

Abstract
MicroRNAs (miRNAs) are a class of small noncoding RNAs that use partial base-pairing to recognize and regulate the expression of messenger RNAs (mRNAs). Mature miRNAs arise from longer primary transcripts (pri-miRNAs) that are processed to a shorter hairpin precursor miRNA (pre-miRNA) by the Microprocessor complex. In Caenorhabditis elegans the primary let-7 (pri-let-7) transcript undergoes trans-splicing, where pri-let-7 is cleaved at a 3′ splice site and the splice-leader-1 (SL1) sequence is appended at the 5′ end. Here we investigate the role of this splicing event in the biogenesis of let-7 miRNA. We hypothesized that splicing changes the secondary structure of the pri-let-7 transcript, creating a more favorable substrate for recognition by the Microprocessor. Supporting this idea, we detected conspicuous structural differences between unspliced and SL1-spliced pri-let-7 transcripts using in vitro ribonuclease (RNase) assays. Through the generation of transgenic worm strains, we found that the RNA secondary structure produced by splicing, as opposed to the act of splicing itself, optimizes processing of pri-let-7 by the Microprocessor in vivo. We also observed that the endogenous spliced, but not the unspliced, pri-let-7 transcripts bind to the Microprocessor and accumulate upon its depletion. We conclude that splicing is a key step in generating pri-let-7 transcripts with a structure that enables downstream processing events to produce appropriate levels of mature let-7.
Keywords: miRNA, pri-miRNA, Drosha, C. elegans, let-7, trans-splicing
INTRODUCTION
MiRNAs are ∼22 nucleotide (nt) noncoding RNAs that use imperfect base-pairing to target mRNAs for down-regulated expression (Pasquinelli 2012). Thousands of miRNAs have been discovered in a wide variety of organisms, including plants, flies, worms, and humans (Kozomara and Griffiths-Jones 2011). Moreover, a single miRNA has the potential to regulate hundreds of different targets (Ha and Kim 2014). Consequently, miRNAs are implicated in almost all biological pathways and their misexpression can lead to developmental impairment and disease. The let-7 miRNA in particular is abnormally expressed in various human cancers and the consequent misregulation of its protein-coding targets has been directly implicated in the disease state (Mondol and Pasquinelli 2012; Gurtan and Sharp 2013). Additionally, the mature sequence of let-7 is perfectly conserved across Bilaterian species (Pasquinelli et al. 2000), making it a relevant model for studying the regulation of miRNA biogenesis.
The general miRNA biogenesis pathway is well understood (Finnegan and Pasquinelli 2013; Ha and Kim 2014). Briefly, long primary miRNAs (pri-miRNAs) are transcribed by RNA polymerase II (Pol II) and, like mRNAs, they are capped and polyadenylated. The Microprocessor, which contains the RNase III enzyme Drosha and the RNA binding protein Pasha (DGCR8 in mammals), cleaves the pri-miRNA into a ∼65-nt imperfect hairpin known as the precursor miRNA (pre-miRNA). A noncanonical pathway exists for a class of miRNAs known as mirtrons, which bypasses the need for Drosha processing (Westholm and Lai 2011). Instead, the pre-miRNA is a ∼65-nt intron that is excised by the spliceosome. In mammals and Drosophila, Exportin-5 then transports the pre-miRNA from the nucleus to the cytoplasm where it is subsequently cleaved by Dicer to produce the mature ∼22-nt miRNA. The mature miRNA sequence, or guide strand, is then loaded onto an Argonaute protein, which is the principle protein in the miRNA Induced Silencing Complex (miRISC). The loaded miRISC targets mRNAs with partial complementarity to induce deadenylation and/or translational repression (Ha and Kim 2014).
The substrate requirements for the Microprocessor have been studied extensively (Lee et al. 2003; Denli et al. 2004; Gregory et al. 2004; Zeng and Cullen 2005; Han et al. 2006; Kim and Kim 2007; Kataoka et al. 2009; Flynt et al. 2010; Janas et al. 2011; Warf et al. 2011; Macias et al. 2012; Auyeung et al. 2013; Conrad et al. 2014; Quick-Cleveland et al. 2014). Through deep sequencing and in vitro processing assays, a general model of the ideal Drosha substrate has emerged. The archetype metazoan pri-miRNA contains a pre-miRNA hairpin ∼65-nt long, or ∼3 helical turns, which features a ∼10-nt terminal loop, internal bulges in the stem every ∼11 nt or one helical turn, and flanking unstructured RNA sequences (known as the basal segment) (Lee et al. 2003; Zeng and Cullen 2005; Han et al. 2006). Sequences adjacent to Drosha cleavage sites tend to fold into 2- to 4-nt symmetrical internal loops. These sites are found on average ∼11-nt away from the unpaired basal segment. Recent work suggests that processing of human pri-miRNAs also involves a sequence motif that is not seen in worms (Auyeung et al. 2013; Conrad et al. 2014). Across species, though, the secondary structure is critical for determining the efficiency of pri-miRNA processing.
While investigating the transcription of primary let-7 (pri-let-7), our laboratory discovered two transcriptional start sites (A and B) that produce nascent pri-let-7 transcripts and one or both give rise to an SL1 trans-spliced isoform (Bracht et al. 2004). Notably, the 3′ splice site (ss) required for trans-splicing is conserved in sequence and position in let-7 genes in other nematode species (Bracht et al. 2004). Trans-splicing is a common event in Caenorhabditis elegans as up to 70% of mRNAs have their 5′ ends replaced by one of two, 22-nt trimethylguanosine-capped RNA leader sequences (SL1 or SL2) (Blumenthal 2012). In some cases, trans-splicing is essential for separating mRNAs in an operon. The function of this event for mRNAs from nonoperonic genes remains elusive, but it is thought to aid in their nuclear export or translation (Wallace et al. 2010). None of these functions seems relevant for pri-miRNAs, as they are noncoding RNAs processed in the nucleus, eliminating the SL1 sequence before export to the cytoplasm. Instead, we predicted that the SL1 sequence, the act of splicing, and/or a resulting structural change in the primary let-7 RNA is important for downstream miRNA processing events. Here, these possibilities were tested through a series of in vitro structural studies, in vivo rescue experiments, and RNA immunoprecipitation assays. Altogether, our results support the conclusion that SL1 trans-splicing of pri-let-7 in C. elegans facilitates structural rearrangements that promote Microprocessor binding and cleavage. This example raises the possibility that splicing may be broadly used to enable structural changes that regulate Microprocessor activity of the many other pri-miRNAs embedded in transcripts subject to cis- or trans-splicing.
RESULTS AND DISCUSSION
Splicing remodels the pri-let-7 secondary structure
Trans-splicing of pri-let-7 occurs when the 3′ ss, which is found 38-nt upstream of the pre-let-7 sequence, is cleaved, and the 22-nt SL1 sequence is appended, replacing the cap and the region between the 3′ ss and transcriptional start sites (Bracht et al. 2004). Secondary structure predictions using lowest free energy thermodynamic computations on the mfold server provided initial evidence that there were differences between unspliced and SL1-spliced pri-let-7 RNAs (Zuker 2003; Bracht et al. 2004). However, previous research on structural features of pre-miRNAs found 8 out of 10 experimentally determined structures differed from those predicted by mfold (Krol et al. 2004), primarily in the terminal loop region and other secondary bulges in the stem. Therefore, we sought to interrogate the potential pri-let-7 secondary structures using biochemical methods. We subjected ∼200 nt of in vitro transcribed spliced and unspliced primary transcripts to RNase secondary structure analysis to identify single-stranded and double-stranded regions in the folded RNA molecules (Supplemental Fig. 1). Figure 1 depicts the structures best supported by RNase structure probing coupled with mfold predictions. Several bases in potential loop regions produced variable patterns of paired and unpaired structures, indicating transient interactions in these regions. These results indicate that the unspliced pri-let-7 transcript features structured regions at the base of the precursor, disrupting what should be unpaired areas critical for Drosha slicing activity (Zeng and Cullen 2005; Han et al. 2006; Warf et al. 2011; Quick-Cleveland et al. 2014). In contrast, the SL1-spliced model takes on a structure that better resembles a canonical pri-miRNA substrate, including a terminal loop, internal loops in the upper and lower stem, and flanking unpaired basal segments.
FIGURE 1.

Secondary structures of unspliced and spliced pri-let-7 RNAs. RNA secondary structure analysis of in vitro transcribed unspliced and SL1-spliced model pri-let-7 transcripts. Numbers indicate positions of bases in the model transcripts. The composite results from four independent experiments are shown. Here we show a close-up view of the area in the red box subject to processing by the Microprocessor. Three shades of color are used to represent “high,” “medium,” and “low” sensitivity of each nucleotide to the RNase enzyme, such that the darker the color the more reactive to cleavage and the more “structured” (blue) or “unpaired” (red) the base appears to be. Nucleotides in shades of blue circles were sensitive to cleavage by RNase V1, denoting double-strandedness. Shades of red circles represent single-stranded nucleotides that were cleaved in response to RNase T1 or RNase A treatment. Nucleotides labeled both red and blue indicate evidence for single- and double-stranded nature, which is expected from G–U pairs and regions of the transcript that are more dynamic. Unlabeled (white) nucleotides were inconclusive. Open circles between nucleotides represent regions of expected pairing that were not detectable by the RNase structure probing. The mature let-7 miRNA sequence is highlighted in yellow. The SL1 sequence is highlighted in green and labeled. Representative PAGE used for data analyses are shown in Supplemental Figure 1.
The splicing-induced structural changes facilitate pri-let-7 processing
We investigated the role of SL1 splicing of pri-let-7 in vivo by introducing let-7 transgenes using Mos-1 transposon-mediated Single Copy Insertion (MosSCI) and crossing the resulting transgenic animals to a let-7 null background, let-7(mn112) (Fig. 2A; Reinhart et al. 2000; Frokjaer-Jensen et al. 2008). The transgenes were engineered to test the splicing, sequence, and structural requirements of pri-let-7 processing. As part of the heterochronic pathway, let-7 is crucial for developmental timing of cell fates during the fourth larval stage (L4) to adult transition (Mondol and Pasquinelli 2012). Pri-let-7 expression begins at the first larval stage (L1) and oscillates throughout development with peaks preceding each larval molt (Van Wynsberghe et al. 2011). During early larval stages, LIN-28 protein cotranscriptionally binds to pri-let-7 and blocks its processing (Van Wynsberghe et al. 2011; Stefani et al. 2015). By the L4 stage, LIN-28 levels have decreased over 10-fold, allowing processing of pri-let-7 to proceed. Worms with mutant alleles of let-7 display various phenotypes ranging from lethality to aberrations in cuticular structures known as alae (Fig. 2B; Reinhart et al. 2000). Accordingly, we examined whether our transgenic animals exhibited phenotypes associated with loss of let-7 (Reinhart et al. 2000). In addition, we analyzed primary and mature let-7 RNA expression from the transgenes at the L4 stage of development using Northern blot and quantitative reverse transcriptase PCR assays (qRT-PCR) (Fig. 2C–F).
FIGURE 2.

Splicing is important for let-7 biogenesis in vivo. (A) Single copy transgenes with altered versions of the trans-splice site were inserted in Chromosome (Chr) II and expressed in the let-7(Δmn112) (Chr X) null mutant background to test for effects on rescue activity and let-7 biogenesis. The arrows represent let-7 A and B transcriptional start sites, the striped area represents the mn112 deletion, and the light rectangle is indicative of the mature let-7 sequence. In addition to the wild-type (WT) rescue construct, which contains the let-7 promoter regions, transcriptional start sites, and the 3′ splice site (3′ss) recognized by the spliceosome, four mutated versions of the let-7 transgene were generated. The “pre-spliced” construct (SL1) replaces the 3′ ss with the 22-nt splice-leader sequence. The “random” construct (RDM) replaces the 3′ ss with a 22-nt sequence that is similar in GC content to the SL1 sequence but is predicted to support a different secondary structure. The splice site knockout (Δ3′ss) is missing 7 nt essential for splicing recognition. The “misfolded” construct (MSF) contains the SL1 sequence in place of the 3′ ss but also includes a 48-nt mutation that creates a hairpin at the base of the pre-let-7 hairpin, similar to the unspliced secondary structure. (B) Analysis of the alae formation phenotype. Alae are a group of three cuticular ridges that form along the length of the adult C. elegans worm. Examples of alae in WT and worms that have insufficient let-7 activity, which results in “patchy” and gapped alae, are shown. Alae were analyzed by high-powered microscopy in transgenic worms and in a mutant that expresses twofold reduced levels of mature let-7 (let-7(mg279)). Results for RDM are presented as the average of two independent experiments where n = 25, while all other strains are presented as the average of three independent experiments where n = 20 in each. (C–F) Total RNA from triplicate L4 staged SL1, RDM, Δ3′ss, and WT transgenic worms was used for Northern blot and qRT-PCR analyses. Worms with the MSF transgene were inviable and, thus, could not be analyzed for let-7 expression. (C) Representative agarose Northern blot of primary let-7 expression. Detection of 18S ribosomal RNA serves as a loading control. (D) Quantitative RT-PCR of transgenic worms where pri-let-7 (all isoforms) levels were normalized to 18S rRNA and relative expression is compared with average WT expression. Standard error is depicted. (*) P < 0.05. (E) Representative PAGE Northern blot of mature let-7 expression. U6 snRNA serves as a loading control. (F) TaqMan qRT-PCR of mature let-7 normalized to 18S rRNA levels relative to WT. Standard error is depicted. (*) P < 0.05.
Although the transgenic worms lacking a 3′ ss (Fig. 2A, Δ3′SS) were viable, ∼25% of the adults exhibited patchy or no alae, indicative of incomplete rescue activity (Fig. 2B). This degree of abnormal alae development is similar to that observed in let-7(mg279) mutants, which produce twofold less mature let-7 miRNA (Reinhart et al. 2000; Bracht et al. 2004). We confirmed that the pri-let-7 RNAs generated in the Δ3′SS transgenic animals do not undergo trans-splicing by RT-PCR assays (data not shown). Consistent with the defective alae phenotype, the Δ3′SS strain exhibited reduced processing of let-7 with a fivefold accumulation of pri-let-7 and a greater than twofold decrease in the level of mature let-7, compared to the amount of these RNAs detected in the wild-type (WT) strain (Fig. 2C–F). These results demonstrate that SL1-splicing is important for let-7 biogenesis in vivo.
Because the Microprocessor has been shown to associate with spliceosomal proteins, we asked whether the act of splicing is needed for let-7 biogenesis (Gregory et al. 2004; Kim and Kim 2007; Kataoka et al. 2009; Janas et al. 2011). The SL1 “pre-spliced” construct replaces the 3′ss with the 22-nt SL1 sequence (Fig. 2A, SL1). These worms still use the annotated A and B transcriptional start sites to produce pri-let-7 transcripts that encode the SL1 sequence, without undergoing splicing; the resulting RNAs are predicted to fold similarly to the WT trans-spliced version in the vicinity of the precursor sequence. These transgenic worms have almost no discernible alae phenotypes, indicating that the “pre-spliced” SL1 pri-let-7 transgene retains near WT levels of rescue activity (Fig. 2B). Consistent with these observations, “pre-spliced” SL1 worms accumulate modest levels of pri-let-7 and produce amounts of mature let-7 that are comparable with those expressed from the WT construct (Fig. 2C–F). Thus, any contribution of the SL1-spliceosome to let-7 biogenesis is minor.
We also examined the necessity of the SL1 sequence by replacing it with a random 22-nt sequence (RDM) that is similar in GC content to the SL1 sequence but unable to support the same structure (Fig. 2A). The transgenic worms expressing the RDM construct displayed defective alae and let-7 expression phenotypes similar to those of the Δ3′SS transgenic animals, indicating limited rescue activity (Fig. 2B–F).
To further differentiate whether the structural change provided by the SL1 sequence, as opposed to the sequence itself, is crucial for facilitating adequate let-7 expression, we generated a “misfolded” SL1-spliced construct (MSF). This transgene includes the 22-nt SL1 sequence in place of the 3′ss and a 48-nt mutagenized region 24 bases downstream from the 3′ end of the pre-let-7 (Fig. 2A). Despite the inclusion of the SL1 sequence, this construct was completely incapable of rescuing the null let-7 mutant, implying that the presence of the SL1 sequence is not sufficient for facilitating pri-let-7 processing. When analyzed by mfold, this transgene creates a highly structured hairpin at the base of pre-let-7, similar to that found in unspliced pri-let-7. Because of the high complementarity in the sequence, this molecule is predicted to take on a very rigid structure, with little room for flexibility. The likely inability of the MSF RNA secondary structure to “breathe” to the same degree observed for the unspliced form suggests that the potential for RNA molecules to be dynamic contributes to the processing of pri-let-7 in transgenic worms lacking a 3′ss. Furthermore, it points to structural remodeling as the primary role for trans-splicing of pri-let-7 transcripts.
The Microprocessor prefers spliced pri-let-7 in vivo
As part of the Microprocessor, Pasha is responsible for recognizing appropriate pri-miRNA substrates and guiding Drosha to cleave them. To test if Pasha differentially binds endogenous spliced versus unspliced pri-let-7 transcripts, we analyzed RNAs that coimmunoprecipitated with a rescuing GFP-tagged Pasha protein (Lehrbach et al. 2012). Immunoprecipitation of extracts from L4-stage worms revealed a ∼15-fold enrichment of SL1-pri-let-7 in the Pasha:GFP expressing strain (+) compared with the nontransgenic control (−) (Fig. 3A). In contrast, association of the unspliced isoform with Pasha:GFP was not detected above background levels. Thus, the enrichment of total pri-let-7 in the Pasha-GFP IP seems to be entirely reflective of SL1-pri-let-7 binding to this Microprocessor factor. This association is specific for the miRNA-containing transcript, as abundant SL1-spliced mRNAs, such as tba-1 and rbm-28, were not detected in the Pasha-GFP IPs.
FIGURE 3.
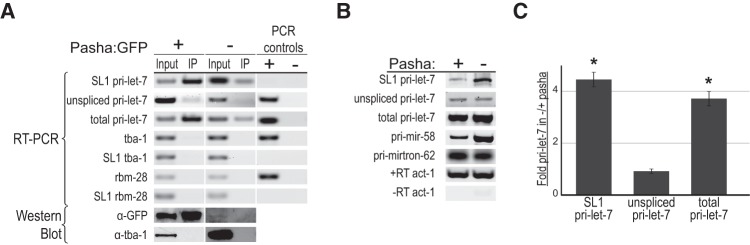
The Microprocessor prefers SL1-spliced pri-let-7 for processing in vivo. (A) Detection of transcripts associated with the Microprocessor in vivo. Extracts from L4 stage worm strains with (+) and without (−) the rescuing Pasha:GFP transgene were subjected to Immunoprecipitation (IP) using an anti-GFP antibody. Total (Input) and Immunoprecipitated (IP) RNAs were detected by RT-PCR followed by Agarose gel electrophoresis. PCR controls show the results using genomic DNA (+), which should not detect SL1-spliced sequences, or water (−) as templates. The bottom panels show the results of Western blot analysis with a GFP antibody to monitor the IP of Pasha:GFP and tubulin (TBA-1) as a loading and specificity control. Results are representative of two independent IP experiments. (B) Total RNA from pasha mutant (−) and rescued (+) worms was collected from L4 staged animals and used for RT-PCR analysis of the indicated transcripts. Results were analyzed by agarose gel electrophoresis and represent three independent experiments. Actin (act-1) served both as a loading control and to control for genomic DNA contamination in –RT samples. Pri-miR-58 is a constitutively expressed miRNA subject to Pasha regulation. Pri-mirtron-62 is not part of the canonical miRNA biogenesis pathway. (C) Average fold increase of pri-let-7 isoforms in pasha mutants (−) relative to rescued (+) worms collected at L4 and analyzed by qRT-PCR. Each pri-let-7 isoform was first normalized to the control transcript Y45F10D.4. The error bars represent standard error. (*) P < 0.05.
To further test if there is a difference in the pri-let-7 isoform utilized by the Microprocessor in vivo, we examined the accumulation of pri-miRNA transcripts in Pasha mutant worms. The temperature-sensitive pash-1(mj100) worms exhibit embryonic lethality when grown at 25°C but develop normally at 15°C (Lehrbach et al. 2012). An extrachromosomal transgene driving ubiquitous expression of a PASH-1:GFP fusion protein is able to rescue the temperature-sensitive allele (Lehrbach et al. 2012). Compared with control worms expressing the PASH-1:GFP transgene (+), there was a fourfold increase in the level of SL1- and total pri-let-7 in the pasha mutants (−) (Fig. 3B,C). The unspliced pri-let-7 transcripts were unaffected by the loss of Pasha, as was pri-mirtron-62, which bypasses the canonical miRNA biogenesis pathway (Ruby et al. 2007). These results demonstrate that the spliced form of pri-let-7 is specifically sensitive to the loss of Pasha and, thus, is the preferred substrate for processing in vivo. This conclusion is also consistent with previous observations that the SL1-spliced pri-let-7 transcript specifically accumulates upon disruption of a let-7 and Argonaute-Like-Gene 1 (ALG-1) auto-regulatory loop that promotes pri-let-7 processing and is the preferred substrate for processing when LIN-28 mediated repression is absent from the first larval stage (Van Wynsberghe et al. 2011; Zisoulis et al. 2012).
Taken together our results demonstrate that a role of trans-splicing in let-7 biogenesis is to remodel the secondary structure of pri-let-7 to promote favorable Drosha processing. Removing the spliceosome requirement by replacing the 3′ ss with the SL1 sequence in our transgene did not have a substantial effect on let-7 biogenesis. Although it is possible that the SL1 sequence itself could help recruit miRNA biogenesis factors, its presence is not sufficient for supporting processing when it is uncoupled from seeding structural remodeling of pri-let-7. Since trans-splicing was not an absolute requirement for let-7 rescue activity and production of the mature miRNA, it is likely that the structure of the unspliced transcript is somewhat flexible or that the Microprocessor has some degree of accessibility to suboptimal processing substrates in vivo. Considering the multiple cis-acting elements and trans-acting factors that regulate pri-let-7 transcription (Johnson et al. 2003; Roush and Slack 2009; Kai et al. 2013), it is also possible that processing of the spliced versus unspliced pri-let-7 transcripts may be under differential control depending on the timing or location of expression.
Although trans-splicing is not common across animal species, pri-miRNAs in other organisms are often found in transcripts subject to cis-splicing. The majority of human miRNAs, including several let-7 family members, are located in introns of host genes (Kim and Kim 2007; Roush and Slack 2008). In some cases, Drosha excises the pre-miRNAs rapidly and independently of pre-mRNA splicing (Kim and Kim 2007). In other studies, the spliceosome and Microprocessor associate together on the pre-mRNA and work mutually to coordinate miRNA processing and pre-mRNA splicing (Kataoka et al. 2009; Janas et al. 2011). In addition, exon–intron junction spanning pre-miRNAs have been found to be subject to regulation by alternative splicing through changes that occur in the pre-miRNA secondary structure (Melamed et al. 2013). Finally, in plants, some pri-miRNAs contain introns that enhance biogenesis when spliced, although the reason for this effect was not clear (Bielewicz et al. 2013; Schwab et al. 2013). Considering the importance of RNA structure for pri-miRNA processing by Drosha, splicing may serve as a mechanism to regulate the folding and, hence, the efficiency of recognition by the Microprocessor for many pri-miRNAs. Thus, the disruption of splicing that accompanies some stress and disease conditions could contribute to the reduced levels of certain miRNAs associated with these states.
MATERIALS AND METHODS
Several methods including worm staging, MosSCI integration, RNA extraction, RNAi treatment, Western blot analysis, Agarose and PAGE Northern Blot analysis, and reverse transcriptase PCR assays have been previously described (Van Wynsberghe et al. 2011; Zisoulis et al. 2012). Wild-type worms were N2 Bristol.
In vitro transcription of RNA
DNA templates for in vitro transcription of unspliced and SL1-spliced pri-let-7 were amplified from wild-type genomic DNA and SL1-spliced pri-let-7 plasmid DNA using the primers listed below. DNA templates were then purified with the QIAquick Gel Extraction Kit (Qiagen). The MEGA Shortscript T7 transcription kit (Ambion) was used to transcribe RNA from these DNA templates and the resulting RNA was phenol chloroform extracted and analyzed by spectrophotometry for purity and quantity. Fifty micrograms of transcribed RNA was treated with Calf Intestinal Alkaline Phosphatase (CIP) to remove the 5′ phosphates and then isolated and purified through 5% TBE-UREA polyacrylamide gel electrophoresis (PAGE) by cutting out the band of interest, crushing it and rocking in 500 μL 0.3 M NaCl at 4°C. CoStar Spin-X columns containing a 0.45 μM filter were used to isolate the supernatant, which was then alcohol precipitated and eluted to 10 μL. The purified transcripts were 5′ end labeled with [γ-32]P in a T4 Polynucleotide Kinase (PNK) reaction and spun through an Illustra Microspin G-50 column (GE Healthcare) to remove unincorporated nucleotides. Afterward, transcripts were run on a test PAGE to ensure homogeneity.
RNase secondary structure analysis
As individual mixes, unspliced and spliced labeled RNA was mixed with 1 μg of Yeast RNA (Ambion) boiled and allowed to refold at room temperature. In separate tubes, samples were treated with either 0.02 Units (U) of RNase V1 (Ambion), 0.02 U of RNase T1 (Ambion), 0.002 U of RNase A (Ambion) or buffer for 15 min at room temperature. An all nucleotide ladder was produced by boiling the RNA mix in an alkaline hydrolysis buffer for 5 min, and Decade Markers (Ambion) were labeled and prepared per instructions. Reactions were stopped by adding formamide loading buffer and analyzed by 20 cm × 40 cm × 0.4 mm 8% TBE-UREA-PAGE. Gels were dried, exposed to phosphor screens, and scanned on a Typhoon phosphorimager.
RNA immunoprecipitations
RNA immunoprecipitations were performed as previously described (Van Wynsberghe et al. 2011; Zisoulis et al. 2012; Broughton and Pasquinelli 2013). Briefly, L4-staged Pasha:GFP and wild-type, nontransgenic, worms were resuspended in Lysis Buffer (100 mM NaCl, 2.5 mM HEPES [pH 7.5], 250 μM EDTA [pH 8.0], 0.1% SDS, 0.1% NP-40, Complete Mini Protease Inhibitor cocktail [Roche], 2 mM DTT, 25 U/mL rRNasin [Promega]) and sonicated in ten second pulses, five times, resting on ice for 1 min in between pulses. Extracts were cleared by spinning at 16,000g for 15 min, 4°C, then snap frozen on dry-ice ethanol and stored at −80°C. Lysates were thawed by rocking at 4°C and protein concentration measured with a Qubit Fluorometer. Equal lysate amounts were precleared with Protein G Dynabeads (Invitrogen), followed by incubation at 4°C either with preconjugated GFP magnetic beads (Clonotek) for 2 h or with GFP polyclonal antibody overnight and 1 h with washed Protein G Dynabeads. Beads were washed twice with high salt wash buffer (50 mM Tris–HCl [pH 7.4], 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate), low salt wash buffer (20 mM Tris–HCl [pH 7.4], 10 mM MgCl2, 0.2% Tween-20), and proteinase K buffer (100 mM Tris–Cl [pH 7.4], 50 mM NaCl, and 10 mM EDTA), before treatment with proteinase K (Invitrogen) and urea. RNA was TRIzol (Invitrogen) extracted, treated with RQ1 DNase (Promega), and re-extracted before cDNA synthesis with random primers and Superscript III (Invitrogen). PCR was performed with the below listed oligos (IDT).
Sequences and primers used in this study
Sequences inserted/deleted in this study: SL1 5′-GGTTTAATTACCCAAGTTTGAG-3′, 3′ss 5′-TTTTCAG-3′, RDM 5′-GTATCCGTAAAGCTCATTAAGC-3′, MSF 5′-AAGACGACGCAGCTTCGAAGAGTTCTGTCTCCTCTACTAATCGCCTGC-3′.
Primers for T7 transcription: unspliced pri-let-7 fwd 5′-TAATACGACTCACTATAGGTTTTTCAGGCAAGCAGGCGAT-3′ and rev 5′-GAAAAACAAAGAGGTGAAAGTAAG-3′, SL1-spliced let-7 fwd 5′-TAATACGACTCACTATAGGTTTAATTACCCAAGTTTGA-3′ and rev 5′-AAAGAAAGTTGTGAGAGCAAGACG-3′. Primers for RT-PCR: unspliced pri-let-7 fwd 5′-GTCTAATTTAACAACAAGTACTAATCCATT-3′, SL1-sequence fwd 5′-GGTTTAATTACCCAAGTTTGAG-3′, total pri-let-7 fwd 5′-CAAGCAGGCGATTGGTGGA-3′ and pri-let-7 rev 5′-GTAAGGTAGAAAATTGCATAGTTC-3′, pri-mir-58 fwd 5′-GGCTTCAGTGGCTCCTCT-3′ and pri-mir-58 rev 5′-CGTTTAGTGCGCACATTCGGCAA-3′, mirtron-62 fwd 5′-CCATGTACTCCGGCTATAGTGAG-3′ and mirtron-62 rev 5′-GATGTTGAACAACCTGTAAGCTAGATT-3′, actin fwd 5′-GTGTTCCCATCCATTGTCGGAAGAC-3′ and actin rev 5′-GTGAGGAGGACTGGGTGCTCTT-3′, tba-1 fwd 5′-ATGCGTGAGGTCATCTCCAT-3′ and tba-1 rev 5′-TGATGGCATAGTTCCATCGG-3′, rbm-28 fed 5′-GATTCAGGAGTTATGGGTATAATTCTTC-3′ and rbm-28 rev 5′-GGCTGTATCCGCCGTAGC-3′.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Finnegan and members of the Pasquinelli laboratory for their suggestions and critical reading of this manuscript. We thank undergraduate helpers A. Weaver and K. Nassiri for helping with phenotype analysis, RNA extractions, and PCRs. We thank Dr. Miska for the Pasha mutant and GFP-tagged strains, Dr. David for use of a Real-Time PCR machine, and Dr. Oegema and Dr. Desai for the polyclonal GFP antibody. V.M. was supported in part by the University of California, San Diego, Cellular and Molecular Genetics Training Program through an institutional training grant from the National Institute of General Medical Sciences (T32 GM007240) and an NIH Ruth L. Kirschstein National Research Service Award Individual Predoctoral Fellowship to Promote Diversity in Health-Related Research (F3194041A). This work was funded by grants from the NIH (GM071654) and the Keck and Peter Gruber Foundations (A.E.P.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.052118.115.
REFERENCES
- Auyeung VC, Ulitsky I, McGeary SE, Bartel DP. 2013. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell 152: 844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielewicz D, Kalak M, Kalyna M, Windels D, Barta A, Vazquez F, Szweykowska-Kulinska Z, Jarmolowski A. 2013. Introns of plant pri-miRNAs enhance miRNA biogenesis. EMBO Rep 14: 622–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal T. 2012. Trans-splicing and operons. In Wormbooks (ed. Community TCeR). WormBook, Pasadena, CA. [Google Scholar]
- Bracht J, Hunter S, Eachus R, Weeks P, Pasquinelli AE. 2004. Trans-splicing and polyadenylation of let-7 microRNA primary transcripts. RNA 10: 1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton JP, Pasquinelli AE. 2013. Identifying Argonaute binding sites in Caenorhabditis elegans using iCLIP. Methods 63: 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad T, Marsico A, Gehre M, Andersson Ørom U. 2014. Microprocessor activity controls differential miRNA biogenesis in vivo. Cell Rep 9: 542–554. [DOI] [PubMed] [Google Scholar]
- Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. 2004. Processing of primary microRNAs by the Microprocessor complex. Nature 432: 231–235. [DOI] [PubMed] [Google Scholar]
- Finnegan EF, Pasquinelli AE. 2013. MicroRNA biogenesis: regulating the regulators. Crit Rev Biochem Mol Biol 48: 51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynt AS, Greimann JC, Chung W-J, Lima CD, Lai EC. 2010. MicroRNA biogenesis via splicing and exosome-mediated trimming in Drosophila. Mol Cell 38: 900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. 2008. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet 40: 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. 2004. The Microprocessor complex mediates the genesis of microRNAs. Nature 432: 235–240. [DOI] [PubMed] [Google Scholar]
- Gurtan AM, Sharp PA. 2013. The role of miRNAs in regulating gene expression networks. J Mol Biol 425: 3582–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, Kim VN. 2014. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524. [DOI] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom K-H, Nam J-W, Heo I, Rhee J-K, Sohn SY, Cho Y, Zhang B-T, Kim VN. 2006. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 125: 887–901. [DOI] [PubMed] [Google Scholar]
- Janas MM, Khaled M, Schubert S, Bernstein JG, Golan D, Veguilla RA, Fisher DE, Shomron N, Levy C, Novina CD. 2011. Feed-forward microprocessing and splicing activities at a microRNA-containing intron. PLoS Genet 7: e1002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Lin S-Y, Slack FJ. 2003. The time of appearance of the C. elegans let-7 microRNA is transcriptionally controlled utilizing a temporal regulatory element in its promoter. Dev Biol 259: 364–379. [DOI] [PubMed] [Google Scholar]
- Kai ZS, Finnegan EF, Huang S, Pasquinelli AE. 2013. Multiple cis-elements and trans-acting factors regulate dynamic spatio-temporal transcription of let-7 in C. elegans. Dev Biol 374: 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka N, Fujita M, Ohno M. 2009. Functional association of the Microprocessor complex with the spliceosome. Mol Cell Biol 29: 3243–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Kim VN. 2007. Processing of intronic microRNAs. EMBO J 26: 775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2011. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39: D152–D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Sobczak K, Wilczynska U, Drath M, Jasinska A, Kaczynska D, Krzyzosiak WJ. 2004. Structural features of microRNA (miRNA) precursors and their relevance to miRNA biogenesis and small interfering RNA/short hairpin RNA design. J Biol Chem 279: 42230–42239. [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, et al. 2003. The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415–419. [DOI] [PubMed] [Google Scholar]
- Lehrbach NJ, Castro C, Murfitt KJ, Abreu-Goodger C, Griffin JL, Miska EA. 2012. Post-developmental microRNA expression is required for normal physiology, and regulates aging in parallel to insulin/IGF-1 signaling in C. elegans. RNA 18: 2220–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias S, Plass M, Stajuda A, Michlewski G, Eyras E, Cáceres JF. 2012. DGCR8 HITS-CLIP reveals novel functions for the Microprocessor. Nat Struct Mol Biol 19: 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed Ze, Levy A, Ashwal-Fluss R, Lev-Maor G, Mekahel K, Atias N, Gilad S, Sharan R, Levy C, Kadener S, et al. 2013. Alternative splicing regulates biogenesis of miRNAs located across exon-intron junctions. Mol Cell 50: 869–881. [DOI] [PubMed] [Google Scholar]
- Mondol V, Pasquinelli AE. 2012. Let's make it happen: the role of let-7 microRNA in development. In Current topics in developmental biology: microRNAs in development (ed. Hornstein E), pp. 1–30. Academic Press, Waltham, MA. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE. 2012. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 13: 271–282. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Muller P, et al. 2000. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408: 86–89. [DOI] [PubMed] [Google Scholar]
- Quick-Cleveland J, Jacob JP, Weitz SH, Shoffner G, Senturia R, Guo F. 2014. The DGCR8 RNA-binding heme domain recognizes primary microRNAs by clamping the hairpin. Cell Rep 7: 1994–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. 2000. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901–906. [DOI] [PubMed] [Google Scholar]
- Roush S, Slack FJ. 2008. The let-7 family of microRNAs. Trends Cell Biol 18: 505–516. [DOI] [PubMed] [Google Scholar]
- Roush SF, Slack FJ. 2009. Transcription of the C. elegans let-7 microRNA is temporally regulated by one of its targets, hbl-1. Dev Biol 334: 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan CH, Bartel DP. 2007. Intronic microRNA precursors that bypass Drosha processing. Nature 448: 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab R, Speth C, Laubinger S, Voinnet O. 2013. Enhanced microRNA accumulation through stemloop-adjacent introns. EMBO Rep 14: 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani G, Chen X, Zhao H, Slack FJ. 2015. A novel mechanism of LIN-28 regulation of let-7 microRNA expression revealed by in vivo HITS-CLIP in C. elegans. RNA 21: 985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wynsberghe PM, Kai ZS, Massirer KB, Burton VH, Yeo GW, Pasquinelli AE. 2011. LIN-28 co-transcriptionally binds primary let-7 to regulate miRNA maturation in Caenorhabditis elegans. Nat Struct Mol Biol 18: 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace A, Filbin ME, Veo B, McFarland C, Stepinski J, Jankowska-Anyszka M, Darzynkiewicz E, Davis RE. 2010. The nematode eukaryotic translation initiation factor 4E/G complex works with a trans-spliced leader stem-loop to enable efficient translation of trimethylguanosine-capped RNAs. Mol Cell Biol 30: 1958–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, Johnson WE, Bass BL. 2011. Improved annotation of C. elegans microRNAs by deep sequencing reveals structures associated with processing by Drosha and Dicer. RNA 17: 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westholm JO, Lai EC. 2011. Mirtrons: microRNA biogenesis via splicing. Biochimie 93: 1897–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Cullen BR. 2005. Efficient processing of primary microRNA hairpins by Drosha requires flanking nonstructured RNA sequences. J Biol Chem 280: 27595–27603. [DOI] [PubMed] [Google Scholar]
- Zisoulis DG, Kai ZS, Chang RK, Pasquinelli AE. 2012. Autoregulation of microRNA biogenesis by let-7 and Argonaute. Nature 486: 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.