also be directly used as therapeutic agents.
Cells are inherently capable of carrying out complex computations and responses, and the immune system in particular is composed of cells designed to perform cytotoxic tasks through careful assessment of targets. Adoptive T cell therapy, the use and engineering of a patient’s T cells as therapeutic agents, has emerged as a promising branch of immunotherapy (Figure 1 A). Much of the current success in adoptive T cell therapy is derived from the genetic engineering of tumor-targeting receptors. However, synthetic sensors, switches, and circuits are also being explored to improve efficacy and safety by providing greater control over the location, duration, and magnitude of T cell activity (Figure 1C). Synthetic biology, an emerging discipline aimed at reprogramming living organisms through the combined use of genetics, engineering principles, and systems and computational analysis [16–18], is primed to deliver the genetic tools necessary to enhance the control of these living therapies and explore T cell behavior [19]. In addition to the introduction of exogenous sensors and circuits, the endogenous machinery of the host cell (chassis) presents numerous opportunities for tinkering and optimization (Figure 1C). Therefore, cellular engineering and genome editing of T cells are also under active investigation [20, 21]. Much akin to the role synthetic chemistry plays in transforming the development of small molecule drugs, synthetic biology approaches are becoming a major engine in driving the progress of adoptive T cell therapy.
Figure 1.
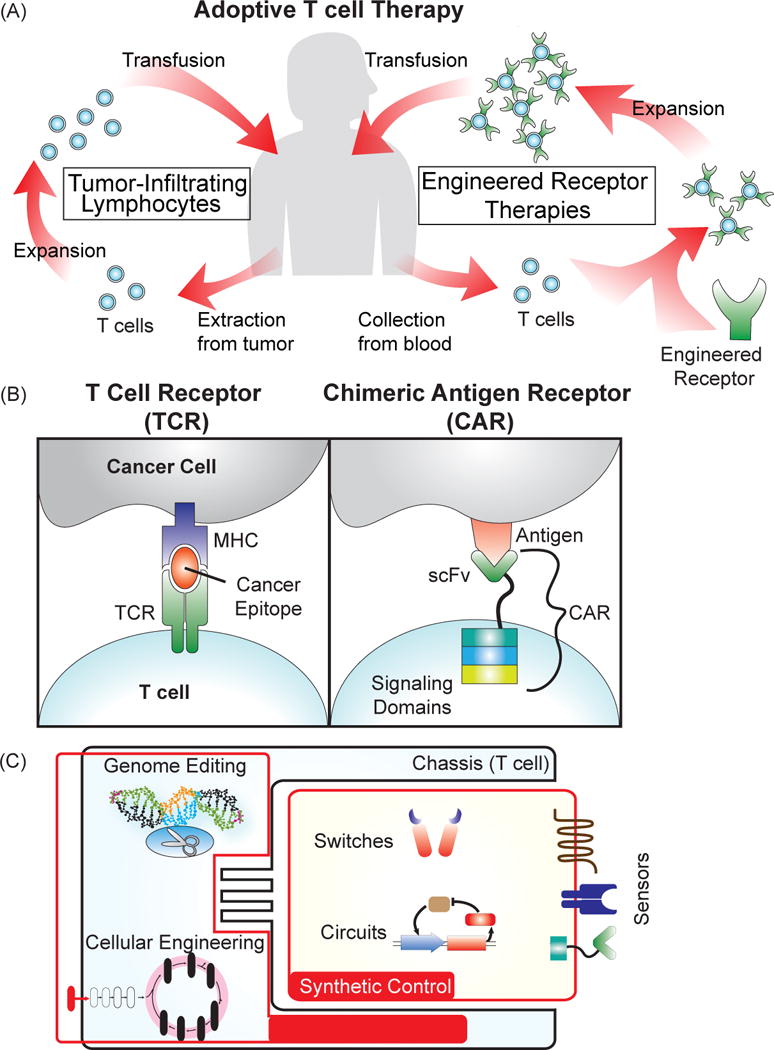
Adoptive T cell therapy for cancer treatment. (A) Several approaches for the adoptive transfer of a patient’s own T cells for cancer therapy. Tumor-infiltrating lymphocytes (TILs) involve extraction of T cells directly from the tumor, ex vivo expansion, and then transfusion back into the patient. For engineered receptor therapies, T cells are collected from the blood, genetically modified to express a cancer-targeting receptor, expanded, and then transfused back into the patient. (B) Receptors engineered to target cancer cells. T cell receptors (TCRs) naturally recognize protein epitopes presented by the major histocompatibility complex of a target cell. Engineering a TCR to detect cancer epitopes “teaches” the T cell to detect cancer cells. Chimeric antigen receptors (CARs) are composed of a single-chain variable fragment (scFv) from an antibody fused to intracellular T cell signaling domains that trigger activation and proliferation of the T cell. CARs recognize markers expressed at the surface of a cell, and by choosing a cancer-specific scFv, can be made to trigger killing of the cancer cell upon binding to the target antigen. (C) Engineering T cells for improvement of adoptive T cell therapy. Generating novel receptors and circuits can enable increased control over cell-based therapies, and techniques to engineer the chassis, such as genome editing and cellular engineering, can drive the development of more powerful treatments.
Genetic engineering and cellular immunotherapy: a potent combination against tumors
One of the most promising and earliest forms of adoptive T cell therapy involves the use of a patient’s tumor-infiltrating lymphocytes (TILs), which are T cells extracted from the tumor. These isolated TILs were expanded ex vivo, and then transfused back into the patient to treat cancer [22]. Due to their inherent ability to locate and traffic to the tumor site, TILs have had some success against melanoma in clinical trials [23, 24]. However, the identification and isolation of TILs in sufficient quantity from a patient is challenging, limiting their potential [25]. The shortcomings of TILs have accelerated efforts to redirect the specificity of T cells towards cancer rather than relying on the isolation of T cells with inherent tumor-targeting capability. A patient’s T cells can be modified with genes that encode tumor-targeting receptors that will “teach” the T cell to bind and kill cancer cells [25]. In this process, T cells (typically CD8 T cells) are collected from the patient, genetically modified ex vivo to express the receptor, and then transfused back into the patient. Two different types of receptors have been used for this purpose. One is a T cell receptor (TCR) that is engineered to detect cancer epitopes [26, 27]. The other is a chimeric antigen receptor (CAR) that is composed of a cancer antigen-specific single chain variable fragment (scFv) fused to T cell signaling domains that trigger activation and proliferation [28, 29] (Figure 1B). The design of CARs has undergone some engineering through the choice and addition of different T cell signaling domains that can drive activation or proliferation, resulting in therapeutic variations between these different designs. TCRs and CARs are distinguished from one another by the type of cancer antigen they recognize. TCRs on CD8 T cells recognize protein epitopes derived from proteins expressed in the cell and presented on the surface by the major histocompatibility complex-1 (MHC-1). CARs bind to markers expressed at the surface of the cell.
Both TCR- and CAR-based therapy have been tested in clinical trials with promising results. In one clinical trial treating 20 patients with melanoma using TCRs targeted towards Melanoma Antigen Recognized by T cells 1 (MART-1), 33% of the patients demonstrated objective responses (Clinical Trials: NCT00509288, NCT00509496) [27]. Treatment of lymphoid leukemia with CD19-specific CARs have shown up to a 90% complete response rates (Clinical Trials: NCT01044069, NCT01626495, NCT01029366, NCT01593696) [30–33], though similar clinical success in CAR-based targeting of myeloid leukemia has not been achieved yet (Clinical Trials: NCT01864902, CTX 08-0002) [34, 35].
Although extremely encouraging and commonly considered to be breakthroughs in the fight against cancer [36], toxicities have been observed in clinical trials associated with both forms of engineered T cell therapy [31, 37]. The selectivity between tumors and vital organs is an especially significant safety issue that has emerged with both TCRs and CARs [37]. The identification of target epitopes and antigens for these therapies is limited by the potential for expression of these targets on non-cancerous cells, which could lead to autoimmune responses against healthy tissue. MART-1 has demonstrated this “on-target, off-tumor” autoimmune toxicity in TCR therapy [27]. And in one trial using an ERBB2-specific CAR to treat a patient with colon cancer, the patient died after the CAR-bearing T cells responded to low levels of ERBB2 in the vital organs (Clinical Trial: NCT00924287) [38].
Another major safety concern is the potential for an excessively strong, life-threatening T cell response. In clinical trials using CARs to treat leukemia, the release of large amounts of cytokines [30], or cytokine release syndrome (CRS), has led to severe symptoms including high fever, hypotension, and hypoxia [30]. CRS has been treated with immunosuppressive steroids and antibodies to temper the response of the immune system [33]. A recent clinical trial was also conducted to determine the maximum load of CAR-bearing T cells that can be given to a patient while minimizing the severity of CRS [33].
Despite these adverse side effects, the promising results of adoptive T cell therapy in clinical trials have generated enormous enthusiasm, which has led to numerous joint ventures, acquisitions, and collaborations within the pharmaceutical industry, as well as between industry and academia (Table 1). In particular, CARs have attracted the most attention because of their extraordinarily positive clinical trial results (Table 1). Both the success of these clinical trials and the significant financial investment from the industry heighten the urgency to engineer a cell-based therapy that is effective and safe, as well as to design practical strategies that will make manufacturing these therapies cheaper and faster.
Table 1.
Advances in adoptive T cell therapy
| Institutions | Date | Technology | Clinical Trials | Details | Ref |
|---|---|---|---|---|---|
| University of Pennsylvania/Novartisa | August, 2012 | CTL019 | NCT02167360, NCT02228096, NCT02030834, NCT01626495, NCT01029366 | CD19-specific CAR for B Cell Acute Lymphoblastic Leukemia (B-ALL) | [32] |
| Juno Therapeuticsb,c | December, 2013 | JCAR015 | NCT01044069, NCT01840566 | CD19-specific CAR for B-ALL and non-Hodgkin lymphoma (NHL) | [30, 109–111] |
| JCAR017 | NCT02028455 | CD19-specific CAR for leukemia | |||
| JCAR014 | NCT01865617 | CD19-specific CAR for chronic lymphocytic leukemia, NHL, ALL | |||
| JTCR016 | NCT01640301, NCT00052520 | WT1-specific TCR for leukemia, myelodysplastic syndrome | |||
|
Exploration of other targetsd |
Preclinical |
L1CAM: neuroblastoma, glioblastoma, lung cancer, pancreatic cancer, and ovarian cancer MUC-16: ovarian cancer ROR-1: lung cancer, triple negative breast cancer, pancreatic cancer, prostate cancer, ALL |
|||
| Juno/Opus Bio, Inc./National Cancer Institute (NCI)e | December, 2014 | CD22-CAR | NCT02315612, NCT02159495 | Leukemia, lymphoma | |
| Celgene/Bluebird Bio/Baylorf | March, 2013 | Hematological/Solid Tumors | |||
| Kite Pharmaceuticals/National Cancer Instituteg,h | October, 2012 | CD19-CAR | NCT00924326, NCT02348216 | B-cell leukemia, lymphoma | [112, 113] |
| EGFRvlll-CAR | NCT01454596 | Glioblastoma | [114, 115] | ||
| NY-ESO-1 TCR | NCT01967823 | ESO-expressing tumors | |||
| HPV-16E6 TCR | NCT0228081 | Cervical/Head & Neck Cancer | |||
| HPV-16E7 TCR | Cervical/Head & Neck Cancer | ||||
| MAGE A3 TCR | NCT02111850 | MAGE-A3-DP4-expressing tumors | |||
| SSX2 TCR | NCT02153905 | MAGE-A3-expressing tumors, metastatic melanoma | |||
| Kite Pharmaceuticals/Amgen | January, 2015 | Exploration of other targets | Preclinical | Hematologic cancers and solid tumors | |
| MD Anderson/Ziopharm Oncology/lntrexonj | January, 2015 | RheoSwitch Therapeutic System | Preclinical | Switches for control of dynamic range, spatial expression, and temporal expression | |
| Non-Viral Integration | Sleeping Beauty transposon system for integration of CARs in T cells | ||||
| Universal donor | Preclinical | ||||
| CD19-specific CAR | NCT01497184, NCT01653717 | Leukemia, lymphoma | |||
| Cellectis/Ohio State Universityk | January, 2015 | CS1-specific CAR | Preclinical | Multiple myeloma | |
| Pfizer/Cellectisl | June, 2014 | Working on several targets for CAR therapy | |||
| GlaxoSmithKline/Adaptimmunem | June, 2014 | NY-ESO-1 TCR | NCT01350401, NCT01567891, NCT01892293 | Multiple myeloma, melanoma, sarcoma, and ovarian cancer | |
| Advanced Designs for Adoptive T Cell Therapy | |||||
| Servier/Cellectisn | February, 2014 | UCART19 | Filing for clinical trial authorization | Off-the shelf T cell for leukemia | |
| Unum Therapeuticso | 2014 | ACTR+anti-CD20 antibody | NCT02315118 | Leukemia, Non-Hodgkin’s lymphoma | [51] |
| Bellicum Pharmaceuticals | 2011 | GoCAR-T | Preclinical | Prostate stem cell antigen-expressing solid tumors | [50] |
| Bellicum Pharmaceuticals/Texas Children’s Hospital/Baylor College of Medicine | 2011 | CaspaCIDe | NCT01494103 | Graft-versus-host disease | [67] |
| Bluebird bio (Acquisition of Pregenen)p | June, 2014 | Homing endonucleases, MegaTALEs | |||
| Novartis/lntelliaq | January, 2015 | CRISPR | |||
Synthetic receptors and circuits for spatiotemporal control of T cell activity
Current T cell therapies, although promising, all share a similar design that triggers the same signaling pathways in response to a single target antigen. In this section, we review the next wave of receptor designs that expand the signaling pathways triggered, enhance specificity, or provide inducible controls over the therapy. Furthermore, we also discuss the development of drug-inducible switches and circuits that will endow additional spatiotemporal control over the T cell response.
Receptors
Next generation receptors
Understanding how receptors affect T cell response is particularly important to the implementation of CAR-based therapy due to the novel nature of CARs. The design of CARs has undergone changes over time, and the second generation of CARs contains an intracellular proliferative domain derived from either CD28 or 4-1BB (Figure 2A). The choice of proliferative domain has led to divergent therapeutic outcomes that have been further explored in vitro [30–33, 39]. In clinical trials, the CD28 domain has been associated with faster short-term expansion, but also shorter persistence compared to trials that use the 4-1BB domain. Developing CARs with different signaling domains could lead to the development of receptors with varied properties and in turn, treatments that are more complex and oriented around a patient’s specific needs. An inhibitory CAR (iCAR) has been developed using signaling domains from inhibitory pathways to suppress T cell activity upon binding to antigens from healthy cells [40], demonstrating the potential to reprogram the functionality of CARs using different signaling domains. A library approach has been used to test for different proliferative domains and their ability to drive anti-tumor activity [39, 41]. Similar approaches to systematically map the effect of parameters that describe different signaling domains to therapeutic outcomes will provide valuable information for the optimal design of T cell therapy.
Figure 2.
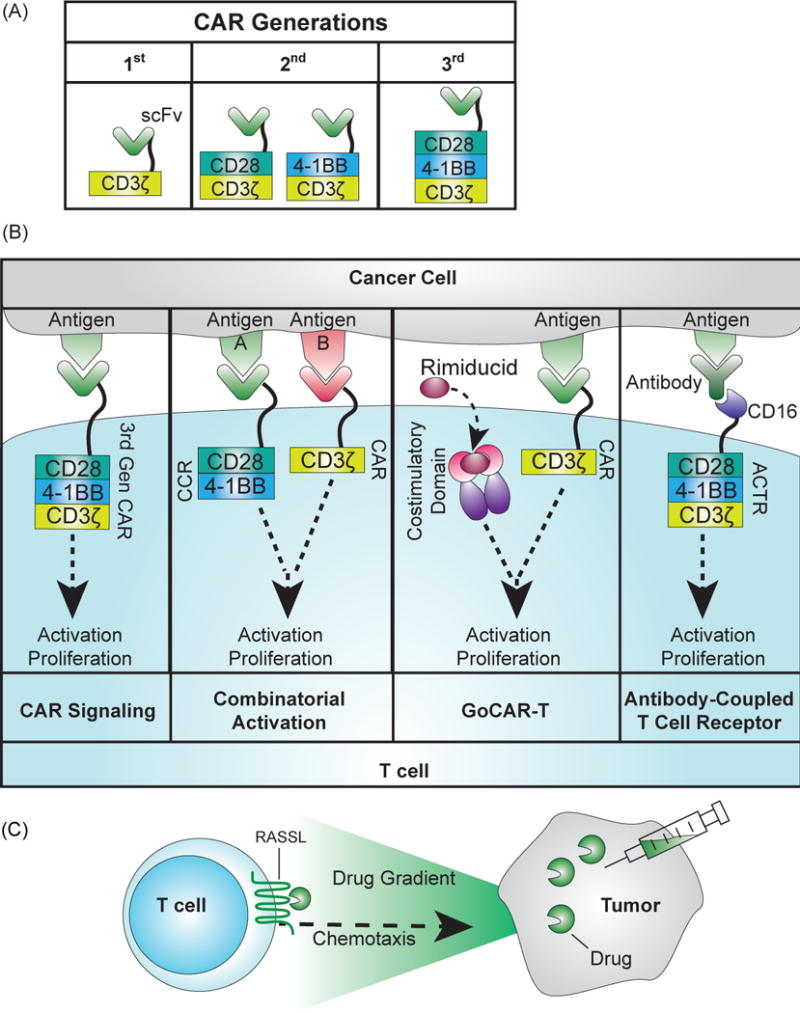
Receptor engineering for adoptive T cell therapy. (A) Development of CARs across three generations. The first generation CAR consisted of a scFv fused to the activating CD3ζ domain that drove activation upon antigen binding, but did not lead to sufficient persistence in clinical trials [116]. Second and third generation CARs have included the intracellular portions of proliferative signaling proteins, CD28 or 4-1BB. (B) Signaling of novel receptors for adoptive T cell therapy. First panel: In second and third generation CARs, binding of the antigen to the scFv triggers proliferation and activation of the T cell. Second panel: With combinatorial activation, binding to the CAR and chimeric costimulatory receptor (CCR) is required to drive both activation and proliferation of the T cell [48]. Third panel: The GoCAR-T system requires binding to the target antigen for activation, but it also requires the addition of the drug rimiducid to dimerize the co-stimulatory domain for activation and proliferation [50] (http://www.bellicum.com/technology/gocart/). Fourth panel: Antibody-coupled T cell receptors (ACTR) express a CD16 domain at the T cell surface instead of a scFv. CD16 binds to antibodies, and choosing antibodies that bind to the surface of cancer cells will drive T cell activity against the cancer cell [51]. (C) Engineered receptor for chemotaxis [59]. RASSLs, an engineered G protein-coupled receptor, are activated at the surface of the T cell by a drug and drive chemotaxis of the cell along the drug gradient. Adding the drug to a tumor can direct T cells towards the tumor site.
Combinatorial receptor system
A major concern in adoptive T cell therapy is the potential for severe side effects due to the inadvertent attack on healthy tissue by engineered T cells [37, 38]. Improving the specificity of the T cell towards cancer represents a high priority in this field, and multiple strategies are being explored to identify tumor-specific antigens [42–47]. However, due to the heterogeneous nature of tumors, a single antigen is unlikely to uniquely distinguish all tumors from healthy tissues. Requiring the T cell to recognize two targets for full activation and proliferation will increase specificity towards the intended cancer cells, which has been achieved using a combinatorial activation system consisting of a low-activating CAR and a chimeric costimulatory receptor (CCR) [48, 49] (Figure 2B). The CAR and CCR each recognize different antigens, and antigen recognition by both receptors is required to drive full activation and proliferation of the T cell. This combinatorial activation system demonstrated promising selectivity in mouse models [48].
Split receptor systems
While specificity is critical to the outcome of T cell therapy, the magnitude and duration of T cell response will also influence the severity of any side effects that arise. Novel split receptor designs where full activation of the engineered T cell requires both the antigen target and an exogenous factor (such as a drug or antibody) are under investigation. These systems provide a method to titrate the response of the T cell through dosage of the secondary activating factors. For example, the GoCAR-T designed by Bellicum Pharmaceuticals contains a CAR that, similar to the combinatorial activation system, is split into the antigen-responsive activation domain and a co-stimulatory domain [50] (http://www.bellicum.com/technology/gocart/) (Figure 2B). The co-stimulatory domain is fused to a rimiducid-inducible homodimerizer domain. For full activation of the T cell, binding to both rimiducid and the antigen is required.
Unum Therapeutics is also developing an alternative “universal CAR” design using their Antibody-Coupled T cell Receptor (ACTR) system (http://www.forbes.com/sites/brucebooth/2014/10/21/cellular-immunotherapy-unum-therapeutics-out-of-many-one/). ACTR contains the same T cell signaling domains as the current CARs, but the scFv is replaced with the extracellular portion of CD16, a receptor that binds to the constant fragment of antibodies [51] (Figure 2B). With ACTRs, any clinically relevant cancer-specific antibody can, in theory, be administered to the patients. The antibody binds to the T cell through CD16, which triggers T cell activation upon antigen binding. With the prevalence of commercially available, cancer-specific antibodies, the ACTR system can rapidly expand the repertoire of potential targets for engineered T cells. These split systems illustrate the potential of separating target recognition from T cell activation, providing a bedside “on-demand” control of therapeutic strength and duration through the addition of a drug or antibody.
Chemokine receptors
A major barrier to the success of T cell-based therapy against solid tumors is the marked reduction of T cell trafficking to the tumor microenvironment [52]. The irregular blood flow and endothelium modification found in a tumor mass restricts T cell adhesion and infiltration [53]. Recent in vivo results have shown that localized delivery to the tumor can improve T cell activity against a solid tumor [54], and a biopolymer implant has been developed to provide localized delivery in conjunction with cytokines to improve T cell proliferation [55]. T cell localization can also be improved by directing their migration machinery toward signals derived from tumors. In T cells, the overexpression of CXCR2, a receptor that binds to the tumor-derived chemokine Growth-Regulated Oncogene-α, drove preferential trafficking to the site of the tumor [56, 57]. Another approach to direct T cell infiltration into tumors is to introduce small molecule-inducible chemotaxis receptors into the T cell, which can be achieved using engineered G protein-coupled receptors called RASSLs that are activated solely by a synthetic ligand [58] (Figure 2C). The induction ligand can be added to the site of the tumor, directing T cell traffic towards the cancerous cells. Using this system, the migration of engineered T cells can be controlled in mice in response to localized delivery of the small molecule clozapine-N-oxide (CNO) [59].
Control switches and circuits
Kill switches
Designed to increase the safety of the therapy, inducible kill switches are simple circuits that provide a mechanism to terminate a T cell therapy if the patient exhibits severe side effects [60, 61]. Several drug-inducible kill switches have been tested in clinical trials for this purpose. In patients who received allogeneic bone marrow transplants (allo-BMT), the transplanted T cells were modified to express herpes simplex virus-thymidine kinase (HSV-TK), which drives apoptosis in the cell upon addition of ganciclovir [62]. Patients who developed GVHD were treated with ganciclovir, triggering apoptosis in the modified T cells. However, induced killing of T cells was not complete, and the viral origins of HSV-TK led to immunogenic responses [63]. CD20 and an inducible Caspase 9 (iCasp9) (Figure 3A) are being explored as possible alternatives due to their human origins [63, 64]. In particular, iCasp9 has been tested in vivo in conjunction with a CD20-specific CAR to demonstrate its potential to eliminate CAR-bearing T cells [65]. In a clinical trial to treat leukemia patients receiving stem-cell transplants, donor T cells were modified to express iCasp9. In patients who developed GVHD, induction of iCasp9 activity killed more than 90% of the transgenic T cells within 30 minutes of induction, successfully controlling GVHD with no observed immunogenicity [66]. This system is under commercial development by Bellicum Pharmaceuticals (Table 1) [67].
Figure 3.
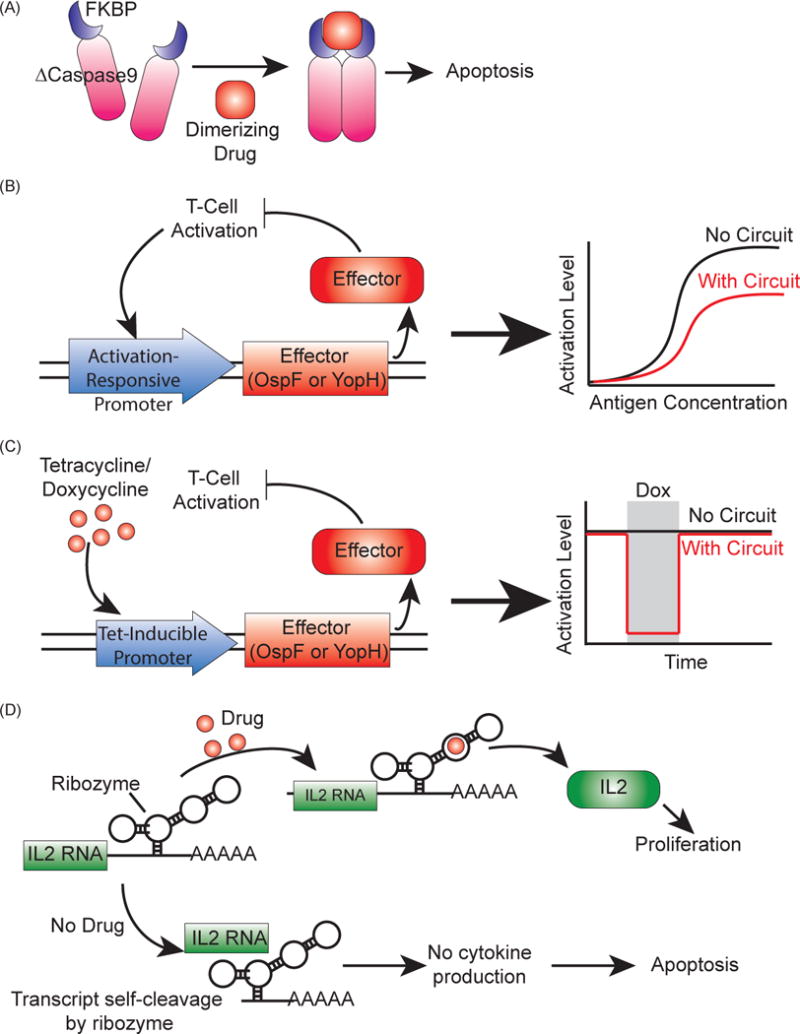
Synthetic genetic circuits to regulate T cell activity in patients. (A) Inducible suicide gene using iCasp9, a Caspase9 mutant (ΔCaspase9) fused to FKBP dimerizing domains [63, 66]. When the dimerizing drug AP1903 is added, ΔCaspase9 dimerizes and drives apoptosis. (B) Amplitude limiter using the bacterial virulence proteins OspF or YopH as effectors [70]. These effectors reduce T cell activation, and expressing them under an activation-responsive promoter creates a negative feedback loop that reduces T cell activation. (C) Pause switches using OspF or YopH as effectors [70]. Using a tetracycline-inducible promoter to control effector expression, the addition of the drug will drive effector production, which will in turn shut off activation until the drug is removed. (D) A ribozyme switch to control T-cell proliferation [71]. Cytokine RNA is expressed with the ribozyme switch, which will drive self-cleavage of the transcript and lead to no cytokine expression without the addition of the appropriate drug. When drug is added, the cytokine transcript is preserved, leading to cytokine production and proliferation.
Pause switch and amplitude limiter
While inducible kill switches provide vital control over the safety of adoptive T cell therapy, they ultimately limit the benefit a patient might receive from the treatment. Modulating the T cell response through other circuits can enable physicians to fine tune the immune response before resorting to termination of the therapy.
The bacterial virulence proteins OspF and YopH can modify the activity of a critical kinase in TCR signaling pathways to reduce T cell activation [68, 69]. With these proteins as effectors in genetic circuit design, the behavior of the T cell can be further controlled. For example, a library expressing OspF or YopH under a series of TCR-responsive promoters were designed as a negative feedback loop to reduce the amplitude of T cell activation [70] (Figure 3B). The amplitude could be furthered adjusted by tagging the OspF or YopH with a degradation tag. This circuit could be used to lower the activation of an engineered T cell, potentially reducing the severity of CRS. OspF and YopH were also used to design an inducible pause switch for T cells [70] (Figure 3C). By expressing the proteins under a doxycycline-inducible promoter, the T cell activity could be paused upon addition of doxycycline. This circuit presents an alternative to the inducible kill switch, allowing for the therapy to stop without completely destroying the cells involved so that they may be used again.
Growth switch
Increasing the growth of T cells will lead to greater persistence of the therapy, while decreasing the growth can potentially limit the severity of CRS. Therefore, a drug-inducible controller for the growth of engineered T cells can provide a powerful “dial” to regulate the efficacy and safety of the therapy. A ribozyme switch to regulate the expression of the cytokines IL2 or IL15 has been developed such that without the addition of a drug, the cytokine-ribozyme mRNA self-cleaves and no cytokine is produced [71] (Figure 3D). The addition of a drug prevents self-cleavage, allowing cytokine production that drives proliferation of the T cell. While T cell expansion can conceivably be controlled by adding cytokines directly to the patient or expressing transgenes under inducible promoters, a large cytokine dosage can lead to systemic toxicity in the patient [72], and the packaging of large amounts of transgenes can be challenging [73]. An RNA-based system allows for specific control that is easy to deliver and non-immunogenic.
Host cell (chassis) engineering and genome editing in adoptive T cell therapy
Another important design criterion of adoptive T cell therapy is deciding the best cell type for therapy as the type of T cell used has a direct role in the efficacy of the treatment. In particular, naïve and early effector T cells are more effective at treating tumors in mice than differentiated effector T cells [74]. This effect was attributed to several characteristics, including the entry of differentiated effector T cells into a proapoptotic state and an inability to produce IL-2. Therefore, using T cells that are less differentiated could potentially increase their efficacy.
Cellular engineering of chassis
Engineering T cells to bias a population towards the naïve phenotype before transfusion back into the patient is an attractive possibility to boost the antitumor effect of the therapy [21]. Increasing evidence illustrates the importance of metabolism on T cell development and differentiation. In particular, differentiation into effector T cells is accompanied with a transition from oxidative phosphorylation to aerobic glycolysis [75]. Limiting the cells’ dependence on glycolysis can prevent T cells from differentiating into effector cells [76]. Hence, a metabolic engineering strategy can be employed to limit glycolysis by lowering the expression of the glucose transporter Glut 1 or by reducing the activation of the protein kinase Akt, a glycolysis enhancer [77] (Figure 4A).
Figure 4.
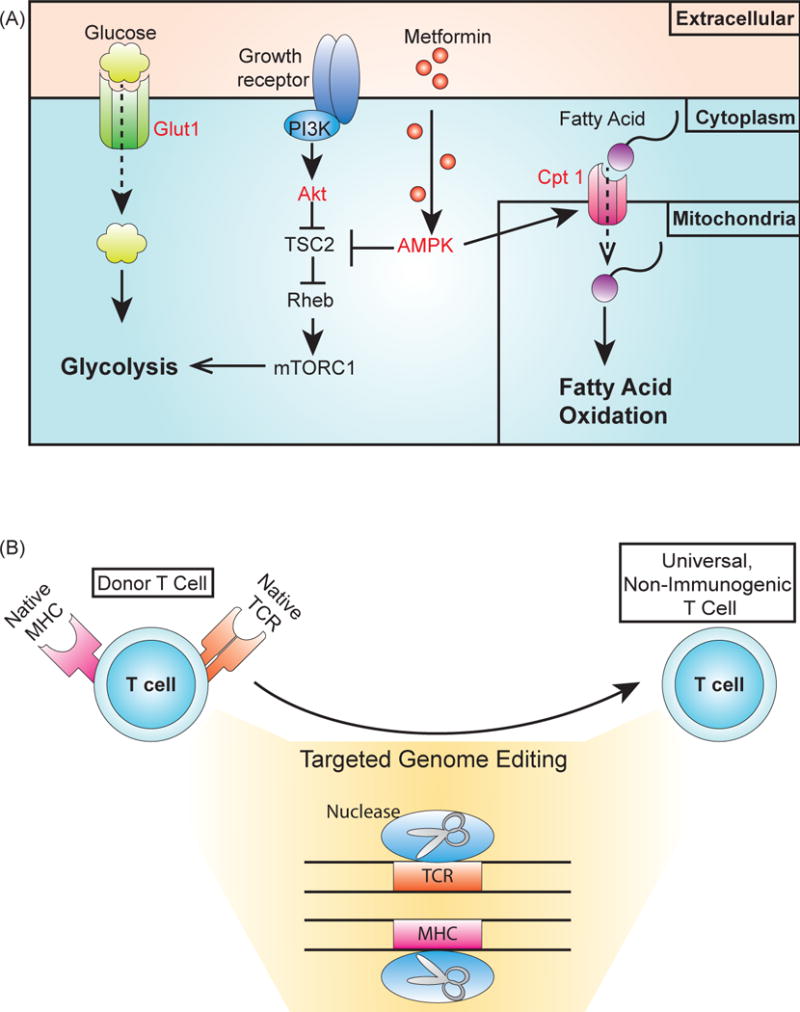
Cellular engineering and genome editing for chassis engineering. (A) Targets to bias T cells towards a naïve state or development of the memory T cell phenotype. Limiting glycolysis can prevent T cells from differentiating into effector cells promote memory T cell development [75, 79]. In addition, memory T cell development can be increased by promoting fatty acid oxidation. Different components of these metabolic pathways can be targeted for these aims (labeled in red). Reducing expression of Glut1, the glucose transporter limits the intake of glucose, while expressing an Akt inhibitor limits glycolysis due to Akt activation of mTOR through TSC2 and Rheb signaling [76, 77, 117]. Fatty acid oxidation can be promoted by overexpressing the fatty acid transporter Cpt-1 [79]. Activating AMPK with the drug metformin promotes fatty acid activation, both by repressing components involved in activating glycolysis and by indirectly overexpressing Cpt-1 [79]. (B) Genome editing for the production of an allogeneic, non-immunogenic T cell. Using targeted nucleases to disrupt expression of the T cell receptor (TCR) renders the T cell unable to detect targets until modification with an engineered receptor [87]. This process could be used to produce universal T cells from healthy donors that could be stored in a “T cell bank” for use in patients, as the lack of endogenous TCR expression would prevent graft-versus-host disease (GVHD). The expression of CARs and genetic circuits can involve the expression of components that elicit an immunogenic response. Disrupting major histocompatibility complex (MHC) expression prevents the T cell from presenting epitopes from these components, which would reduce the risk of an immunogenic response [88].
In addition to using more naïve T cells, promoting the development of the memory T cell phenotype (particularly central or stem cell memory) may help prevent relapse of the disease [78]. Limiting glycolysis can also help to promote memory T cell formation, as can the enhancement of fatty acid oxidation (FAO) [79, 80]. For instance, the addition of metformin, an activator of AMPK that promotes fatty acid oxidation, limits CD8+ effector T cell differentiation while increasing CD8+ memory T cell development (Figure 4A) [79]. Moreover, the overexpression of CPT 1, a critical fatty acid transporter, had been shown to increase FAO and promoter memory T cell formation in mice [79].
RNA interference and genome editing
Clinical trials with CARs have demonstrated significant promise against hematological cancers such as leukemia, but solid tumors remain a major challenge due to their immunosuppressive microenvironment [2]. For example, tumors can deplete the microenvironment of tryptophan [7] while also expressing factors to promote regulatory T cell development [81] and effector T cell death [82]. Antibody drugs such as ipilimumab (anti-CTLA-4) and pembrolizumab (anti-PD-1) attempt to address the challenges presented by the microenvironment by blocking inhibitory or apoptotic signals in T cells. However, given the complexity of the tumor microenvironment, a greater understanding of the T cell’s response to these challenges can help in the development of novel strategies to improve their performance in adoptive T cell therapy.
Analyzing how the disruption of certain genes within a T cell affects survival in a tumor can provide insight into the development of a stronger tumor-penetrating T cell. Given the large number of genes within the T cell, a library approach to gene disruption would be especially valuable for identifying novel factors at play in the T cell. One such shRNA knockdown library was used in an in vivo screen, revealing the potential of the Ppp2r2d knockdown to promote anti-tumor activity [83]. Similar approaches using gene activation libraries could reveal important factors to promote T cell activity, and exploring tools for multiplexed activation and knockdown libraries could also illustrate the more complex responses in a T cell that would be beneficial to target in therapy [84–86].
In addition to making T cells more adept at navigating the tumor microenvironment, T cells can be engineered to be both safer and easier to produce for therapy. Acquiring and modifying a patient’s T cells is a very involved process, and potential costs are estimated to be as high as $500,000 (http://www.wsj.com/articles/new-costly-cancer-treatments-face-hurdles-getting-to-patients-1412627150). Currently, using a patient’s own T cells is important to prevent GVHD. However, a cell-based therapy could mitigate some of the challenges of large scale, personalized therapy if, similar to a blood bank, healthy donors could provide T cells. This sort of T cell bank would ideally provide “off-the-shelf” cancer-killing cell products that can be manufactured at a large scale and implemented on demand.
T cells could be modified for this universal cell-based therapy by disrupting the donor T cells’ endogenous TCR, rendering them responsive only to targets programmed by the chosen cancer-targeting receptor (Figure 4B). A zinc finger nuclease has been used to eliminate endogenous TCR chain expression in a T cell that also expressed a CD19-specific CAR [87] (Box 1). This potential for an “off-the-shelf” therapy is valuable for pharmaceutical companies, and several companies with adoptive T cell portfolios are investing in companies with expertise in DNA nucleases technologies (Table 1).
Box 1. Genome editing for adoptive immunotherapy.
Genome editing for targeted disruption of genes, such as HLAs or inhibitory receptors [95, 96], can render T cells safer and more powerful for adoptive immunotherapy. Several genome editing systems are available that rely on the same underlying mechanism: a nuclease targets a sequence and creates a double stranded break. The break is then repaired using either the error-prone non-homologous end joining (NHEJ) or homology directed repair (HDR), which will disrupt expression of the gene [95]. To contend with the potential of off-target cutting [97, 98], tools have been developed to predict off-target cleavage for several of these systems [99, 100].
Meganucleases are nucleases that belong to one of five families characterized by their sequence and structure motifs [101]. Due to their large recognition sites, meganucleases are very specific. They can also be engineered for new targets by combining domains of other meganucleases or mutating residues [101], but these techniques and other approaches to expand the meganuclease repertoire require large screens to find the optimal meganuclease. To facilitate this process, computational tools are being developed to predict meganuclease design for new targets [102].
Transcription activator-like effector nucleases (TALENs) are derived from the DNA-binding domain of TAL effectors, bacterial proteins whose base specificity can be altered by changing two specific amino acids [95]. An array of these proteins can be generated to target a desired sequence and then fused to the catalytic domain of the Fok1 nuclease to confer the ability to cut the target sequence. [103].
Zinc Finger Nucleases (ZFNs) are composed of zinc finger proteins that bind to DNA nucleotide triplets. By combining zinc fingers with known binding sequences, a larger desired sequence can be targeted. Similar to TALENs, the zinc finger protein is fused to the Fok1 catalytic domain to enable targeted cutting [104].
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) is an RNA-guided nuclease system derived from bacterial immune defenses. The nuclease Cas9 is targeted to a sequence through short complementary RNA sequences. The RNA guides Cas9 to the complementary target sequence, where it can then cut the DNA. RNA design with this system is very straightforward, making CRISPR a promising tool for genome editing [105].
A similar approach could be used to reduce the risk of immunogenicity of an engineer T cell. By disrupting expression of the T cell’s MHC, or human leukocyte antigen (HLA), which is involved in presenting potential antigens on the surface of a cell for detection by other T cells [88] (Figure 4B). By removing HLA expression, a T cell would no longer be able to present potential epitopes at the surface, which would prevent an immunogenic response to any of the components involved in CAR expression or other circuitry.
Concluding remarks and future perspectives
Living cells are increasingly viewed as an attractive platform for designing the ultimate smart therapeutics [19] due to their extraordinarily sophisticated systems to sense and respond to challenges as well as their flexibility in accommodating genetic modification. Given that the genetic engineering strategies discussed in this review for improving adoptive immunotherapy are not mutually exclusive, an intriguing possibility is to combine several, or even all, of the technologies together to generate extremely sophisticated therapeutic agents for controlling when, where, how long, and how strong the therapeutic agents will engage tumors.
Designing and implementing genetic circuits in immunotherapy may provide powerful tools for control over the therapy. However, there are several challenges in designing genetic circuits for this purpose. One of the potential limitations is the need for methods that can efficiently integrate large amounts of DNA in T cells. Viral integration can become inefficient as the size of the insert increases [73], and circuits that require the expression of several components can become large, making efficient transduction of these genes very challenging. Transposon-based systems such as PiggyBac and Sleeping Beauty, which can integrate large sequences of DNA, are a potential alternative to viral transduction [106, 107]. Sleeping Beauty has been used to integrate CARs into T cells in a clinical setting, making it a viable option for integrating larger circuits into cells for immunotherapy [108].
Many genetic circuits rely on proteins that are derived from other organisms. The expression of these foreign proteins in a cell has the potential to elicit an immune response against the engineered T cell, leading to the death of the cell and potentially reduced efficacy of the treatment. This immunogenic response was observed when HSK-KT was used as an inducible suicide gene in patients [63], and components in other circuits could also elicit this response. One option to avoid an immunogenic response is to disrupt the antigen presentation process in the T cell so that it can no longer present the foreign protein as a potential threat to other immune cells. This disruption might be achievable through genome editing to knock out expression of the major histocompatibility complex (MHC) [88], which is used to present protein epitopes to other T cells. With MHC expression disrupted, the engineered T cell can effectively hide the presence of foreign proteins from other immune cells, preventing an immunogenic response.
Although T cells, which already possess powerful machinery to kill cancer cells or foreign invading organisms, have captured the most attention in cell-based cancer therapy, other cell types and organisms are also being explored as potential therapeutic agents. For instance, adoptive T cell therapy is not a viable option for patients with a T cell deficiency. Induced pluripotent stem cells can potentially be used to derive cancer targeting T cells [89], and NK cells are a possible alternative to T cells. NK cells expressing a Her2-specific CAR are able to eliminate tumor cells in vivo [90, 91]. These cells can also trigger cytotoxicity to tumor cells through multiple receptors, and their limited life span may make them less dangerous for patients. The availability of the NK-92 cell line from Conkwest provides a platform for an “off-the-shelf” form of cell-based therapy that might provide further advantages to using NK cells for CAR-based therapy. In addition to NK cells, oncolytic viruses are showing promise in clinical trials against glioblastomas and multiple myeloma [92, 93]. Their specificity toward tumors may also be improved with micro-RNA-based classifiers [94]. Similar to the engineering strategies outlined here to advance T cell therapy, these other cells and organisms can be further modified to improve their performance, and this parallel emergence of immunotherapy, cellular engineering, and synthetic biology is creating a unique interface to usher in a new era of cell-based cancer therapy.
Highlight.
Synthetic biology will play an important role in advancing adoptive T cell therapy.
Engineered receptors and genetic circuits can make cell-based therapies safer and more powerful.
Cellular engineering and genome editing can further improve the T cell as a chassis for therapy.
Acknowledgments
D.C. acknowledges funding from Boston University Cross-disciplinary Training in Nanotechnology for Cancer (XTNC) Program. W. W. acknowledges funding from the NIH Director’s New Innovator Award (DP2CA186574) and Boston University College of Engineering Dean’s Catalyst Award. The authors regret that many important publications were not included in this review due to space constrains.
Glossary
- Chimeric antigen receptor (CAR)
Engineered receptor that fuses an extracellular single chain variable fragment (scFv) of an antibody to intracellular T cell signaling domains
- Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)
An inhibitory receptor that downregulates T cell response
- Epitope
Fragments of proteins expressed in a cell that are presented on the surface by the major histocompatibility complex (MHC) for detection by T cells. Epitopes that represent pathogenic organisms to the T cell trigger T cell activation upon binding
- Fatty Acid Oxidation
Cascade of β-oxidation reactions that converts fatty acids in the mitochondria to produce Acetyl-CoA
- Hematopoietic stem cell transplant (HSCT)
Transplant of blood cells from the bone marrow that give rise to all other blood cells. HSCT is usually performed in patients with blood or bone marrow cancers
- Immunogenicity
The potential for a molecule to elicit an immune response. Proteins expressed in a cell can be processed into smaller fragments called epitopes to be presented at the surface of the cell as potential antigens by the MHC. T cells assess these MHC-peptide complexes through their T cell receptor (TCR), which are selected to distinguish epitopes derived from self-proteins and those derived from foreign organisms. If the TCR recognizes an epitope as a foreign antigen, it will activate the T cell and drive the death of the antigen-presenting cell. In T cell therapy, engineered T cells can be targeted by other immune cells due to the expression of foreign proteins as part of the receptors or circuit components of non-human origin
- Programmed Cell Death 1 (PD-1)
A cell surface receptor that negatively modulates T cell response by promoting apoptosis
- Ribozyme
RNA molecules that can act as catalytic agents in a reaction
- T cell receptor (TCR)
Receptors expressed on the surface of T cells to drive recognition of pathogenic organisms through epitope-MHC binding
- Tumor-infiltrating lymphocytes (TILs)
T cells that have been able to penetrate the tumor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–409. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes Dev. 2011;25:2559–2572. doi: 10.1101/gad.169029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouchlaka MN, Redelman D, Murphy WJ. Immunotherapy following hematopoietic stem cell transplantation: potential for synergistic effects. Immunotherapy. 2010;2:399–418. doi: 10.2217/imt.10.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, Yu Z, Rosenberg SA, Restifo NP. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother. 2010;33:1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wrzesinski C, Paulos CM, Gattinoni L, Palmer DC, Kaiser A, Yu Z, Rosenberg SA, Restifo NP. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. The Journal of clinical investigation. 2007;117:492–501. doi: 10.1172/JCI30414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72:5435–5440. doi: 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- 8.Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, Tagaya Y, Rosenberg SA, Waldmann TA, Restifo NP. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci U S A. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melero I, Mazzolini G, Narvaiza I, Qian C, Chen L, Prieto J. IL-12 gene therapy for cancer: in synergy with other immunotherapies. Trends Immunol. 2001;22:113–115. doi: 10.1016/s1471-4906(00)01824-x. [DOI] [PubMed] [Google Scholar]
- 10.Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, Anichini A. Interleukin-12: biological properties and clinical application. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:4677–4685. doi: 10.1158/1078-0432.CCR-07-0776. [DOI] [PubMed] [Google Scholar]
- 11.Lee CS, Cragg M, Glennie M, Johnson P. Novel antibodies targeting immune regulatory checkpoints for cancer therapy. British journal of clinical pharmacology. 2013;76:233–247. doi: 10.1111/bcp.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spigel DR, Socinski MA. Rationale for chemotherapy, immunotherapy, and checkpoint blockade in SCLC: beyond traditional treatment approaches. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2013;8:587–598. doi: 10.1097/JTO.0b013e318286cf88. [DOI] [PubMed] [Google Scholar]
- 13.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moser JC, Pulido JS, Dronca RS, McWilliams RR, Markovic SN, Mansfield AS. The Mayo Clinic experience with the use of kinase inhibitors, ipilimumab, bevacizumab, and local therapies in the treatment of metastatic uveal melanoma. Melanoma research. 2015;25:59–63. doi: 10.1097/CMR.0000000000000125. [DOI] [PubMed] [Google Scholar]
- 15.Philips GK, Atkins M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. International immunology. 2015;27:39–46. doi: 10.1093/intimm/dxu095. [DOI] [PubMed] [Google Scholar]
- 16.Way JC, Collins JJ, Keasling JD, Silver PA. Integrating biological redesign: where synthetic biology came from and where it needs to go. Cell. 2014;157:151–161. doi: 10.1016/j.cell.2014.02.039. [DOI] [PubMed] [Google Scholar]
- 17.Auslander S, Fussenegger M. From gene switches to mammalian designer cells: present and future prospects. Trends Biotechnol. 2013;31:155–168. doi: 10.1016/j.tibtech.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Bashor CJ, Horwitz AA, Peisajovich SG, Lim WA. Rewiring cells: synthetic biology as a tool to interrogate the organizational principles of living systems. Annu Rev Biophys. 2010;39:515–537. doi: 10.1146/annurev.biophys.050708.133652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischbach MA, Bluestone JA, Lim WA. Cell-based therapeutics: the next pillar of medicine. Science translational medicine. 2013;5:179ps177. doi: 10.1126/scitranslmed.3005568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol. 2015;36:71–80. doi: 10.1016/j.it.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo Presti E, Dieli F, Meraviglia S. Tumor-Infiltrating gammadelta T Lymphocytes: Pathogenic Role, Clinical Significance, and Differential Programing in the Tumor Microenvironment. Front Immunol. 2014;5:607. doi: 10.3389/fimmu.2014.00607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dudley ME, Gross CA, Somerville RP, Hong Y, Schaub NP, Rosati SF, White DE, Nathan D, Restifo NP, Steinberg SM, Wunderlich JR, Kammula US, Sherry RM, Yang JC, Phan GQ, Hughes MS, Laurencot CM, Rosenberg SA. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:2152–2159. doi: 10.1200/JCO.2012.46.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khammari A, Knol AC, Nguyen JM, Bossard C, Denis MG, Pandolfino MC, Quereux G, Bercegeay S, Dreno B. Adoptive TIL transfer in the adjuvant setting for melanoma: long-term patient survival. Journal of immunology research. 2014;2014:186212. doi: 10.1155/2014/186212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duong CP, Yong CS, Kershaw MH, Slaney CY, Darcy PK. Cancer immunotherapy utilizing gene-modified T cells: From the bench to the clinic. Molecular immunology. 2015 doi: 10.1016/j.molimm.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Thaxton JE, Li Z. To affinity and beyond: Harnessing the T Cell receptor for cancer immunotherapy. Human vaccines & immunotherapeutics. 2014;10:3313–3321. doi: 10.4161/21645515.2014.973314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CC, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. 2004;173:7647–7653. doi: 10.4049/jimmunol.173.12.7647. [DOI] [PubMed] [Google Scholar]
- 29.Jena B, Dotti G, Cooper LJ. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. 2010;116:1035–1044. doi: 10.1182/blood-2010-01-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DC, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, Brentjens R. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Science translational medicine. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, Wang LL, Han WD. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23:184–191. doi: 10.1038/mt.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, Chen K, Shin M, Wall DM, Honemann D, Gambell P, Westerman DA, Haurat J, Westwood JA, Scott AM, Kravets L, Dickinson M, Trapani JA, Smyth MJ, Darcy PK, Kershaw MH, Prince HM. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21:2122–2129. doi: 10.1038/mt.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Couzin-Frankel J. Cancer Immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 37.Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. 2013;31:999–1008. doi: 10.1038/nbt.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frigault MJ, Lee J, Basil M, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan S, Posey A, Jr, Ang S, Cooper LJ, Platt J, Johnson FB, Paulos CM, Zhao Y, Kalos M, Milone M, June CH. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. 2015 doi: 10.1158/2326-6066.cir-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Science translational medicine. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duong CP, Westwood JA, Yong CS, Murphy A, Devaud C, John LB, Darcy PK, Kershaw MH. Engineering T cell function using chimeric antigen receptors identified using a DNA library approach. PLoS One. 2013;8:e63037. doi: 10.1371/journal.pone.0063037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hacohen N, Fritsch EF, Carter TA, Lander ES, Wu CJ. Getting personal with neoantigen-based therapeutic cancer vaccines. Cancer Immunol Res. 2013;1:11–15. doi: 10.1158/2326-6066.CIR-13-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, Zhang W, Sougnez C, Cibulskis K, Sidney J, Stevenson K, Ritz J, Neuberg D, Brusic V, Gabriel S, Lander ES, Getz G, Hacohen N, Wu CJ. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;124:453–462. doi: 10.1182/blood-2014-04-567933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D, Nieuwland M, Stratton MR, Kerkhoven RM, Kesmir C, Haanen JB, Kvistborg P, Schumacher TN. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:e439–442. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24:743–750. doi: 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, Visser M, Stratton MR, Haanen JB, Spits H, van der Burg SH, Schumacher TN. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21:81–85. doi: 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- 47.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, Samuels Y, Rosenberg SA. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19:747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fedorov VD, Sadelain M, Kloss CC. Novel approaches to enhance the specificity and safety of engineered T cells. Cancer J. 2014;20:160–165. doi: 10.1097/PPO.0000000000000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Narayanan P, Lapteva N, Seethammagari M, Levitt JM, Slawin KM, Spencer DM. A composite MyD88/CD40 switch synergistically activates mouse and human dendritic cells for enhanced antitumor efficacy. The Journal of clinical investigation. 2011;121:1524–1534. doi: 10.1172/JCI44327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, Chng WJ, Campana D. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014;74:93–103. doi: 10.1158/0008-5472.CAN-13-1365. [DOI] [PubMed] [Google Scholar]
- 52.Bellone M, Calcinotto A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Frontiers in oncology. 2013;3:231. doi: 10.3389/fonc.2013.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, Kreuger J, Dimberg A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-kappaB-induced endothelial activation. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2015;29:227–238. doi: 10.1096/fj.14-250985. [DOI] [PubMed] [Google Scholar]
- 54.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Science translational medicine. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stephan SB, Taber AM, Jileaeva I, Pegues EP, Sentman CL, Stephan MT. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat Biotechnol. 2015;33:97–101. doi: 10.1038/nbt.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, Wang E, Young HA, Murphy PM, Hwu P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. 2002;13:1971–1980. doi: 10.1089/10430340260355374. [DOI] [PubMed] [Google Scholar]
- 57.Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, Whittington M, Yang Y, Overwijk WW, Lizee G, Hwu P. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:5458–5468. doi: 10.1158/1078-0432.CCR-10-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Conklin BR, Hsiao EC, Claeysen S, Dumuis A, Srinivasan S, Forsayeth JR, Guettier JM, Chang WC, Pei Y, McCarthy KD, Nissenson RA, Wess J, Bockaert J, Roth BL. Engineering GPCR signaling pathways with RASSLs. Nat Methods. 2008;5:673–678. doi: 10.1038/nmeth.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park JS, Rhau B, Hermann A, McNally KA, Zhou C, Gong D, Weiner OD, Conklin BR, Onuffer J, Lim WA. Synthetic control of mammalian-cell motility by engineering chemotaxis to an orthogonal bioinert chemical signal. Proc Natl Acad Sci U S A. 2014;111:5896–5901. doi: 10.1073/pnas.1402087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tey SK. Adoptive T-cell therapy: adverse events and safety switches. Clinical & translational immunology. 2014;3:e17. doi: 10.1038/cti.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Frontiers in pharmacology. 2014;5:254. doi: 10.3389/fphar.2014.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F, Traversari C, Bordignon C. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 63.Marin V, Cribioli E, Philip B, Tettamanti S, Pizzitola I, Biondi A, Biagi E, Pule M. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum Gene Ther Methods. 2012;23:376–386. doi: 10.1089/hgtb.2012.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, Heslop HE, Spencer DM, Rooney CM. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–4254. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, Brouns SA, Spencer DM, Till BG, Jensen MC, Riddell SR, Press OW. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One. 2013;8:e82742. doi: 10.1371/journal.pone.0082742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou X, Di Stasi A, Tey SK, Krance RA, Martinez C, Leung KS, Durett AG, Wu MF, Liu H, Leen AM, Savoldo B, Lin YF, Grilley BJ, Gee AP, Spencer DM, Rooney CM, Heslop HE, Brenner MK, Dotti G. Long-term outcome and immune reconstitution after haploidentical stem cell transplant in recipients of allodepleted-T-cells expressing the inducible caspase-9 safety transgene. 2014 doi: 10.1182/blood-2014-01-551671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C, Parsot C, Sansonetti PJ. An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat Immunol. 2007;8:47–56. doi: 10.1038/ni1423. [DOI] [PubMed] [Google Scholar]
- 69.Yao T, Mecsas J, Healy JI, Falkow S, Chien Y. Suppression of T and B lymphocyte activation by a Yersinia pseudotuberculosis virulence factor, yopH. The Journal of experimental medicine. 1999;190:1343–1350. doi: 10.1084/jem.190.9.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei P, Wong WW, Park JS, Corcoran EE, Peisajovich SG, Onuffer JJ, Weiss A, Lim WA. Bacterial virulence proteins as tools to rewire kinase pathways in yeast and immune cells. Nature. 2012;488:384–388. doi: 10.1038/nature11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen YY, Jensen MC, Smolke CD. Genetic control of mammalian T-cell proliferation with synthetic RNA regulatory systems. Proc Natl Acad Sci U S A. 2010;107:8531–8536. doi: 10.1073/pnas.1001721107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 73.Atkinson H, Chalmers R. Delivering the goods: viral and non-viral gene therapy systems and the inherent limits on cargo DNA and internal sequences. Genetica. 2010;138:485–498. doi: 10.1007/s10709-009-9434-3. [DOI] [PubMed] [Google Scholar]
- 74.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. The Journal of clinical investigation. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB. Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur J Immunol. 2003;33:2223–2232. doi: 10.1002/eji.200324048. [DOI] [PubMed] [Google Scholar]
- 77.Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180:4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. The Journal of clinical investigation. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Landskron J, Helland O, Torgersen KM, Aandahl EM, Gjertsen BT, Bjorge L, Tasken K. Activated regulatory and memory T-cells accumulate in malignant ascites from ovarian carcinoma patients. Cancer immunology immunotherapy: CII. 2015;64:337–347. doi: 10.1007/s00262-014-1636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ostrand-Rosenberg S, Horn LA, Haile ST. The programmed death-1 immune-suppressive pathway: barrier to antitumor immunity. J Immunol. 2014;193:3835–3841. doi: 10.4049/jimmunol.1401572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou P, Shaffer DR, Alvarez Arias DA, Nakazaki Y, Pos W, Torres AJ, Cremasco V, Dougan SK, Cowley GS, Elpek K, Brogdon J, Lamb J, Turley SJ, Ploegh HL, Root DE, Love JC, Dranoff G, Hacohen N, Cantor H, Wucherpfennig KW. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature. 2014;506:52–57. doi: 10.1038/nature12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Malina A, Katigbak A, Cencic R, Maiga RI, Robert F, Miura H, Pelletier J. Adapting CRISPR/Cas9 for functional genomics screens. Methods in enzymology. 2014;546:193–213. doi: 10.1016/B978-0-12-801185-0.00010-6. [DOI] [PubMed] [Google Scholar]
- 87.Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, Huls H, Miller JC, Kebriaei P, Rabinovitch B, Lee DA, Champlin RE, Bonini C, Naldini L, Rebar EJ, Gregory PD, Holmes MC, Cooper LJ. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–5705. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reyes LM, Estrada JL, Wang ZY, Blosser RJ, Smith RF, Sidner RA, Paris LL, Blankenship RL, Ray CN, Miner AC, Tector M, Tector AJ. Creating class I MHC-null pigs using guide RNA and the Cas9 endonuclease. J Immunol. 2014;193:5751–5757. doi: 10.4049/jimmunol.1402059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, Sadelain M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. 2013;31:928–933. doi: 10.1038/nbt.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3:e28147. doi: 10.4161/onci.28147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kruschinski A, Moosmann A, Poschke I, Norell H, Chmielewski M, Seliger B, Kiessling R, Blankenstein T, Abken H, Charo J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc Natl Acad Sci U S A. 2008;105:17481–17486. doi: 10.1073/pnas.0804788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Freeman AI, Zakay-Rones Z, Gomori JM, Linetsky E, Rasooly L, Greenbaum E, Rozenman-Yair S, Panet A, Libson E, Irving CS, Galun E, Siegal T. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13:221–228. doi: 10.1016/j.ymthe.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 93.Russell SJ, Federspiel MJ, Peng KW, Tong C, Dingli D, Morice WG, Lowe V, O’Connor MK, Kyle RA, Leung N, Buadi FK, Rajkumar SV, Gertz MA, Lacy MQ, Dispenzieri A. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clinic proceedings. 2014;89:926–933. doi: 10.1016/j.mayocp.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xie Z, Wroblewska L, Prochazka L, Weiss R, Benenson Y. Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science. 2011;333:1307–1311. doi: 10.1126/science.1205527. [DOI] [PubMed] [Google Scholar]
- 95.Lloyd A, Vickery ON, Laugel B. Beyond the antigen receptor: editing the genome of T-cells for cancer adoptive cellular therapies. Front Immunol. 2013;4:221. doi: 10.3389/fimmu.2013.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. The Journal of experimental medicine. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of offtarget DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fine EJ, Cradick TJ, Zhao CL, Lin Y, Bao G. An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res. 2014;42:e42. doi: 10.1093/nar/gkt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42:W401–407. doi: 10.1093/nar/gku410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, Paques F. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Current gene therapy. 2011;11:11–27. doi: 10.2174/156652311794520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zaslavskiy M, Bertonati C, Duchateau P, Duclert A, Silva GH. Efficient design of meganucleases using a machine learning approach. BMC bioinformatics. 2014;15:191. doi: 10.1186/1471-2105-15-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nature reviews Genetics. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 105.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nakazawa Y, Huye LE, Dotti G, Foster AE, Vera JF, Manuri PR, June CH, Rooney CM, Wilson MH. Optimization of the PiggyBac transposon system for the sustained genetic modification of human T lymphocytes. J Immunother. 2009;32:826–836. doi: 10.1097/CJI.0b013e3181ad762b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wu SC, Meir YJ, Coates CJ, Handler AM, Pelczar P, Moisyadi S, Kaminski JM. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci U S A. 2006;103:15008–15013. doi: 10.1073/pnas.0606979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Singh H, Figliola MJ, Dawson MJ, Olivares S, Zhang L, Yang G, Maiti S, Manuri P, Senyukov V, Jena B, Kebriaei P, Champlin RE, Huls H, Cooper LJ. Manufacture of clinical-grade CD19-specific T cells stably expressing chimeric antigen receptor using Sleeping Beauty system and artificial antigen presenting cells. PLoS One. 2013;8:e64138. doi: 10.1371/journal.pone.0064138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Davila ML, Kloss CC, Gunset G, Sadelain M. CD19 CAR-targeted T cells induce long-term remission and B Cell Aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS One. 2013;8:e61338. doi: 10.1371/journal.pone.0061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Science translational medicine. 2013;5:177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, Duerkopp N, Roberts IM, Pogosov GL, Ho WY, Ochsenreither S, Wolfl M, Bar M, Radich JP, Yee C, Greenberg PD. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Science translational medicine. 2013;5:174ra127. doi: 10.1126/scitranslmed.3004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Badhiwala J, Decker WK, Berens ME, Bhardwaj RD. Clinical trials in cellular immunotherapy for brain/CNS tumors. Expert review of neurotherapeutics. 2013;13:405–424. doi: 10.1586/ern.13.23. [DOI] [PubMed] [Google Scholar]
- 115.Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H, Zhang W, Fine HA, Rosenberg SA. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23:1043–1053. doi: 10.1089/hum.2012.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, Chen CC, Yang JC, Rosenberg SA, Hwu P. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, Tran E, Hanada K, Yu Z, Palmer DC, Kerkar SP, Michalek RD, Upham T, Leonardi A, Acquavella N, Wang E, Marincola FM, Gattinoni L, Muranski P, Sundrud MS, Klebanoff CA, Rosenberg SA, Fearon DT, Restifo NP. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015;75:296–305. doi: 10.1158/0008-5472.CAN-14-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]