

Abstract
The in vivo assembly of ribosomal subunits requires assistance by maturation proteins that are not part of mature ribosomes. One such protein, RbfA, associates with the 30S ribosomal subunits. Loss of RbfA causes cold sensitivity and defects of the 30S subunit biogenesis and its overexpression partially suppresses the dominant cold sensitivity caused by a C23U mutation in the central pseudoknot of 16S rRNA, a structure essential for ribosome function. We have isolated suppressor mutations that restore partially the growth of an RbfA-lacking strain. Most of the strongest suppressor mutations alter one out of three distinct positions in the carboxy-terminal domain of ribosomal protein S5 (S5) in direct contact with helix 1 and helix 2 of the central pseudoknot. Their effect is to increase the translational capacity of the RbfA-lacking strain as evidenced by an increase in polysomes in the suppressed strains. Overexpression of RimP, a protein factor that along with RbfA regulates formation of the ribosome's central pseudoknot, was lethal to the RbfA-lacking strain but not to a wild-type strain and this lethality was suppressed by the alterations in S5. The S5 mutants alter translational fidelity but these changes do not explain consistently their effect on the RbfA-lacking strain. Our genetic results support a role for the region of S5 modified in the suppressors in the formation of the central pseudoknot in 16S rRNA.
Keywords: ribosome assembly, 16S rRNA central pseudoknot, RbfA, RimP, translational accuracy
INTRODUCTION
Biogenesis of ribosomes, the cellular components responsible for protein synthesis, is the most energy intensive part of cellular metabolism consuming up to 80% of cellular energy in rapidly dividing bacteria, yeast, and human cells (Bremer and Patrick 1996; Warner 1999; Tschochner and Hurt 2003). Ribosomes also constitute by far the largest fraction of cellular RNA and protein in all cells in order to support production of thousands of ribosomes per minute in rapidly growing cells. Ribosomes are quite complex in structure, involving multiple RNAs and many integral ribosomal proteins as well as both RNAs and proteins undergoing many covalent modifications. The process of biogenesis, therefore, must be highly coordinated to achieve the required rate of production. Bacterial, but not eukaryotic ribosomal subunits can be reconstituted in vitro into active ribosomes from their components of rRNA and ribosomal proteins (r-proteins) but the process is slow and requires nonphysiological conditions (Nierhaus 1991) whereas biogenesis in vivo takes as little as 3 min (Champney 1977). This difference in efficiency can be attributed to the presence of accessory proteins that assist during cellular assembly of ribosomal subunits. Hundreds of factors facilitate biogenesis of eukaryotic ribosomes (Woolford and Baserga 2013), which along with their inability to be reconstituted in vitro argues for the complexity of their structure. In contrast, only around a dozen of biogenesis factors have been identified in bacteria but others are suspected to exist (Connolly and Culver 2009). However, even with so few bacterial factors, their functions are diverse including endonucleases that process the rRNA transcripts to mature rRNAs, enzymes that modify rRNA and r-proteins, DEAD box RNA helicases, GTPases, and many proteins with unknown functions (Connolly and Culver 2009). Known proteins with a role in assembly of the 30S subunit in Escherichia coli include the GTPases Era (Nashimoto et al. 1985; Nashimoto 1993; Sayed et al. 1999; Inoue et al. 2003) and YjeQ/RsgA (Himeno et al. 2004; Campbell and Brown 2008), the rRNA and r-protein modifying enzymes KsgA (Connolly et al. 2008) and RimJ (Roy-Chaudhuri et al. 2010), the ribosome maturation factors RimM (Lövgren et al. 2004) and RimP (Nord et al. 2009), and the cold-shock protein RbfA (Dammel and Noller 1993). In addition, the DnaK chaperone system and the tRNA modifying enzyme YrdC (RimN) may participate in 30S maturation (Maki et al. 2002, 2003; Kaczanowska and Ryden-Aulin 2005), although their role is controversial (Alix and Nierhaus 2003; El Yacoubi et al. 2009). The complexity of the folding of the rRNAs (Stern et al. 1989) and the need to insert ribosomal proteins into this folded structure (Recht and Williamson 2004) present significant thermodynamic barriers to ribosome assembly. Although the precise role of individual factors is frequently unknown, the factors Era, RimM, and RimP have been shown to increase the rate of incorporation of specific r-proteins during in vitro reconstitution of the 30S ribosomal subunits (Bunner et al. 2010).
RbfA (ribosome binding factor A) was initially identified as a high-copy suppressor of the dominant cold-sensitive C23U mutation in 16S rRNA. The protein associates with free 30S ribosomal subunits but not with 70S ribosomes or polysomes (Dammel and Noller 1993, 1995). Thermus thermophilus RbfA binds to the 30S subunit near C23 in a position that overlaps with the binding sites for the A and P site tRNA (Datta et al. 2007) suggesting that the cold sensitivity of the C23U mutant might result from slowed recruitment of RbfA to this binding site. Consistent with that idea, a mutant lacking RbfA is also cold-sensitive and shows a constitutive cold-shock response (Jones and Inouye 1996). Mutations in both 16S rRNA and rbfA affect ribosome function and biogenesis in similar ways, with increased levels of free 50S and 30S subunits and decreased levels of 70S ribosomes and polysomes (Dammel and Noller 1993, 1995). An rbfA null mutant also showed slowed conversion of 17S rRNA to mature 16S rRNA (Bylund et al. 1998), which was exacerbated at lower temperatures (Xia et al. 2003). All of these phenotypes are shared by mutants lacking the 30S ribosomal subunit biogenesis factors Era (Nashimoto et al. 1985), KsgA (Connolly et al. 2008), RimM (Lövgren et al. 2004), RimP (Nord et al. 2009), and YjeQ/RsgA (Guo et al. 2011). Genetic interactions suggest that these factors act together to complete the final steps in 30S maturation (Shajani et al. 2011). For example, overexpressing RbfA improves the growth and translational efficiency at 37°C of a strain lacking the ribosome maturation factor RimM (Bylund et al. 1998) and overexpression of the GTPase Era suppresses partially the slow growth and cold-sensitive 30S subunit maturation deficiency of the mutant lacking RbfA (Inoue et al. 2003).
Ribosome assembly begins cotranscriptionally with local secondary structures folding in a 5′–3′ direction and many ribosomal proteins binding before the transcript is complete (for review, see Shajani et al. 2011). Assembly progresses through multiple parallel pathways, some of which may lead to kinetically trapped structures that cannot progress by further rRNA folding or ribosomal protein binding (Sykes and Williamson 2009). The role of the RimM and RbfA factors appears to be to bind nascent rRNAs and block the formation of trapped structures (Williamson 2003; Clatterbuck Soper et al. 2013).
Clatterbuck Soper et al. (2013) used in vivo footprinting to demonstrate that presumed chaperones RbfA and RimM are required for mature folding of the 3′ end of 16S rRNA including the “head” domain and helix 44, which forms important intersubunit bridges in the mature structure (Yusupov et al. 2001; Schuwirth et al. 2005; Selmer et al. 2006). Pre-30S subunits formed in the absence of these proteins lacked tertiary binding ribosomal proteins (S2, S3, and S21) and helices 1 and 2 (h1 and h2), which comprise the central pseudoknot, and helix 44 were significantly destabilized. These results were corroborated by Sashital et al. (2014) who used quantitative mass spectrometry and electron microscopy to demonstrate roles for several late biogenesis factors in formation of the central pseudoknot and anchoring the head domain in its mature position. These large scale assembly events are coordinated with the addition of individual ribosomal proteins, beginning with S5, which forms part of the interface with the head domain in its tethered position.
The last steps of rRNA processing occur during the final stage of 30S assembly. Mutants lacking the late-30S ribosome biogenesis factors RbfA (Inoue et al. 2003), Era (Nashimoto et al. 1985), RimM (Lövgren et al. 2004), KsgA (Connolly et al. 2008), RimP (Nord et al. 2009), and RsgA (Guo et al. 2011) accumulate an immature 17S rRNA carrying 5′ and 3′ extensions that are removed in generating mature 16S rRNA. Retaining as little as 10 nt of the 5′ leader is predicted to result in formation of an alternative structure lacking h1 of the central pseudoknot rRNA (Young and Steitz 1978; Dammel and Noller 1993), a structure located in the ribosomal A site near other structures associated with fidelity. In addition to accumulating 17S rRNA a mutant form of ribosomal protein S5 (G28D) and in rimM and ksgA mutants also cause reduced translational fidelity (Roy-Chaudhuri et al. 2010). Because overexpression of the biogenesis protein RimJ in the mutant expressing S5-G28D both restores translational fidelity and increases 16S maturation, Roy-Chaudhuri et al. (2010) have suggested that the rRNA extension is the cause of decreased fidelity. Clatterbuck Soper et al. (2013) showed that the 17S-containing pre-30S subunits can catalyze dipeptide formation in vitro but at a rate substantially lower than mature 30S subunits; whether the immature subunits participate in translation in vivo, however, has not been demonstrated. A recent structural study showed that immature 30S subunits isolated from an ΔrbfA ΔrsgA mutant strain that lack of S5 also show increased mobility of the head domain and flexibility of h1 and the 5′ end of the rRNA, suggesting an active role for S5 in both tethering of the head domain and formation of the central pseudoknot (Yang et al. 2014). This correlation, however, does not establish an active role for S5 in that process other than to comprise part of the stable structure formed under the control of late biogenesis factors like RbfA.
To further explore the relationship between RbfA and ribosome biogenesis and function we identified chromosomal mutations that suppress the cold sensitivity and slow growth of an rbfA::KmR null mutant. Several of these mutations target the rpsE gene, encoding S5. These suppressor mutations also increased the rate of translation and suppressed the lethality of overexpressing a second ribosome assembly protein, RimP, in the rbfA::KmR background. The rpsE suppressors increased the amount of polysomes in the rbfA::KmR background; this result is consistent with improved growth resulting from increased translational output. We found that the immature 17S-contatining 30S subunits do not participate in translation since they were absent from the polysomes. The rpsE suppressors did not significantly reduce the proportion of 17S rRNA in the 30S or 70S monosomes but they did increase the amount of mature or near-mature 30S subunits engaged in translation in polysomes. Because of the suggested connection between late ribosomal biogenesis events and translational fidelity, we tested the effect of rbfA::KmR and the rpsE mutations on translational misreading. The rbfA::KmR mutation caused decreased fidelity as did four of the five rpsE suppressor mutations; the fifth suppressor actually increased fidelity. However, there was no consistent effect on accuracy of combining rbfA::KmR with the rpsE suppressors. This result suggests that the improved growth effect seen in the double mutants does not reflect a change in accuracy. Rather, the rpsE mutations appear to improve the ability of mature 30S subunits to engage in translation, probably by reducing the formation of improperly matured subunits.
RESULTS
Overexpression of RimP in the absence of RbfA causes synthetic lethality
In order to further characterize the function of RbfA we identified high-copy suppressors of the slow growth of a strain lacking RbfA. Plasmids from the NIG collection (Saka et al. 2005), from which the expression of the cloned genes could be induced with IPTG, were introduced into the rbfA::KmR mutant GOB162 and tested for the ability to increase growth rate. Consistent with the work of others (Bylund et al. 1998; Inoue et al. 2003), we found high-copy suppressor interactions between the rbfA::KmR mutant and only three other ribosome biogenesis protein genes. A plasmid carrying the era gene improved growth of the mutant (data not shown) as expected (Inoue et al. 2003). Overexpressing RimM did not alter growth of the rbfA::KmR mutant (Fig. 1) despite the fact that overexpressing RbfA suppresses the slow growth of a strain lacking RimM (Bylund et al. 1998). Surprisingly, overproducing the RimP ribosome maturation factor actually prevented growth of the rbfA::KmR mutant GOB162 at 30°C, 37°C, and 44°C (Fig. 1). All of these factors—Era, RbfA, RimM, and RimP—function late during the cotranscriptional association of r-proteins with the 16S rRNA (Bunner et al. 2010; Sashital et al. 2014) and RbfA, RimM, and RimP are all implicated in formation and stabilization of the central pseudoknot (Sashital et al. 2014). The synthetic lethality from overexpressing RimP in the absence of RbfA is particularly striking, perhaps suggesting that RimP can force bypass of an RbfA “checkpoint” (Connolly and Culver 2013) that coordinates other morphogenetic events with formation of the central pseudoknot.
FIGURE 1.
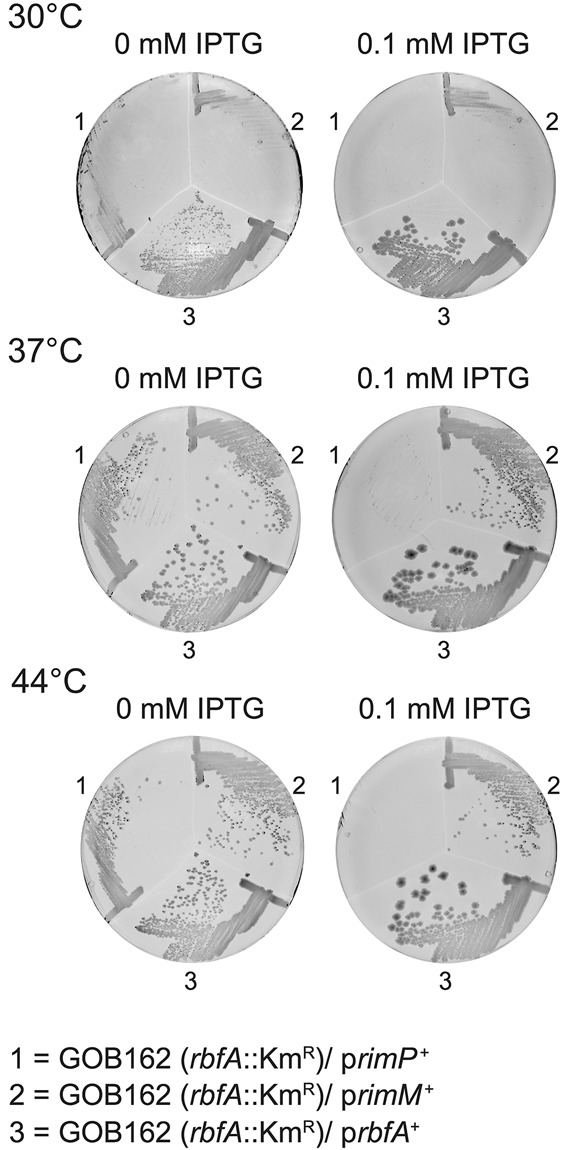
The effect of plasmid-mediated expression of the ribosome maturation proteins RimP, RimM, and RbfA on the growth of the rbfA::KmR mutant GOB162. The plates were incubated for 48 h at 30°C, 37°C, and 44°C, respectively. The plasmids used were pNTR-SD-rbfA (519#5), pNTR-SD-rimM (439#9), and pNT3-yhbC (rimP) (520#2) from the NIG collection and contain an IPTG-inducible promoter that directs the expression of the cloned genes.
Identifying rpsE mutations that suppress the slow growth phenotype of an rbfA rimP double mutant
To investigate the function of ribosome assembly proteins we identified second site mutations that suppressed the slow growth of mutants lacking the biogenesis proteins RbfA and RimP. The rbfA::KmR insertion mutant grows most slowly at low temperature (2.5-fold at 26°C), while a strain carrying an in-frame deletion of rimP (ΔrimP135) grows most slowly at high temperature (2.5-fold at 44°C) (Dammel and Noller 1995; Nord et al. 2009). Attempts to isolate suppressors of the ΔrimP135 mutation were unsuccessful (data not shown) and there are no reports of chromosomal suppressors of the rbfA::KmR mutation. We chose to select suppressors of a rbfA::KmR ΔrimP135 double mutant because it grows more slowly than either single mutant and we reasoned that this more intense phenotype might make it easier to isolate suppressor mutations. To isolate faster-growing derivatives of the rbfA::KmR ΔrimP135 double mutant STN133, cultures were grown overnight in rich medium at 30°C, 37°C, and 44°C and samples were tested for colony size on rich medium plates incubated at the same temperature. Cells were subcultured repeatedly for 7 d to allow faster-growing clones to arise. In this way, 12, 11, and 9 independently isolated faster-growing derivatives were obtained at 30°C, 37°C, and 44°C, respectively. Different classes of clones were identified based on colony size on rich medium plates (Table 1). In general, the difference in growth between the suppressor-containing clones and the rbfA::KmR ΔrimP135 parental strain was more pronounced for clones isolated at 30°C than for those isolated at the other two temperatures.
TABLE 1.
Growth of suppressor containing strains isolated at different temperatures

It seemed probable that some of the suppressor mutations (srar = suppressor to rbfA and rimP) would affect genes encoding small subunit ribosomal proteins, specifically, those dependent on RbfA for binding to the preribosome. Since two-thirds of ribosomal protein genes are found in a cluster at the 74th minute of the E. coli chromosome (Berlyn 1998), we tested whether introducing this region from a suppressor-free strain could replace suppressor mutation in one of the faster-growing suppressor containing clones, STN174 (rbfA::KmR ΔrimP135 srar022). We grew the generalized transducing phage P1 on strain PW078, which contains a Tn10 insertion, zhc-2421::Tn10, conferring tetracycline resistance (TcR) downstream from the last gene of this r-protein gene cluster, rplQ. We used this stock of phage to transduce strain STN174 to TcR. rbfA is located at 71.4, too distant to be cotransduced with zhc-2421::Tn10. The majority of individual transductant colonies examined on rich medium plates at 37°C showed the slow growth characteristics of the original rbfA::KmR ΔrimP135 double mutant STN133 suggesting that the suppressor mutation in strain STN174 had been crossed out and therefore was located in this region of the chromosome. The linkage between zhc-2421::Tn10 and the suppressor mutation was determined to be 95% (data not shown). In order to identify the suppressor mutation, regions containing the genes for r-proteins S4, S11, S13, S5, S8, S14, and S19 were amplified by PCR and sequenced. Strain STN174 contained a C to T transition in position 380 of rpsE, which encodes S5, changing an alanine (GCC) to a valine (GTC) at codon 127; no other sequence changes were found in the other genes sequenced. To determine if the other suppressor mutations also occur in rpsE, the gene was sequenced from all strains listed in Table 1, except STN155, STN156, STN173, and STN185. Strain STN161 (rbfA::KmR ΔrimP135 srar012) contained the identical C to T transition in codon 127 as in STN174, while in STN187 (rbfA::KmR ΔrimP135 srar032), the same GCC alanine codon had been changed to ACC for threonine. STN176 (rbfA::KmR ΔrimP135 srar024) and STN186 (rbfA::KmR ΔrimP135 srar031) both contained a transition from G to A in position 259 changing codon 87 from GGT for glycine to AGT for serine, while in STN181 (rbfA::KmR ΔrimP135 srar027) the same codon was changed to GCT for alanine. STN182 (rbfA::KmR ΔrimP135 srar028) contained a transversion of G to C in position 272 changing codon 91, GGT, for glycine to GCT for alanine. Thus, two mutations were found twice and for two codon positions there were different mutations resulting in different amino acid substitutions. Interestingly, the three different amino acid positions that were altered in the suppressor containing strain are clustered in the structure of S5 (Fig. 2). The three amino acids are located in a small region of S5 within 10–11.3 Å of each other. This region comprises three closely packed structures, two adjacent β sheets and a random coil region. Gly 91 is located on the surface of S5 in a turn between β sheets 5 and 6 that is in close opposition to the first base pair of helix 2 (h2) of the central pseudoknot. Ala 127 is located in an unstructured region carboxy-terminal to sheet 7 on the surface of S5 close to the first base pair of h1 of 16S rRNA. Gly 87 is in sheet 6 and is not directly in contact with the rRNA.
FIGURE 2.
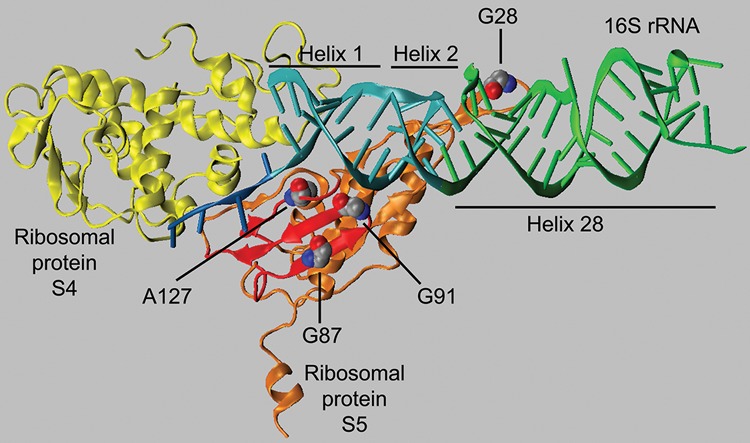
The position of the mutant amino acids of S5 (in orange) in relation to S4 (in yellow) and the central pseudoknot (helices 1 and 2; in cyan) and the neck helix (helix 28; in green) in 30S subunit (PDB ID 2AVY). The three amino acids altered in the S5 mutants described here (G87, G91, and A127) are shown in space-filling mode. The residues are located in a domain of S5 (in red) including a β sheet, composed of two β strands, and an adjoining strand. This domain is in close proximity to the central pseudoknot. The position of the spectinomycin-resistance mutant G28 is also shown in space-filling and is located near the region of h28 that is stacked on h2. The structure was modeled using VMD (Humphrey et al. 1996).
The rpsE mutations suppress the slow growth of the rbfA::KmR but not the ΔrimP135 mutant
Since both the rbfA and the rimP mutations confer slow growth, we investigated whether the rpsE mutations, isolated as suppressors of the slow growth of the double mutant, would improve the growth of either single mutant or have any negative effect on the growth of an rbfA+ rimP+ strain. The suppressors were transferred to strains GOB162 (rbfA::KmR), MW187 (ΔrimP135), and MW100 (wild type) by P1 transduction using the linked marker mutations. None of the five different rpsE mutations suppressed the slow growth of the ΔrimP135 mutant MW187 on solid or liquid rich medium at 44°C or had any detectable effect on the growth of the wild-type strain MW100 on solid rich medium at 21°C, 30°C, or 37°C (data not shown). However, all the rpsE mutations improved the growth on solid rich medium at 30°C, 37°C, and 44°C of the mutant lacking RbfA with the greatest effect at 30°C (data not shown). In liquid medium at 30°C, the rpsE mutations increased the growth rate of the rbfA::KmR mutant 1.3- to 1.6-fold (Fig. 3). Thus, the different rpsE mutations suppressed the growth deficit caused by the lack of RbfA and not RimP.
FIGURE 3.
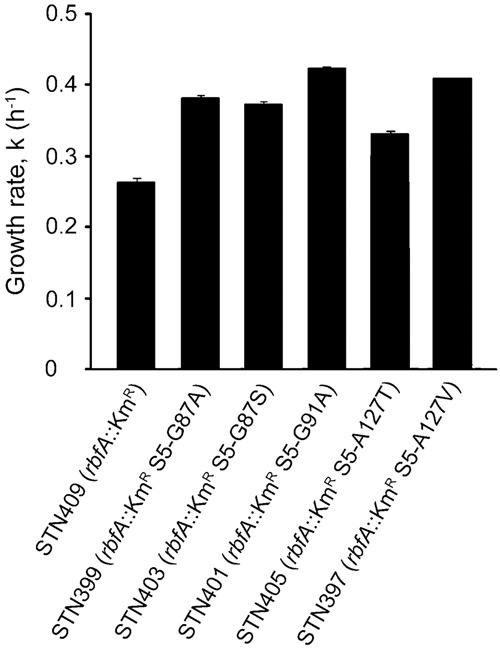
Growth at 30°C in LB medium of an rbfA::KmR mutant and its derivatives expressing variants of r-protein S5 containing different amino acid substitutions. The mean of the growth rate shown is from three independent experiments for all strains except for strain STN399 for which it is from two independent experiments. The growth rate, k, is shown, which is determined from the equation k = ln2/g, where g is the mass doubling time in hours. The error bars represent the standard deviation.
Lack of the cold-shock protein RbfA impairs the response to a shift from 37°C to 15°C causing slower growth than wild type after the cold shock (Jones and Inouye 1996). We tested the ability of the rpsE mutations to suppress the slow growth of the rbfA::KmR mutant after cold shock by comparing the growth of suppressor-free and suppressor-containing derivatives of the rbfA::KmR mutant after shifts from 37°C to 15°C in rich medium. The rbfA::KmR strain resumed growth soon after the shift, but at a rate several-fold lower than for the wild-type strain MW00 (data not shown). The rpsE mutations improved growth at 15°C after shock from 1.2- to 1.6-fold. The fact that the suppressors had the same effect on growth with or without cold-shock suggests that they may generally improve the growth rate rather than altering the response to cold shock per se.
The rpsE mutations increase the proportion of 30S subunits that take part in translation
A strain that lacks RbfA is deficient in the maturation of the 30S ribosomal subunits and shows an accumulation of free 50S and 30S ribosomal subunits and much less translating ribosomes than a wild-type strain probably as a result of this maturation deficiency (Dammel and Noller 1995; Bylund et al. 1998; Inoue et al. 2003; Xia et al. 2003). Polysome profiles obtained after centrifugation through sucrose gradients of total cell extracts of relevant strains grown at 30°C confirmed this observation. For the rbfA::KmR strain the size of the peaks corresponding to 70S ribosomes and polysomes were strongly reduced, whereas those for free 50S and 30S subunits were strongly increased relative to wild type (cf. Fig. 4A,B). The presence of the rpsE suppressors in the rbfA::KmR background resulted in increased amounts of 70S ribosomes and polysomes and reduced amounts of 30S and 50S subunits than in their absence (cf. Fig. 4A–F). To quantify the effect of the suppressors we calculated the ratio of the area of the 30S peak to the area of the peaks for 70S ribosomes and polysomes. The obtained ratio [30S/total 70S] was lower for the suppressor containing strains compared with the rbfA::KmR mutant by an average of 2.2-fold (Fig. 4H). The difference was greatest (2.8-fold) for the rbfA::KmR rpsE-G91A strain and least (1.8-fold) for the rbfA::KmR rpsE-A127T strain. The change in this ratio results from a reduction in the total amount of free subunits (30S reduced by an average of 28% and 50S by 19%) and an increase in associated subunits (70S increasing by 46% and polysomes by 85%). Thus, the presence of the rpsE suppressors improved the fraction of 30S subunits competent to participate in translation but not to wild-type levels. The strongest suppressor (rpsE-G91A), which caused the greatest growth rate, had the greatest improvement and the weakest (rpsE-A127T) had the least; the three with intermediate growth phenotypes were also intermediate for this phenotype. The correlation between the two phenotypes suggests that improvement in growth results from improvement in ability of 30S subunits to participate in translation.
FIGURE 4.

Polysome profiles of an rbfA::KmR mutant (A) and its derivatives expressing variants of r-protein S5 containing different amino acid substitutions (B–F), as well as of a wild-type strain (G). Different ribosomal particles are marked above corresponding peaks. 2x, 3x, and 4x indicate two, three, and four 70S ribosomes per mRNA, respectively. In H, the ratio is shown of the area of the peak for the 70S and 2x, 3x, and 4x polysomes to the total area of total ribosomal particles (“Associated subunits”) and the ratio of the area of the peak for the 30S subunit to the areas of the peaks for the 70S, 2x, 3x, and 4x (“30S/total associated”). The error bars represent the standard error of the mean from three independent experiments as shown for strains STN397, STN399, STN401, and STN409, and from two independent experiments for strains STN403 and STN405.
The rpsE suppressor mutations do not reduce accumulation of 17S rRNA-containing immature 30S subunits
The RbfA factor is required for formation of the mature 5′ and 3′ ends of 16S rRNA. A strain lacking RbfA accumulates 17S rRNA, which is generated by cleavage by RNase III in a stem–loop formed by pairing of the region upstream of the mature 5′ end (the 5′ leader) and downstream from the mature 3′ (the 3′ trailer). This cleavage leaves a 115-nt region upstream of and a 33-nt region downstream from the mature 16S rRNA; further maturation removing these regions converts the larger pre-rRNA that sediments as 17S, to a completely matured 16S rRNA (Gupta and Culver 2014). The strain lacking RbfA performs this further maturation inefficiently so the normally rare 17S form accumulates. The failure of rbfA::KmR to complete processing could slow or block the resulting 30S from participating in translation and the rpsE suppressors could promote 70S formation by accelerating processing.
To determine if the gross phenotypic effect of the rpsE suppressors reflects an increased rate of rRNA maturation, the relative amounts of 5′ ends corresponding to 17S and 16S rRNA were determined for suppressor-containing and suppressor-free rbfA::KmR mutant grown at 30°C using fluorescence-based primer extension on total RNA with FAM-labeled primers specific for 16S rRNA. The primer extension products were mixed with internal lane molecular weight markers before electrophoresis after which the fluorescence in relevant peaks was quantified. The relative amount of 17S rRNA [17S/(16S + 17S)] was 5.4-fold higher in the rbfA::KmR mutant STN409 than in the wild-type strain MW100 (Fig. 5) in agreement with previous findings (Bylund et al. 1998; Xia et al. 2003). The rpsE mutations did reduce the relative amount of 17S rRNA but the effect was rather small, 1.1- to 1.2-fold (Fig. 5). This increase was not statistically significant in any case (P > 0.05 by Student's t-test) and was insufficient to explain the much larger (average 1.5-fold) and statistically significant (P < 0.005) increase in amount of 70S ribosomes plus polysomes formed in the presence of the suppressors. A comparison of the two effects suggests that the rpsE suppressors improve the ability of mature 30S subunits to engage in translation without affecting the probability of maturation of the 5′ end of the rRNA.
FIGURE 5.
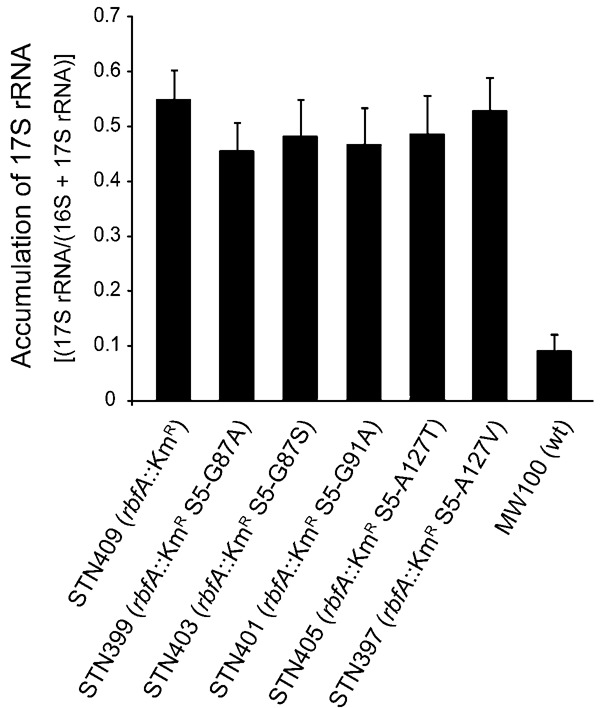
16S rRNA processing in an rbfA::KmR mutant and its derivatives expressing variants of r-protein S5 containing different amino acid substitutions. The graph shows the average accumulation of 17S rRNA to total (16S plus 17S) in total cellular RNA. The error bars represent the standard error of the mean.
Immature 17S rRNA-containing 30S subunits do not participate in translation
The data on 5′ rRNA processing shows that under the conditions of growth used approximately half of the small subunit rRNA is mature 16S and half immature 17S and that this ratio is essentially unaffected by the presence of the suppressor mutant forms of S5. There are two ways that the rpsE mutants might increase total translation given their failing to alter the efficiency of 16S rRNA maturation. The effect might be entirely independent of the maturation state of the rRNA, so the rpsE mutant S5 proteins might allow 17S-containing 30S subunits to associate with 50S subunits and to participate in translation despite being incompletely processed. Translation by ribosomes comprised of incompletely processed 30S subunits has been invoked to explain the error-prone nature of mutants carrying the spectinomycin-resistant mutation of S5, G28D (Roy-Chaudhuri et al. 2010). But there is no evidence showing that 17S-containing 30S subunits participate in translation in vivo. The fact that the proportion of polysomes in the rbfA::KmR strain is reduced much more than 50% suggests that in the absence of RbfA even subunits with mature 16S rRNA may be defective in translation. Under this hypothesis, the increase in translation in the presence of the S5 mutant proteins might result from improvement in some step(s) in biogenesis independent of rRNA 5′ end maturation.
To determine if the 17S rRNA can participate in translation, we performed fluorescence-based primer extension on samples of rRNA isolated after separation by sucrose gradient centrifugation. The results fail to support this conclusion. The primer extension experiments revealed a third form of rRNA in addition to 16S mature and 17S immature in which the nearly mature 16S has short 1–8-nt 5′ extensions (labeled “NM” in Table 2). These extensions probably tend to be too short to interfere with formation of h1 (Young and Steitz 1978; Dammel and Noller 1993) so subunits with this rRNA might be expected to be largely functional. The clearest result from this analysis is that the 17S immature rRNA was entirely excluded from the polysomes (Table 2, column 9) despite the extremely large increase in all the mutant strains. While it remains possible that these subunits can translate in monosomes, the most likely conclusion is that they are not translationally competent. The very low dipeptide formation in vitro by ribosomes containing 17S rRNA would predict a very slow elongation rate, and a slowly elongating ribosome would block the progress of upstream wild-type ribosomes, which would tend to cause a shift toward higher polysomes. If these immature subunits are indeed incompetent for translation then the 70S ribosomes carrying 17S rRNA must be inactive couples or result from abortive attempts by such a ribosome to initiate translation, leading to an mRNA bound by a single such ribosome, which blocks further initiation events. We have not been able to distinguish between these two hypotheses.
TABLE 2.
Presence of immature 16S rRNA in subunits, 70S monosomes, and polysomes
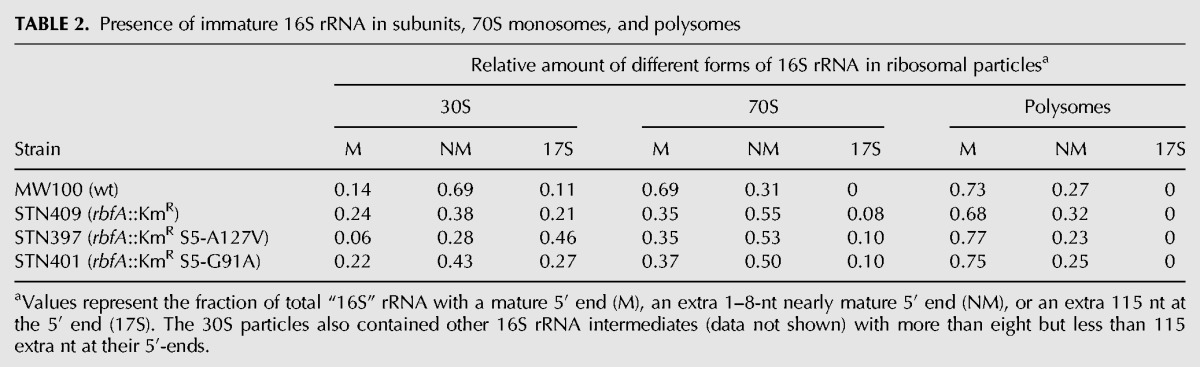
The nearly mature (nm) 16S rRNA can participate in translation, as shown by its presence in polysomes. The fact that the nm16S is the predominant small subunit rRNA in nonpolysomal 70S ribosomes but in polysomes 16S predominates suggests that a proportion of these nm16S-containing 30S subunits is in inactive couples or result from abortive initiation. This could result from two populations of nm16S-containing 30S subunits with alternative 5′ structures with those with slightly longer 5′ extensions not being able to form h1 and being inactive.
The composition of polysomal small subunit rRNAs is similar in wild-type and in the rbfA::KmR mutant strain with and without the S5 suppressor protein. This underscores the concept that the difference among these strains is unrelated to 5′ end maturation. The quantity of polysomal ribosomes is greatest where RbfA is present. In the absence of RbfA the quantity is reduced a 7.2-fold but the presence of the S5 suppressor proteins the quantity increases an average of 85%, still very low compared with wild type, but apparently sufficient to substantially improve growth.
The rpsE mutations prevent the lethality of overexpressing RimP in the rbfA::KmR mutant
The current understanding of the role of ribosome biogenesis factors RbfA and RimM is that they superintend events late in ribosome biogenesis, possibly even slowing the process down to allow critical events to occur to complete biogenesis of translationally competent 30S subunits (Clatterbuck Soper et al. 2013). The need to slow down the last steps in biogenesis may explain the vulnerability of strains lacking RbfA to overexpression of other late-30S biogenesis factors. Our finding that overexpression of RimP is lethal in the absence of RbfA suggests that excess RimP might force pre-30S subunits through a step subsequent to RbfA action, producing defective 30S subunits. The rpsE suppressor mutations on the other hand, improve growth in the absence of RbfA suggesting that they might replace RbfA in slowing the progress through the RimP-sensitive step. Alternatively, the rpsE mutants might affect an RbfA-dependent step that is unrelated to sensitivity to RimP overexpression.
We tested the effect of the rpsE suppressors on the lethal effect of overexpressing the 30S maturation factor RimP in the rbfA::KmR mutant by introducing a rimP+ plasmid from the NIG collection (Saka et al. 2005) into the rbfA::KmR mutant containing the rpsE suppressor mutations. The expression of RimP was induced by addition of 0.03 mM IPTG, a concentration that in the rbfA::KmR background allowed some growth at 30°C but none at 37°C or 44°C (cf. Figs. 6, 1). Growth was improved at 30°C by all five rpsE suppressor mutations although the extent of improvement varied with the greatest effect being caused by the rpsE2342 (S5-G91A) mutant. Four of the rpsE mutants suppressed lethality at 37°C and 44°C, the exception being rpsE2344 (S5-A127T), which was also the weakest suppressor at 30°C. At 44°C only the strongest suppressor, rpsE2342 (S5-G91A) conferred strong growth (Fig. 6).
FIGURE 6.
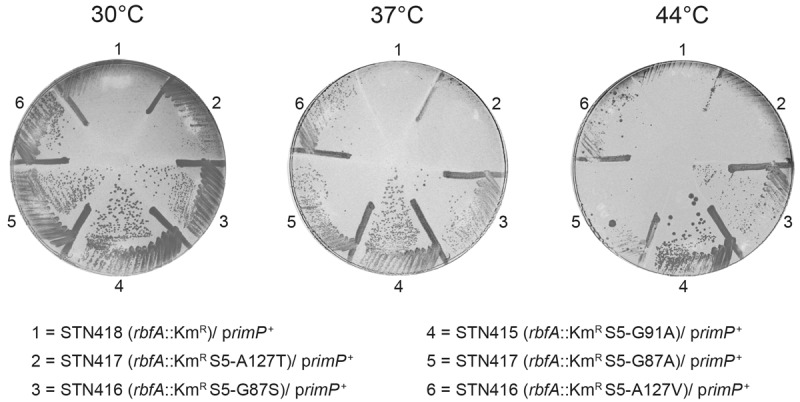
The ability of variants of r-protein S5 containing different amino acids substitutions to abolish the lethal effect of overexpressing the ribosome maturation protein RimP in an rbfA::KmR mutant. The plates were incubated at 30°C, 37°C, and 44°C, for two, one, and two day(s), respectively. The rimP+ plasmid used is described in the legends to Figure 1.
The similarity in the strength of improvement of growth of the strain lacking RbfA and of the suppression of the deleterious effect of overexpressing RimP in the absence of RbfA imply that the role of the S5 suppressor mutant proteins is to slow some event promoted by RimP. Given that the residues altered in S5 are close to the central pseudoknot and that RimP helps form that structure (Sashital et al. 2014), it is a reasonable conjecture that the S5 mutant proteins replace RbfA to an extent in modulating RimP activity in forming that structure. However, RimP also accelerates binding of the set of late binding ribosomal proteins (Bunner et al. 2010), including S5, which is required for binding of the other proteins. The S5 mutant proteins may, therefore bind especially slowly, thus retarding binding of the later binding proteins, which could preclude the formation of a kinetically trapped complex.
Effect of rpsE suppressors on translational accuracy
As we have explained above, various defects in late-30S assembly have been associated with decreased translational fidelity. It is logical, therefore, to suppose that one of the effects of the rpsE mutations is to alter translational accuracy. This is a reasonable hypothesis because mutations of rpsE were among the first to be shown to alter accuracy (Gorini 1974). The rpsE mutations were identified as suppressors of mutations affecting ribosomal protein S12 that confer hyperaccuracy as well as dependence on the error-inducing antibiotic, streptomycin (Gorini 1974). The hyperaccurate mutations encode amino acids in the ribosomal A site, some of which participate directly in cognate tRNA recognition (Ogle et al. 2001). Ribosomal ambiguity (ram) mutations that alter S4 and S5 and suppress S12 streptomycin-dependence change amino acids at an interface between the two proteins. Ogle et al. (2002) proposed that the mutations destabilize the interface and lessen the energy barrier to formation of a high-affinity amino acid-tRNA•ribosome complex, however the validity of the model was questioned by the fact that the phenotype of the mutants corresponds poorly with their effect on the stability of the S4•S5 interface (Vallabhaneni and Farabaugh 2009). It may be significant that an rRNA nucleotide near the 5′ end of the 16S rRNA intercalates within the S4–S5 interface, connecting the S4–S5 interface with the structure of the 5′ end and central pseudoknot, implicating these structures in translational accuracy as well. More recently, a ram mutation outside the S4–S5 interface was identified that changes Gly 28 of S5 to Asp (G28D), which is near the central pseudoknot (Roy-Chaudhuri et al. 2010). This mutation is also cold-sensitive and causes accumulation of the immature 17S rRNA.
We quantified translational accuracy using a system that exploits mutations of an active site codon from the gene encoding Photinus pyralis (firefly) luciferase (Fluc) (Kramer and Farabaugh 2007). Lys 529 (K529) coordinates the reactants, ATP and luciferin, in the active site of Fluc (Branchini et al. 2000) and mutations of that codon cause up to 5000-fold reductions in activity (Kramer and Farabaugh 2007). Several single nucleotide mutations of the K529 codon allow it to be misread frequently by , which normally decodes both Lys codons, thereby inserting the wild-type amino acid while decoding the mutant codon. These misreading events result in the expression of fully functional, wild-type Fluc and the frequency of the events is equal to the ratio of activity from the mutant gene to the wild type. Most near cognate codon mutations (those differing from one of the two Lys codons by a single base change) result in activities averaging ∼3 × 10−4 times wild type; several codons result in greater activities, including the stop codons UAA and UAG, the Arg codons AGA and AGG, and the Asn codon AAU (Kramer and Farabaugh 2007). The greater activity when codons are present reflects more frequent near cognate decoding of these codons by .
To measure accuracy, four error-prone codon mutant codon reporters were tested with the K529 codon changed either to UAG (stop), AGA or AGG (Arg) or AAU (Asn). Consistent with previous results (Kramer and Farabaugh 2007) each of these was misread in a wild-type background at rates above the background of 3 × 10−4 per codon (Fig. 7A, black columns). The activity of the four Fluc mutant reporters increased significantly (P < 0.005) in the rbfA::KmR mutant (Fig. 7B, white columns), which demonstrates that the lack of RbfA causes translation to be more error prone. The increase in errors averaged 2.5-fold for the four Fluc reporters and varied from 1.8- to 3.8-fold. This result is comparable with the effect of the classical ram mutation, rpsD12 (Olsson and Isaksson 1979), which increased errors on these same reporter variants with an average of 3.5-fold varying from 1.4- to 8.8-fold (Kramer and Farabaugh 2007). Thus, the rbfA::KmR induces errors similarly to a classically defined error-prone mutation. This could be as a direct effect of the role of RbfA in ribosome biogenesis or it could be a pleiotropic effect.
FIGURE 7.
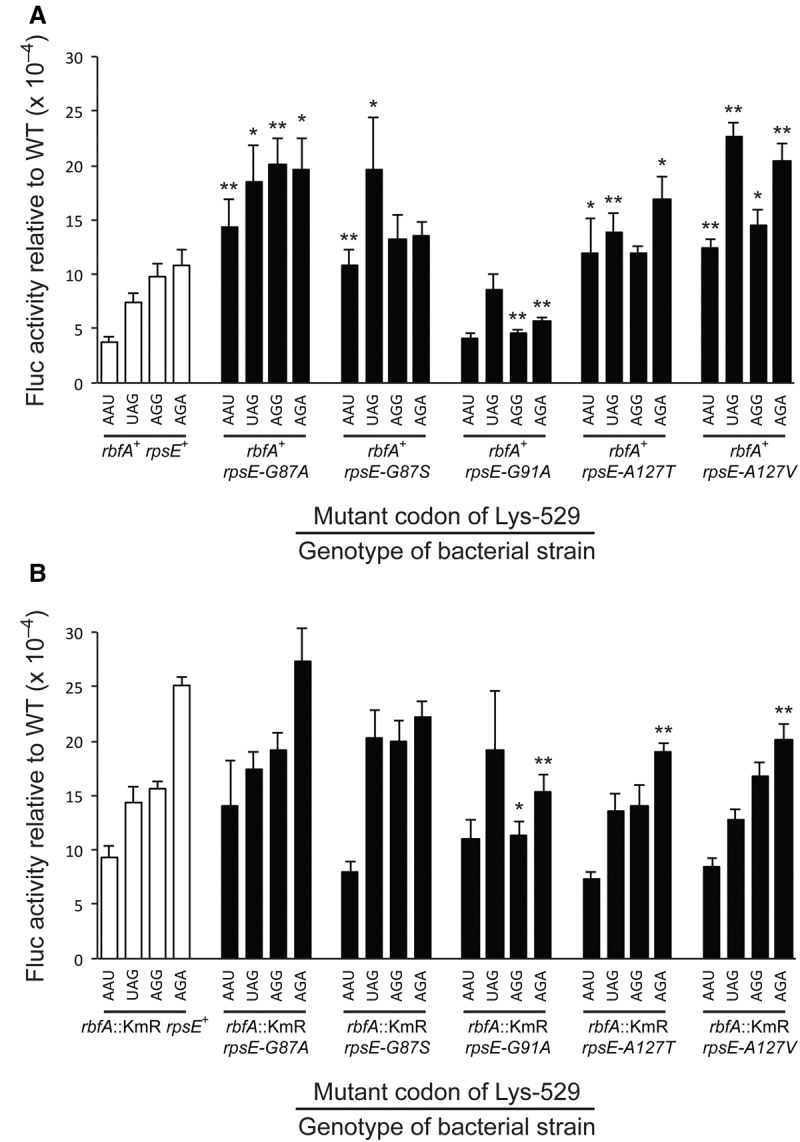
The activity of K529 mutants of firefly luciferase (Fluc) expressed in strains carrying various rpsE suppressor mutations in (A) the wild-type background or (B) in the presence of the rbfA::KmR mutation. The Fluc activities produced for each of the mutants of Fluc K529 are expressed as a ratio to that of the wild-type enzyme. Activities are shown for the four known error-prone mutants replacing the wild-type Lys codon with AAU (Asn), UAG (stop), AGG (Arg), or AGA (Arg). Error bars are standard errors of the mean. Significance is indicated: (*) P ≤ 0.05; (**) P ≤ 0.005.
All but one of the rpsE suppressor mutations similarly tended to increase translational errors. In the presence of RbfA four mutations—rpsE-G87A, rpsE-G87S, rpsE-A127T, and rpsE-A127V—each caused significantly increased enzyme activity from at least two of the misreading Fluc reporter constructs, indicating that they increased translational misreading. The average increase in misreading in these cases was 2.4-fold, with a range from 1.5- to 3.8-fold. These mutations therefore result in an error-prone phenotype that is very similar to that of an rpsD12 mutant. The rpsE mutations increased significantly errors in 13 of 16 cases tested; for the three exceptional cases although enzyme activity increased the fold increase was not judged statistically significant. The exceptional mutation was rpsE-G91A, which did not have a significant effect on the activity of the AAU and UAG mutant reporters but which caused a twofold reduction for AGA and AGG. We conclude that the rpsE-G91A mutant rather than being error prone is actually hyperaccurate.
The rpsE suppressors when tested in the rbfA::KmR background had a very different effect (Fig. 7B). We saw no significant increase compared with the rbfA::KmR rpsE+ parental strain in activity for any of the Fluc mutant reporters and actually saw significant decreases in four cases—the AGA mutant reporter in rpsE-G91A, rpsE-A127T, and rpsE-A127V and the AGG reporter in rpsE-G91A. The fact that rpsE-G91A on its own appeared to be hyperaccurate might suggest that the reduced activity of the AGA and AGG reporter mutants might result from rpsE-G91A being epistatic to rbfA::KmR, but that does not explain the reductions in the presence of the other two rpsE suppressors. If the rbfA::KmR and rpsE suppressors affected translational accuracy through distinct mechanistic pathways we would have expected to see an additive or perhaps even synergistic increase in errors by the double mutants. Not having seen that suggests that rbfA::KmR and rpsE suppressors function through a single molecular pathway to reduce translational accuracy. The reason that the rpsE-G91A mutant shows an opposite phenotype is not clear. The lack of a phenotypic interaction between them in terms of translational accuracy is inconsistent with the observed interaction in terms of cold sensitivity and growth rate, which suggests that their accuracy phenotypes cannot explain their observable growth phenotypes.
DISCUSSION
We have exploited the extremely slow growth phenotype of a double mutant lacking two ribosome biogenesis proteins, RbfA and RimP, to identify a large set of suppressor mutations that restore growth although not to wild-type rates. We had expected that some of these suppressors would affect ribosomal proteins since one of the roles of these two factors is to aid assembly of a set of late binding proteins. By screening for mutations in a ribosomal protein operon we identified a set of seven mutations comprising five unique sequence changes in ribosomal protein S5. The identity of the genes mutated in the other suppressor mutants is actively under investigation using whole-genome sequencing.
Finding suppressor mutants of S5 is significant since it is the first of the late assembly ribosomal proteins whose binding is promoted by RbfA and RimP (Bunner et al. 2010; Sashital et al. 2014). It is also significant that S5 binds to a portion of the central pseudoknot (Schuwirth et al. 2005), a structure whose assembly depends on RbfA and RimP (Sashital et al. 2014). The binding site for RimP on the 30S subunit is not known, but RbfA binds to three domains of the ribosome, the head (h28, h29, h30), body (h18 and ribosome protein S12) and 3′ minor (h44, h45) domains (Datta et al. 2007). By binding, RbfA deforms many of these structures and blocks both the A and P sites to exclude participation in translation. RbfA does not appear to be in contact with S5 or the central pseudoknot, implying that its effect on binding of S5 and stabilizing h1 of the pseudoknot is indirect. It is possible that RimP contacts both S5 and the pseudoknot but it is unclear why, then, the S5 mutations would suppress the defects associated with loss of RbfA, rather than RimP. As shown in Figure 2, S5 directly contacts both h1 and h2 of the central pseudoknot and also h28, which stacks on h2. The previously identified rpsE-G28D mutation alters an amino acid in close contact with h28, the nearest approach of S5 to the site of RbfA binding. The mutations identified here alter residues G87, G91, and A127, all of which are within a triple β-stranded domain that contacts the central pseudoknot and distant from RbfA. Gly-91, which is altered in the strongest suppressor mutant, is in the loop between β-strands 5 and 6 of S5 very close to the phosphodiester bond between nucleotides 19 and 20 of h2. The other two amino acids, G87 and A127, are in the other two β-strands of the domain. The substitution of the small Gly and Ala amino acids by bulkier amino acids could perturb packing of these strands with adjacent structures of the hydrophobic core of S5, altering the conformation of the interface between S5 and the central pseudoknot. Only one of the mutations introduces a more substantial structural change, altering Ala 127 to Val; the rather conservative nature of most of the changes suggests that less conservative changes might result in lethality or poor growth phenotypes.
Mutants lacking RbfA accumulate a small subunit rRNA, 17S, with extensions on both the 5′ and 3′ ends as do mutants lacking several other late-30S biogenesis factors, the S5-G28D mutant and the destabilizing C23U mutant of h1. The presence of 17S in mature 30S subunits has been identified as a possible cause of the decreased fidelity of translation and decreased cellular fitness of the C23U mutant and by extension the others (Roy-Chaudhuri et al. 2010). One possibility for the mechanism by which the S5 suppressors improve growth in the absence of RbfA is by improving the maturation of 17S to 16S rRNA. If that were true, the suppressor mutants would also be expected to increase the fidelity of translation. Our results show that the S5 suppressors have no significant effect on 17S maturation; in the rbfA::KmR strain either with or without the rpsE suppressor mutations ∼50% of the 30S subunits had 17S rRNA with the rest consisting of 16S mature or nearly mature 16S with short 1 to 8 nt 5′ extensions. Since the proportion of 17S did not change in the suppressed strains the mechanism of suppression cannot be through improved 5′ terminal processing. An alternative model would suggest that the suppressors are bypass mutations, improving the fidelity of ribosomes with the 17S-containing 30S subunit. We showed that this is also not the case since the 17S-containing ribosomes do not participate in translation. Thus, the model in which 17S reduces fidelity is not viable.
If the S5 suppressors do not change the proportion of immature rRNA in the 30S subunits, then what could they do to improve growth and translational fidelity? The remaining model would suggest that the S5 suppressors improve some other step in the maturation of pre-30S subunits. We have observed that the suppressors increase the proportion of these subunits that participate in translation; the pool of ribosomes in polysomes improves in all suppressors and the effect is greatest in the strongest suppressors. The fact that apparently mature 30S subunits fail to participate as fully in translation as they do in a wild-type strain suggests that in the strain lacking RbfA a large proportion of apparently mature 30S subunits are in some way defective in translation. This defect could reflect the absence of one or more essential ribosomal proteins or the formation of inappropriate rRNA secondary or tertiary structures in the absence of RbfA. This model is consistent with the role for RbfA in avoiding kinetically trapped complexes. In its absence during late-30S assembly some rRNA folding may occur prematurely, excluding one or more of the last ribosomal proteins to bind. Sashital et al. (2014) showed that in a ΔrimP strain, the 30S peak of a sucrose gradient largely consisted of subunits lacking one or more of the last four proteins, S2, S3, S12, and S21, but that using a pulse-chase approach they showed that these complexes are only delayed in assembly and not dead-end complexes. Experiments to test for deficits in late ribosomal proteins in the rbfA::KmR strain with and without the suppressors are planned and should resolve this issue.
The third alternative is that the absence of RbfA results in misfolding of the rRNA that is not reflected in lack of binding of ribosomal proteins. It is possible that, in the absence of RimP or RbfA chaperones, the rRNA may adopt a noncanonical structure that produces functional complexes with reduced fidelity. The S5 suppressors, through their interaction with the central pseudoknot, could limit the formation of those structures. The fact that the mutant S5 proteins reverse the lethal effect of overexpressing RimP in the absence of RbfA is consistent with this concept. RimP is known to stabilize the central pseudoknot (Sashital et al. 2014) and accelerate binding of S5 and S12 (Bunner et al. 2010). The lethality of overexpressing RimP may result from these processes proceeding too quickly, creating kinetically trapped complexes. The S5 suppressors presumably block the downstream effects that result subsequent to its binding, or slow the binding itself in mimicry to the presumed effect of RbfA.
We also show here that in addition to their effect on assembly the S5 suppressors alter translational accuracy. The evidence shows that both the rbfA::KmR and four of the five rpsE suppressor mutants are error prone, with an effect of a similar magnitude as the canonical ram mutations (Gorini 1974; Kramer and Farabaugh 2007). Interestingly, the rbfA::KmR rpsE double mutants do not show greater error frequencies, which suggests that both lack of RbfA and the mutant forms of S5 contribute to increasing error in the same mechanistic pathway. This similar error-prone effect is despite the fact that the rbfA::KmR and rpsE mutants have opposite gross phenotypes and that the suppressor with the strongest effect on growth of the rbfA::KmR strain actually exhibits the opposite phenotype of hyperaccuracy. Thus, increased inaccuracy is not a necessary and direct cause of improving the growth of the strain lacking RbfA.
The observed increase in translational errors in a G28D 16S rRNA mutant has been proposed to result from 30S subunits carrying the 17S immature rRNA (Roy-Chaudhuri et al. 2010); the argument was that the extra rRNA perturbs the function of the ribosomal A site within which it resides so as to increase the frequency of acceptance of incorrect aminoacyl-tRNAs. Since the 17S-containing ribosomes appear to be inactive in translation, incomplete 5′ processing must not be the cause of the error phenotype. The two effects are largely associated among mutants of several ribosome biogenesis proteins, the mutant expressing the S5-G28D and the mutant expressing the unstable C23U for of 16S rRNA. The diversity of primary defects in these biogenesis mutants—lacking GTPases, chaperones, and a protein methylase—suggest that the accuracy effect, like the 5′ maturation, must be distant from the site of action of the primary defect. This suggests that both 17S rRNA accumulation and inaccuracy result from incomplete or inaccurate assembly of the mature 30S subunit. Direct biochemical characterization of mature 30S subunits produced in these mutant strains could identify the mechanism underlying this effect.
MATERIALS AND METHODS
Strains, phages, plasmids, oligonucleotides
Relevant strains, phages, plasmids, and oligonucleotides used are listed in Table 3. Strain STN133 (Hfr P4X rbfA::KmR ΔrimP135) was constructed by transfer of rbfA::KmR from GOB162 (Hfr P4X rbfA::KmR) into strain MW187 (Hfr P4X ΔrimP135) (Nord et al. 2009) by P1 transduction selecting for KmR and screening for slow growth at 37°C. The slow-growing clones were confirmed by PCR analysis to contain rimP135ΔrimP135.
TABLE 3.
Bacterial strains, bacteriophages, plasmids, and oligonucleotides

Strains having zhc2421::Tn10 linked to the rpsE mutations were constructed by P1 transduction using PW078 (Hfr P4X zhc2421::Tn10) (G Bylund, O Persson, M Lövgren, M Wikström, unpubl.) as donor and the rpsE mutants STN174, STN176, STN181, STN182, and STN187 as recipients, selecting for TcR and screening for fast growth. The obtained clones were used as donors in P1 transductions when zhc2421::Tn10 was transferred together with the rpsE mutations to STN133 (Hfr P4X rbfA::KmR ΔrimP135) selecting for TcR and screening for faster growth than that of STN133 yielding strains STN360, STN363, STN366, STN369, and STN372. The rpsE mutations in these five strains were transferred to strains MW100 (Hfr P4X), MW187 (Hfr P4X ΔrimP135), and GOB162 (Hfr P4X rbfA::KmR) by P1 transduction selecting for TcR. The obtained strains STN377, STN379, STN381, STN383, STN385, STN387, STN389, STN391, STN393, STN395, STN397, STN401, STN403, and STN405 (Table 3) were verified by DNA sequencing to contain the rpsE mutations.
The rbfA::KmR ΔrimP135 mutations of the strains STN174, STN181, STN182, STN186, and STN187 containing rpsE mutations were replaced by the corresponding rbfA+ and rimP+ genes linked to argG2424::miniTn10Cm by P1 transduction using GOB375 (Hfr P4X argG2424::miniTn10Cm) (Bylund et al. 2001) as donor, selecting for CmR and screening for KmS. The resulting strains STN419-423 and strain MW100 were made recA by P1 transduction using strain JC10289 (thr-1 leuB6 Δ(srlR-recA)306::Tn10) (Csonka and Clark 1979; Willis et al. 1981) selecting for TcR, yielding strains STN424-428 and STN410, respectively.
For the tests of translational missense error frequencies, the rbfA::KmR and rpsE suppressor mutations were transferred by P1 transduction to the Xac genetic background (ara Δ[lac-proAB] gyrA(nalR) rpoB(rifR) argE[amber]) (Andersson et al. 1982; Dahlgren and Ryden-Aulin 2000).
Growth conditions
Rich medium used was LB (Bertani 1951). Cultures were grown at indicated temperatures and growth was monitored at 600 or 650 nm using a Beckman Coulter DU 730 spectrophotometer.
Polysome profiles after sucrose gradient centrifugation
Cell cultures were grown in LB at 30°C to A650 = 0.5. Preparation of polysome extracts and fractionation by sucrose gradient centrifugation were as described previously (Nord et al. 2009).
Primer extension on rRNA
Primer extension on 16S rRNA using a FAM-labeled primer and analysis of the obtained products were as described previously (Nord et al. 2009). Briefly, the amount of immature relative to mature 5′-ends of 16S rRNA was determined by running fluorescent-based primer extension on total RNA preparations with the primer FAM16S-R4 binding downstream from the 5′-end of mature 16S rRNA and analyzing the fluorescing extension products using a DNA sequencer. The fluorescence in the primer extension products corresponding to 16S and 17S rRNA was quantified using the GeneScan Analysis Software version 3.1 (Applied Biosystems).
Translational misreading error frequency measurement
Assays of the expression of dual-luciferase reporters cultures were performed as described (Kramer and Farabaugh 2007). Cultures of individual clones were grown to OD600 of from 0.5 to 0.8 in liquid LB medium with 100 µg/mL ampicillin at 37°C and then pelleted by centrifugation and resuspended in 200 µL 1 mg/mL lysozyme/10 mM Tris–HCl, pH 8.0/1 mM EDTA. Cells were incubated on ice for 10 min, frozen on dry ice and thawed on ice. Five microliter samples of this extract were assayed for Photinus pyralis (firefly) luciferase (Fluc) and Renilla reniformis (sea pansy) luciferase (Rluc) activities using the Dual-Luciferase Reporter Assay System (Promega). Luminescence was measured using the luminometer function of a Modulus II Microplate Multimode Reader (Promega). Three transformants for each construct were assayed, each in triplicate. The Rluc activity was used as an internal standard and standardized Fluc activity was calculated as the ratio of the Fluc to Rluc activity expressed in relative light units (RLU). The significance of differences in activity were determined by comparing values obtained in each rpsE suppressor mutant strain and the congenic rpsE+ strain using a two-tailed, homoscedastic Student's t-test.
ACKNOWLEDGMENTS
We thank Dr. Masayori Inouye for providing plasmids pU23 and pSTL102. The E. coli mobile plasmid ORF library was a generous gift from the National Institute of Genetics, Japan (http://www.shigen.nig.ac.jp/ecoli/strain/top/top.jsp). We thank Drs. Glenn Björk and Marcus Johansson for critical reading of the manuscript. P.M.W. was supported by the Carl Trygger Foundation and the Magnus Bergvall Foundation. This work was supported in part by a grant from the US National Institutes of Health (National Institute of General Medical Sciences) (R01 GM029480) to P.J.F.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.051383.115.
REFERENCES
- Alix JH, Nierhaus KH. 2003. DnaK-facilitated ribosome assembly in Escherichia coli revisited. RNA 9: 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DI, Bohman K, Isaksson LA, Kurland CG. 1982. Translation rates and misreading characteristics of rpsD mutants in Escherichia coli. Mol Gen Genet 187: 467–472. [DOI] [PubMed] [Google Scholar]
- Berlyn MK. 1998. Linkage map of Escherichia coli K-12, edition 10: the traditional map. Microbiol Mol Biol Rev 62: 814–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62: 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchini BR, Murtiashaw MH, Magyar RA, Anderson SM. 2000. The role of lysine 529, a conserved residue of the acyl-adenylate-forming enzyme superfamily, in firefly luciferase. Biochemistry 39: 5433–5440. [DOI] [PubMed] [Google Scholar]
- Bremer H, Patrick P. 1996. Modulation of chemical composition and other parameters of the cell by growth rate. In Escherichia coli and Salmonella typhimurium: cellular and molecular biology, 2nd ed (ed. Curtiss R III, et al. ), pp. 1553–1569. American Society for Microbiology, Washington, DC. [Google Scholar]
- Bunner AE, Nord S, Wikström PM, Williamson JR. 2010. The effect of ribosome assembly cofactors on in vitro 30S subunit reconstitution. J Mol Biol 398: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylund GO, Wipemo LC, Lundberg LAC, Wikström PM. 1998. RimM and RbfA are essential for efficient processing of 16S rRNA in Escherichia coli. J Bacteriol 180: 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylund GO, Lövgren JM, Wikström PM. 2001. Characterization of mutations in the metY-nusA-infB operon that suppress the slow growth of a ΔrimM mutant. J Bacteriol 183: 6095–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell TL, Brown ED. 2008. Genetic interaction screens with ordered overexpression and deletion clone sets implicate the Escherichia coli GTPase YjeQ in late ribosome biogenesis. J Bacteriol 190: 2537–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champney WS. 1977. Kinetics of ribosome synthesis during a nutritional shift-up in Escherichia coli K-12. Mol Gen Genet 152: 259–266. [DOI] [PubMed] [Google Scholar]
- Clatterbuck Soper SF, Dator RP, Limbach PA, Woodson SA. 2013. In vivo X-ray footprinting of pre-30S ribosomes reveals chaperone-dependent remodeling of late assembly intermediates. Mol Cell 52: 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly K, Culver G. 2009. Deconstructing ribosome construction. Trends Biochem Sci 34: 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly K, Culver G. 2013. Overexpression of RbfA in the absence of the KsgA checkpoint results in impaired translation initiation. Mol Microbiol 87: 968–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly K, Rife JP, Culver G. 2008. Mechanistic insight into the ribosome biogenesis functions of the ancient protein KsgA. Mol Microbiol 70: 1062–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csonka LN, Clark AJ. 1979. Deletions generated by the transposon Tn10 in the srl recA region of the Escherichia coli K-12 chromosome. Genetics 93: 321–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren A, Ryden-Aulin M. 2000. A novel mutation in ribosomal protein S4 that affects the function of a mutated RF1. Biochimie 82: 683–691. [DOI] [PubMed] [Google Scholar]
- Dammel CS, Noller HF. 1993. A cold-sensitive mutation in 16S rRNA provides evidence for helical switching in ribosome assembly. Genes Dev 7: 660–670. [DOI] [PubMed] [Google Scholar]
- Dammel CS, Noller HF. 1995. Suppression of a cold-sensitive mutation in 16S rRNA by overexpression of a novel ribosome-binding factor, RbfA. Genes Dev 9: 626–637. [DOI] [PubMed] [Google Scholar]
- Datta PP, Wilson DN, Kawazoe M, Swami NK, Kaminishi T, Sharma MR, Booth TM, Takemoto C, Fucini P, Yokoyama S, et al. 2007. Structural aspects of RbfA action during small ribosomal subunit assembly. Mol Cell 28: 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yacoubi B, Lyons B, Cruz Y, Reddy R, Nordin B, Agnelli F, Williamson JR, Schimmel P, Swairjo MA, de Crecy-Lagard V. 2009. The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res 37: 2894–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorini L. 1974. Streptomycin and misreading of the genetic code. In Ribosomes (ed. Nomura M, et al. ), pp. 791–803. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- Guo Q, Yuan Y, Xu Y, Feng B, Liu L, Chen K, Sun M, Yang Z, Lei J, Gao N. 2011. Structural basis for the function of a small GTPase RsgA on the 30S ribosomal subunit maturation revealed by cryoelectron microscopy. Proc Natl Acad Sci 108: 13100–13105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Culver GM. 2014. Multiple in vivo pathways for Escherichia coli small ribosomal subunit assembly occur on one pre-rRNA. Nat Struct Mol Biol 21: 937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeno H, Hanawa-Suetsugu K, Kimura T, Takagi K, Sugiyama W, Shirata S, Mikami T, Odagiri F, Osanai Y, Watanabe D, et al. 2004. A novel GTPase activated by the small subunit of ribosome. Nucleic Acids Res 32: 5303–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14: 33–38. [DOI] [PubMed] [Google Scholar]
- Inoue K, Alsina J, Chen J, Inouye M. 2003. Suppression of defective ribosome assembly in a rbfA deletion mutant by overexpression of Era, an essential GTPase in Escherichia coli. Mol Microbiol 48: 1005–1016. [DOI] [PubMed] [Google Scholar]
- Jones PG, Inouye M. 1996. RbfA, a 30S ribosomal binding factor, is a cold-shock protein whose absence triggers the cold-shock response. Mol Microbiol 21: 1207–1218. [DOI] [PubMed] [Google Scholar]
- Kaczanowska M, Ryden-Aulin M. 2005. The YrdC protein—a putative ribosome maturation factor. Biochim Biophys Acta 1727: 87–96. [DOI] [PubMed] [Google Scholar]
- Kramer E, Farabaugh P. 2007. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 13: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lövgren JM, Bylund GO, Srivastava MK, Lundberg LA, Persson OP, Wingsle G, Wikström PM. 2004. The PRC-barrel domain of the ribosome maturation protein RimM mediates binding to ribosomal protein S19 in the 30S ribosomal subunits. RNA 10: 1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki JA, Schnobrich DJ, Culver GM. 2002. The DnaK chaperone system facilitates 30S ribosomal subunit assembly. Mol Cell 10: 129–138. [DOI] [PubMed] [Google Scholar]
- Maki JA, Southworth DR, Culver GM. 2003. Demonstration of the role of the DnaK chaperone system in assembly of 30S ribosomal subunits using a purified in vitro system. RNA 9: 1418–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashimoto H. 1993. Non-ribosomal proteins affecting the assembly of ribosomes in Escherichia coli. In The translational apparatus (ed. Nierhaus KH, et al. ), pp. 185–195. Plenum Press, New York. [Google Scholar]
- Nashimoto H, Miura A, Saito H, Uchida H. 1985. Suppressors of temperature-sensitive mutations in a ribosomal protein gene, rpsL (S12), of Escherichia coli K12. Mol Gen Genet 199: 381–387. [DOI] [PubMed] [Google Scholar]
- Nierhaus KH. 1991. The assembly of prokaryotic ribosomes. Biochimie 73: 739–755. [DOI] [PubMed] [Google Scholar]
- Nord S, Bylund GO, Lövgren JM, Wikström PM. 2009. The RimP protein is important for maturation of the 30S ribosomal subunit. J Mol Biol 386: 742–753. [DOI] [PubMed] [Google Scholar]
- Ogle J, Brodersen D, Clemons WM Jr, Tarry M, Carter A, Ramakrishnan V. 2001. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292: 897–902. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. 2002. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111: 721–732. [DOI] [PubMed] [Google Scholar]
- Olsson MO, Isaksson LA. 1979. Analysis of rpsD mutations in Escherichia coli. I. Comparison of mutants with various alterations in ribosomal protein S4. Mol Gen Genet 169: 251–257. [DOI] [PubMed] [Google Scholar]
- Recht MI, Williamson JR. 2004. RNA tertiary structure and cooperative assembly of a large ribonucleoprotein complex. J Mol Biol 344: 395–407. [DOI] [PubMed] [Google Scholar]
- Roy-Chaudhuri B, Kirthi N, Culver GM. 2010. Appropriate maturation and folding of 16S rRNA during 30S subunit biogenesis are critical for translational fidelity. Proc Natl Acad Sci 107: 4567–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka K, Tadenuma M, Nakade S, Tanaka N, Sugawara H, Nishikawa K, Ichiyoshi N, Kitagawa M, Mori H, Ogasawara N, et al. 2005. A complete set of Escherichia coli open reading frames in mobile plasmids facilitating genetic studies. DNA Res 12: 63–68. [DOI] [PubMed] [Google Scholar]
- Sashital DG, Greeman CA, Lyumkis D, Potter CS, Carragher B, Williamson JR. 2014. A combined quantitative mass spectrometry and electron microscopy analysis of ribosomal 30S subunit assembly in E. coli. Elife 3: e04491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed A, Matsuyama S, Inouye M. 1999. Era, an essential Escherichia coli small G-protein, binds to the 30S ribosomal subunit. Biochem Biophys Res Commun 264: 51–54. [DOI] [PubMed] [Google Scholar]
- Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. 2005. Structures of the bacterial ribosome at 3.5 Å resolution. Science 310: 827–834. [DOI] [PubMed] [Google Scholar]
- Selmer M, Dunham CM, Murphy FVt, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. 2006. Structure of the 70S ribosome complexed with mRNA and tRNA. Science 313: 1935–1942. [DOI] [PubMed] [Google Scholar]
- Shajani Z, Sykes MT, Williamson JR. 2011. Assembly of bacterial ribosomes. Annu Rev Biochem 80: 501–526. [DOI] [PubMed] [Google Scholar]
- Stern S, Powers T, Changchien LM, Noller HF. 1989. RNA-protein interactions in 30S ribosomal subunits: folding and function of 16S rRNA. Science 244: 783–790. [DOI] [PubMed] [Google Scholar]
- Sykes MT, Williamson JR. 2009. A complex assembly landscape for the 30S ribosomal subunit. Annu Rev Biophys 38: 197–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triman K, Becker E, Dammel C, Katz J, Mori H, Douthwaite S, Yapijakis C, Yoast S, Noller HF. 1989. Isolation of temperature-sensitive mutants of 16 S rRNA in Escherichia coli. J Mol Biol 209: 645–653. [DOI] [PubMed] [Google Scholar]
- Tschochner H, Hurt E. 2003. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol 13: 255–263. [DOI] [PubMed] [Google Scholar]
- Vallabhaneni H, Farabaugh PJ. 2009. Accuracy modulating mutations of the ribosomal protein S4-S5 interface do not necessarily destabilize the rps4-rps5 protein–protein interaction. RNA 15: 1100–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR. 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24: 437–440. [DOI] [PubMed] [Google Scholar]
- Wikström PM, Byström AS, Björk GR. 1988. Non-autogenous control of ribosomal protein synthesis from the trmD operon in Escherichia coli. J Mol Biol 203: 141–152. [DOI] [PubMed] [Google Scholar]
- Williamson JR. 2003. After the ribosome structures: how are the subunits assembled? RNA 9: 165–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis DK, Uhlin BE, Amini KS, Clark AJ. 1981. Physical mapping of the srl recA region of Escherichia coli: analysis of Tn10 generated insertions and deletions. Mol Gen Genet 183: 497–504. [DOI] [PubMed] [Google Scholar]
- Woolford JL Jr, Baserga SJ. 2013. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195: 643–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B, Ke H, Shinde U, Inouye M. 2003. The role of RbfA in 16S rRNA processing and cell growth at low temperature in Escherichia coli. J Mol Biol 332: 575–584. [DOI] [PubMed] [Google Scholar]
- Yang Z, Guo Q, Goto S, Chen Y, Li N, Yan K, Zhang Y, Muto A, Deng H, Himeno H, et al. 2014. Structural insights into the assembly of the 30S ribosomal subunit in vivo: functional role of S5 and location of the 17S rRNA precursor sequence. Protein Cell 5: 394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RA, Steitz JA. 1978. Complementary sequences 1700 nucleotides apart form a ribonuclease III cleavage site in Escherichia coli ribosomal precursor RNA. Proc Natl Acad Sci 75: 3593–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. 2001. Crystal structure of the ribosome at 5.5 Å resolution. Science 292: 883–896. [DOI] [PubMed] [Google Scholar]