

Abstract
Hydrogen exchange (HX) mass spectrometry (MS) is valuable for providing conformational information for proteins/peptides that are very difficult to analyze with other methods such as peripheral membrane proteins and peptides that interact with membranes. We developed a new type of HX MS measurement that integrates Langmuir monolayers. A lipid monolayer was generated, a peptide or protein associated with it, and then the monolayer-associated peptide or protein was exposed to deuterium. The deuterated species was recovered from the monolayer, digested, and deuterium incorporation monitored by MS. Test peptides showed that deuterium recovery in an optimized protocol was equivalent to deuterium recovery in conventional solution HX MS. The reproducibility of the measurements was high despite the requirement of generating a new monolayer for each deuterium labeling time. We validated that known conformational changes in the presence of a monolayer/membrane could be observed with the peptide melittin and the myristoylated protein Arf-1. Results in an accompanying paper show that the method can reveal details of conformational changes in a protein (HIV-1 Nef) which adopts a different conformation depending on if it can insert into the lipid layer. Overall, the HX MS Langmuir monolayer method provided new and meaningful conformational information for proteins that associate with lipid layers. The combination of HX MS results with neutron or X-ray reflection of the same proteins in Langmuir monolayers can be more informative than isolated use of either method.
Keywords: Arf-1, deuterium, melittin, dynamics, membrane
Membrane proteins are involved in many cellular processes ranging from regulation, recognition, metabolism, transport, and signaling1. Recent accounts indicate that ∼58% of utilized drug targets were membrane proteins2,3. Despite much effort focused on both transmembrane and peripheral membrane proteins, it has been difficult to obtain high resolution structural information for many membrane proteins. Membrane protein structures account for ∼2.5% of all coordinate files deposited in the Protein Data Bank4 (www.rcsb.org5). The major obstacle to structural characterization of membrane proteins is often the membrane itself, which is generally not compatible with structural studies and many biophysical measurements. Solubilizing membrane proteins with detergents to make them compatible with aqueous buffers and methodologies is one alternative but this is not always successful, and even when successful, questions can linger as to how detergents may alter protein structure6. Analytical methods that make use of membrane mimetics (i.e., artificial membranes) are attractive alternatives because the structure of the membrane protein is more likely to be preserved in the mimetic.
We have previously applied hydrogen exchange (HX) mass spectrometry (MS) to membrane proteins7-14, primarily using liposomes or nanodiscs as the membrane mimetic. Other groups have also used HX MS for membrane proteins, utilizing detergents [e.g. 15-17], and liposomes [e.g. 18-20]. Each membrane mimetic has advantages and disadvantages. Detergents may force the protein into a non-native conformation6 and can suppress peptide ionization if not properly removed prior to electrospray7,21. Creation of both liposomes and nanodiscs can be challenging. For liposomes, there can be issues with reproducibility, lipid membrane curvature effects (especially in vesicles, see Refs. 22,23 for review), and protein directionality while nanodiscs have a background, undesirable protein component (the membrane stabilizing protein) and lipid packing density is not easily modified.
Lipid packing density, the number of lipid molecules per unit area, is fluid in cellular membranes with some regions packed more tightly than other regions, often dependent on the lipid composition, degree of hydrocarbon chain saturation, percentage of cholesterol, and other factors24-27. Some proteins may alter the packing density of surrounding lipids, perhaps as a result of conformational changes during function, membrane insertion, or other reasons. To study membrane protein/peptide conformational changes, therefore, control over lipid packing density can sometimes be critical28, including the ability to reproducibly create, measure and change lipid packing density. Nanodiscs have a fixed density and a fixed number of lipid molecules; packing density can be modulated during nanodisc creation but cannot be altered once the nanodisc is formed. As a result, protein/peptide interactions with nanodiscs that require insertion or conformational changes may be restricted. Liposomes can change size and therefore offer the least control over packing density. We have explored29,30 Langmuir monolayers in neutron reflection studies as an alternative because lipid packing density can be monitored, changed and reproduced in real time.
Langmuir monolayers are formed from amphiphilic molecules spread at an air-liquid interface31. Monolayers comprised of lipids are analogous to one leaflet of a lipid bilayer membrane; that is, two monolayers make a membrane. When these monolayers are generated and maintained within a Langmuir trough, the user is able to finely control and change the lipid packing density throughout the experiment with a high degree of reproducibility. The advantage of fine control over the monolayer was recently utilized with neutron reflectometry (NR) to investigate the conformational changes in HIV-1 Nef upon association with monolayers of different lipid packing density29. NR is capable of resolving and modeling an overall, monolayer-associated shape profile, but is silent to the finer details of dynamics and local conformation. The opportunity to combine local information provided by HX with global structural information by neutron or X-ray reflectivity is a great advantage. However, a disadvantage of Langmuir monolayers is that they are only half of a biological membrane and therefore applicable only for peripheral membrane proteins or peptides that interact with one leaflet of the membrane bilayer.
In the current work, we report on development of a new method for studying peripheral membrane proteins and peptides that interact with lipid layers using HX MS and Langmuir monolayers. We show that this method is capable of investigating protein/peptide conformations over a range of lipid packing density and compositions, and that it can be applied to proteins/peptides that anchor or interact with one face of a lipid bilayer. The method was validated by comparing solution HX and monolayer HX of membrane-binding peptides and proteins known to exhibit structural changes when associated with lipid layers. In an accompanying paper32, we used the HX MS Langmuir monolayer method to monitor the conformational changes of the HIV-1 Nef protein upon lipid interaction. Previous NR experiments29 identified a conformational change in HIV-1 Nef that is dependent on lipid packing density and the ability of the Nef protein to insert into the lipid layer. The Langmuir HX MS method proved to be particularly valuable for characterizing this lipid insertion event. Overall, we expect Langmuir monolayer HX MS methodology will be applicable to a large number of other interesting proteins and systems.
Experimental Procedures
Chemicals and Materials
Melittin (≥85%, part# M2272), pepsin (porcine gastric mucosa, part# P6887), aspergillopepsin (Protease from Aspergillus saitoi, part# P2143), angiotensin I (≥90%, part# A9650), bradykinin (≥98%, part# B3259) and leucine enkephalin (≥95%, part# L9133) were purchased from Sigma-Aldrich (Saint Louis, MO). 1,2-dipalmitoyl-sn-glycero-3-phosphoserine (DPPS) 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (DPPG) was purchased from Avani Polar Lipids (Alabaster, AL). Deuterium oxide (99.8%) and sodium deuteroxide (40%) were purchased from Cambridge Isotope Laboratories (Andover, MA). Other common lab chemicals were purchased from Research Products International (Mount Prospect, IL).
Myristoylated Arf-1 Expression and Purification
Myristoylated human Arf-1 (myrArf-1)33 expression and purification was similar to that described previously for myristoylated HIV-1 Nef9. The Arf-1 gene was purchased from Addgene (plasmid number 28168) and sub-cloned into the pET-Duet vector. The pET-Deut vector contained human N-myristoyltransferase 1 (hNMT1) in the first multiple cloning site and myrArf-1 with a C-terminal polyhistidine tag in the second multiple cloning site. Protein was isolated using Ni-NTA agarose (QIAGEN, Valencia, CA), washed and eluted as described for HIV-1 Nef9. Purity and proper myristoylation were confirmed by polyacrylamide gel electrophoresis (SDS-PAGE) and electrospray mass spectrometry.
Solution Hydrogen Exchange
Solution hydrogen exchange experiments were carried out at room temperature (22 °C) by diluting peptide or protein stock solutions in equilibration buffer (50 mM citric acid, 50 mM sodium phosphate, 150 mM NaCl, pH 6.0, H2O) 15-fold with labeling buffer (50 mM citric acid, 50 mM sodium phosphate, 150 mM NaCl, pD 6.0, 99.8% D2O). Melittin buffers (both H2O and D2O) also contained 5 mM EDTA. Both Arf-1 buffers were adjusted to pH 7.0 and 2 mM magnesium chloride was added. Following dilution into D2O, samples were continuously labeled for predetermined times ranging from 10 seconds to 4 hours before being quenched to pH 2.6 using a 0 °C solution of quench buffer (0.8% formic acid and 0.8M guanidine hydrochloride in H2O). Quenched samples were digested on ice for 5 minutes by adding pepsin and aspergillopepsin (60 μg and 70 μg, respectively, dissolved in water). We found this digestion strategy and enzyme combination maximized the digestion of protein in the presence of lipid (further described below) so it was also used for solution digestion. Digested samples were injected into a Waters nanoAcquity UPLC with HX technology34 (Milford, MA) for desalting, separation, and mass analysis (details below).
Monolayer Hydrogen Exchange Experiments
Monolayer hydrogen exchange experiments utilized a modified Langmuir trough system (FIGURE 1) and the previously described protocol for generating the monolayer29,30. The total volume of the trough (blue in FIGURE 1) was 18 mL. All experiments were performed at room temperature (22 °C). For melittin experiments, 1,2-dipalmitoyl-sn-glycero-3-phosphoserine (DPPS) was used for the monolayer; for all other experiments, 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (DPPG) was used. Lipid was spread from a mixture of chloroform/methanol (70/30 by volume) onto the surface of aqueous subphase (equilibration buffer for each type of peptide/protein, as described for solution experiments) that had been placed in the trough. Once the chloroform and methanol had evaporated, the deposited lipids were compressed to a set pressure using a motorized, computer-controlled movable barrier. By controlling the position of the barrier via computer, the monolayer pressure could be fixed at any value. After the pressure had stabilized, peptide/protein was injected into the subphase beneath the lipid monolayer (to a final concentration of ∼1 μM) using gel-loading pipette tips. If peptide/protein interacted with the lipids in the monolayer and inserted hydrophobic residues into the lipids tails, thereby crowding the monolayer and increasing the molecular packing density, the barrier position automatically adjusted to maintain constant pressure (see Supporting Information FIGURE S1). The resulting barrier movement increased the surface area of the monolayer. Interaction of the protein with the Langmuir monolayer was allowed to proceed until the relative barrier position was less than 5 mm from the back edge of the trough. Then, 100 mL of labeling buffer were rapidly circulated through the trough using a peristaltic pump (FIGURE 1). The subphase exchange process took 10 seconds, after which the timing for continuous labeling experiments began. Adsorbed samples were deuterated for predetermined times ranging from 10 seconds to 4 hours. After the labeling time, approximately 300 μL of monolayer, subphase buffer and the monolayer-associated protein were quickly vacuum aspirated into a sample tube at 0 °C (FIGURE 1). Quench buffer (as used in solution labeling above) was quickly added (in less than 5 seconds) to drop the pH of the aspirated sample to 2.5-2.6. The quenched sample was digested for 5 minutes at 0 °C (all parameters identical to solution HX experiments) before injection into the UPLC-MS system.
Figure 1.

Diagram of the Langmuir trough used for labeling experiments from the top (A) and the side (B). This design was adapted from the trough used for neutron reflection studies29 where the neutron beam footprint was a square of 25 mm on each side. The areas in blue represent the buffered aqueous subphase (18 mL total volume) upon which the monolayer (orange in panel B) floats. After lipid is spread on the subphase, a motor-driven barrier compresses the monolayer to a set lipid packing density as monitored with the pressure sensor. The computer control system (not shown) integrates the pressure sensor and barrier position and can be set to move the barrier to maintain constant pressure, if desired. Protein samples are injected underneath the monolayer after it is in place. Red arrows indicate the flow of buffer through the trough, driven by the peristaltic pump, during subphase exchange and deuterium labeling. A vacuum pump is used to aspirate labeled samples directly from the monolayer into a cold collection tube.
Mass Analyses and Data Processing
Peptide separation was performed at 0 °C using a Waters nanoAcquity UPLC with HX technology34 (Milford, MA). Peptides were trapped on a Waters UPLC BEH C18 1.7 μm VanGuard BEH column and desalted with 0.1% formic acid in water for 3 minutes at 100 μL/min. To reduce the amount of lipids entering the mass spectrometer, an additional Vanguard BEH column was placed directly in line with the analytical column (Waters HSS T3 1.8 μm C18, 1.0 mm × 50 mm). This first trap captures the majority of lipids present in the sample and is replaced frequently or washed with chloroform overnight21 to remove lipid. Peptides were eluted over 6 minutes using a 5-35% gradient of water:acetonitrile with 0.1% formic acid flowing at a rate of 60 μL/min. Lipid that did elute only appeared at high acetonitrile concentrations when, more most samples, the LC system was disconnected from the mass spectrometer. Mass measurements were performed with a Waters Synapt G2 HDMS equipped with a standard ESI source and lock-mass correction using (+2) Glu-fibrinogen peptide. All mass spectra were acquired in MSE mode35 and spanned a range of 50-2000 m/z. The peptic peptides from myrArf-1 were identified (FIGURE S2) with a combination of exact mass measurements and MS/MS. Data processing to extract deuteration levels for each peptide was performed with DynamX software (Waters). All deuterium levels are reported as relative36 and there were no corrections for back-exchange. Relative deuterium incorporation curves for identified peptides of myrArf-1 are provided in FIGURE S3.
Results and Discussion
Langmuir Trough Design and Circulation Testing
To perform HX MS with a Langmuir monolayer, the Langmuir trough used previously for neutron reflection studies29 was adapted to facilitate hydrogen exchange labeling experiments. The major modification allowed for efficient and quick exchange of the aqueous subphase (the buffer under the monolayer) for a deuterated labeling buffer. Several ports were machined into opposite sides of the trough and additional Teflon tubing was connected to a high speed peristaltic pump to rapidly exchange the subphase with fresh buffer (as shown in FIGURE 1). Circulation tests were performed to determine the smallest volume of subphase buffer required for complete subphase exchange. For these tests, buffer containing dye was used and absorbance measurements of the subphase buffer were taken before and after circulation. The efficiency of subphase exchange was calculated using a ratio of the two absorbance measurements. Subphase exchange with 50 mL of buffer (approximately 2× the total volume of the trough and tubing) produced 83.8% exchange of the subphase. Circulating 100 mL of fresh buffer (4× the volume of the trough and tubing) yielded 96.3% exchange of the subphase. With the peristaltic pump used, the minimum time required to circulate 100 mL of buffer without disturbing the monolayer was 10 seconds. For each labeling experiment (deuteration time point), a new monolayer was generated and 100 mL of new labeling buffer were circulated for 10 seconds. Thus, every data point was a biological replicate37 with a new lipid monolayer. This strategy required experiments to test the reproducibility of monolayer creation, lipid interaction, deuterium labeling, and quenching of the exchange reaction, as outlined below.
Reproducibility and Deuterium Recovery of the Trough-Labeling Protocol
After buffer exchange, the labeled species (peptide or protein) was aspirated directly from the trough into a cold sample tube and deuterium labeling was quenched. Some lipid molecules from the monolayer and subphase buffer were also present in each aspirated sample. The variables associated with aspiration were tested to determine the deuterium recovery as a function of protocol, including D2O circulation, time, temperature and quench efficiency. For this purpose, we utilized a solution of fully deuterated peptides to monitor deuterium loss, in a similar manner to that commonly used to monitor deuterium loss in HX MS protocols (e.g., during sample handling, LC separation or mass analysis). A monolayer was prepared on equilibrium buffer and 100 mL of a mixture of deuterated leucine enkephalin, angiotensin I and bradykinin (in labeling buffer) were circulated under the monolayer. The peptide mixture/monolayer was aspirated into the collection tube, quench buffer added, and then the solution immediately injected into the UPLC system (termed trough labeling). In parallel, the same deuterated peptide mixture was added to quench buffer directly without trough circulation or aspiration (termed solution labeling), or directly injected into the UPLC without quenching (termed direct injection). Note that due to differences in timing, species in the trough could have been exposed for deuterium for up to 20 seconds at what we called a 10 seconds labeling-time point (up to 10 seconds of exposure during the 100 mL deuterated buffer circulation + 10 seconds of exchange before aspiration and quenching) while exchange in solution for 10 seconds was, in fact, exposure to deuterium for 10 seconds before quenching. We did not make a correction for this timing difference in any of the experiments. Using the trough protocol or a conventional solution protocol (FIGURE 2), there was little difference in the measured deuterium levels in leucine enkephalin, angiotensin I or bradykinin indicating that the additional sample handling steps in the trough protocol did not lead to more back-exchange of deuterium than a conventional solution labeling protocol. The error of triplicate measurements of deuterium uptake for the trough protocol was also very low (less than ± 1%), indicating that reproducible monolayer creation (recall that each experiment was a new, unique monolayer), buffer exchange/circulation, aspiration, and quenching can all be achieved. These results show that the Langmuir monolayer trough system and associated protocol can provide both reproducible and reliable measurements of HX for peptides/proteins in the presence of a Langmuir monolayer.
Figure 2.

Relative deuterium recovery for a mixture of totally deuterated peptides exposed to the Langmuir monolayer trough labeling protocol (red) or conventional solution labeling in a tube (blue). Deuterium recovery % was calculated using the value of 100% deuterated for each peptide as full backbone amide hydrogen deuteration minus one for deuterium loss at the N-terminus and minus one for each proline residue. Recovery was also measured for the same totally deuterated peptide mixture when directly injected into the UPLC without addition of quench buffer (gray). The average of three independent measurements is shown with error bars indicating the spread of the measurements. For angiotensin I, the +2 charge state is shown to the left, the +3 charge state to the right.
Validation with an Amphipathic Peptide
A major incentive for developing this Langmuir monolayer method for HX MS was to investigate membrane-related conformational changes in proteins and peptides. To test the experimental system with a simple model compound, we measured HX in the peptide melittin in the presence of a monolayer (trough labeling) and compared with HX in the absence of lipids (solution labeling). Melittin is a well-characterized, 26 residue peptide that is known to interact with lipid membranes38. The amphipathic nature of melittin allows it to associate with lipid membranes despite being soluble in water39. Melittin is unstructured in solution40 while its membrane-bound conformation (FIGURE 3A) is largely α-helical. At low concentration it is partially embedded in one monolayer a lipid bilayer41. At higher concentrations, melittin can form pores in bilayers42-45 and also in monolayers46. Due to the conformational changes melittin undergoes in the presence of lipids, which include creation of hydrogen bonds and secondary structure, it was an ideal candidate to test lipid-associated protection from HX.
Figure 3.
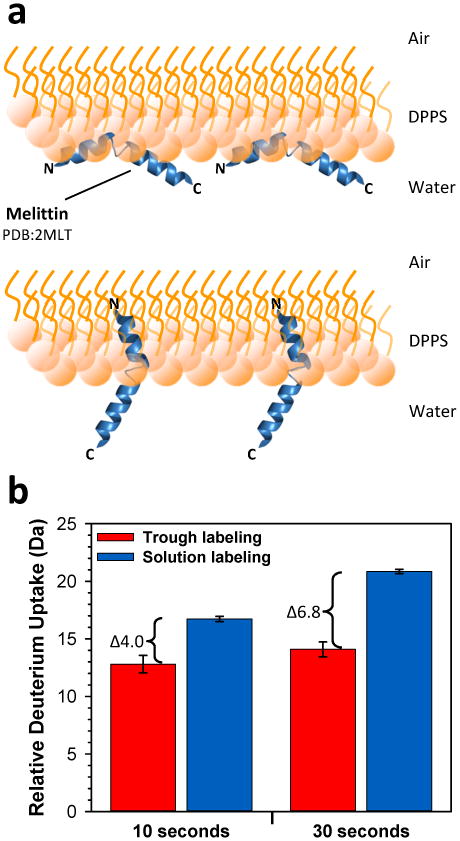
Changes to HX when melittin interacts with monolayers. (A) Cartoon representation of melittin interacting with a DPPS monolayer in a parallel position (top, adapted from Ref.39) or in a perpendicular position (bottom, Ref. 45,46). (B) Relative deuterium incorporation at the times indicated for melittin when monolayer associated and labeled within the trough (red) versus when labeled in solution with a conventional HX protocol (blue). The average of three independent measurements is shown with error bars indicating the spread of the measurements.
We compared the deuterium incorporation for melittin in solution and associated with a monolayer of DPPS (historically, DPPS has been used for monolayer/membrane studies of melittin and therefore we used it rather than DPPG as for all other studies reported here). First, melittin was labeled in solution for 10 and 30 seconds and the relative deuterium levels measured. Then, following these solution HX measurements, melittin was injected underneath a Langmuir monolayer at a pressure of 20 mN/m and allowed to adsorb for approximately 15 minutes. Melittin insertion into the monolayer was evidenced by the backward movement of the barrier indicating an increase in surface area of the monolayer (data not shown). Once the barrier had migrated ∼25 mm, deuterium was circulated, labeling allowed to proceed for 10 seconds, and the monolayer/melittin aspirated, quenched and analyzed by UPLC-MS. A new monolayer was created and all steps were repeated for a 30 second deuteration time point. We obtained triplicate measurements for both the 10 second and 30 second deuterium labeling times (FIGURE 3B). As predicted, in the absence of a lipids the peptide was heavily deuterated in only 30 seconds, agreeing with observations that indicate a lack of structure for melittin in solution. However, when melittin was adsorbed to the Langmuir monolayer, there was an average 4.0 and 6.8 Dalton reduction in deuterium incorporation for the peptide at 10 and 30 seconds of labeling, respectively. These results, along with the trough barrier movement backwards during adsorption, are consistent with creation of the amphipathic α-helix form of melittin and insertion into the lipid monolayer, both of which would induce protection from deuterium labeling. As the concentration of melittin we used was higher than that used in the studies by Gimenez et al.46, we expect that melittin formed pores in the monolayer in these trough experiments and was oriented perpendicular to the monolayer (as in FIGURE 3A, bottom). Our results showing melittin protection and conformational changes upon monolayer association validate this Langmuir monolayer HX MS method for detecting peptide:lipid interactions.
Validation with a Protein
After validating that our Langmuir monolayer HX MS method could be used to monitor conformational changes and lipid-induced protection from exchange in a peptide, we sought to monitor a protein known to undergo significant conformational changes in the presence of membranes. We elected to use ADP-ribosylation factor 1 (Arf-1), a 20 kDa guanine-nucleotide-binding protein that is involved in vesicle formation and trafficking47,48. Proper function of Arf-1 requires lipid association via membrane binding and nucleotide exchange. In addition to membrane anchoring via an N-terminal amphipathic helix, Arf-1 is myristoylated on the glycine residue at position 2, and this modification is crucial for membrane association49. MyrArf-1 is a prototypical protein for a myristoyl switch mechanism where the transition between two different conformations moves the myristoyl group from a position hidden within a hydrophobic pocket of Arf-1 when the protein is cytosolic and GDP-bound, to a position where the myristoyl group is inserted into the membrane when Arf-1 is GTP-bound and membrane-anchored50. Nucleotide exchange in Arf-1 by a guanine exchange factor accompanies the switch mechanism33,48,49. In the GDP-bound form, myrArf-1 associates with the membrane49 with the N-terminal portion (residues 1-16) interacting directly with the membrane and residues 17-177 facing the solvent33,51. Other conformational changes can occur upon the switch from GDP-bound to GTP bound, as high-resolution structures of the GDP-bound soluble and GTP-bound membrane-associated forms show. The well-defined membrane association, clear conformational changes and available structural details for myrArf-1 made it a good model to validate our Langmuir monolayer HX system for studies of protein conformational changes upon membrane association.
HX of monolayer-bound myristoylated Arf-1 bound to GDP was performed in the trough system with a lipid packing pressure of 20 mN/m and deuterium incorporation was compared to HX of myrArf-1 in solution. Digestion of labeled and quenched myrArf-1 produced 32 peptides resulting in 92% coverage of the protein backbone (see Supporting Information FIGURE S2). Overlapping peptides were found in nearly all areas of the protein, and similar HX trends were observed in redundant peptides (full dataset in Supporting Information, FIGURE S3). All deuterium incorporation measurements were made in triplicate, again with a new monolayer (DPPG) spread for every data point. There were distinct differences in exchange as a result of monolayer association (FIGURE 4A). In solution, residues 1-21 incorporated a high amount of deuterium at ten seconds and deuteration remained high for all time points (FIGURE 4Bi), indicative of a lack of structure in this region of myrArf-1. In the presence of the DPPG monolayer, this region of myrArf-1 showed a reduction greater than 5 Daltons in relative deuterium incorporation at ten seconds, which is consistent with stabilization of or creation of structure. However, this region remained dynamic and incorporated as much deuterium as the solution state after 10 minutes (FIGURE 4Bi). Similar HX results (early protection that changed to high deuteration) were obtained upon ordering of unstructured α-helical sequences through stabilization with hydrocarbon staples52 suggesting that the N-terminus of myrArf-1 is unstructured and highly dynamic in solution but becomes partially ordered as a helix upon monolayer interaction, yet still remaining somewhat dynamic. This hypothesis is consistent with published myrArf-1 data describing the formation and insertion of an N-terminal alpha helix33,51.
Figure 4.
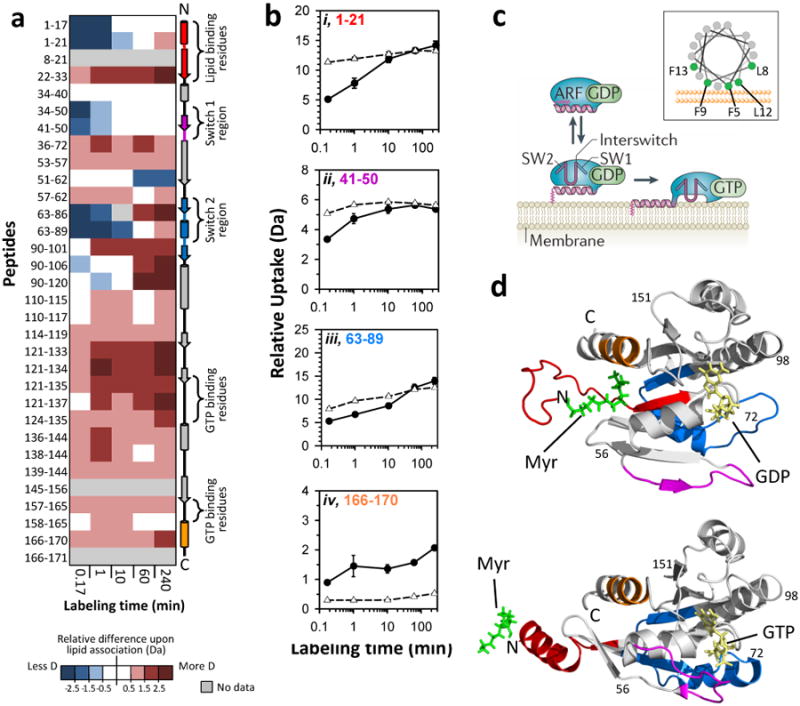
Effects of monolayer association on myristoylated Arf-1. (A) Difference map comparing myrArf-1 HX in the trough (monolayer associated) versus myrArf-1 HX in solution. Deuterium levels for the peptides indicated at the left were obtained from triplicate experiments (data in FIGURE S3). The average amount of deuterium after HX in the trough (monolayer associated) was subtracted from the average amount of deuterium for HX in solution and the value colored (positive values in reds, negative values in blues, as indicated). Secondary structural elements in myrArf-1 are displayed on the right. (B) Deuterium incorporation in four selected myrArf-1 peptides for monolayer associated (-●-) HX and solution (-Δ-) HX. The residues of each peptide are colored to match colored secondary structural elements in panel A. Error bars represent the spread of triplicate measurements. (C) Cartoon model (from Ref. 49) showing the reversible association of myrArf-1*GDP with membranes via myristoylated N-terminal helix prior to nucleotide exchange. A helical wheel of this region is shown in the inset, with the membrane interacting residues highlighted in green. Adapted with permission from Macmillan Publishers Ltd, Ref. 49, Copyright 2011. (D) Structural location in myrArf-1*GDP (PDB:2K5U33) and myrArf-1*GTP (PDB:2KSQ51) of peptides highlighted and color coded (red, pink, blue, orange) in panels A and B. The myristoyl moiety, bound nucleotide and switch regions are indicated.
The myrArf-1 used here was bound to GDP, and we did not investigate the additional conformational changes that occur as a result of nucleotide exchange to GTP after membrane interaction by the myristoyl group33. We nonetheless did observe that peptides in the switch 1 region of myrArf-1, residues 41-50 (FIGURE 4Bii), and peptides encompassing the switch 2 region, residues 63-89 (FIGURE 4Biii), incorporated less deuterium at early labeling times in the monolayer-associated form. Peptides in areas involved in GTP binding showed increases in deuterium when myrArf-1 was monolayer associated: residues 121-135 showed greater than a 2.0 Daltons increase while residues 166-170 showed greater than 1.5 Dalton increase in deuterium uptake after 4 hours (FIGURE 4A,Biv). In solution these regions were more protected, exchanging at most one or two amides hydrogens for peptides between residues 121-135 and less than one for residues 166-170 at ten seconds (see also FIGURE S3). Our results are in agreement with prior studies where it has been shown that much of the GTP binding residues, both switch regions and the C-terminal portion of myrArf-1 are exposed when membrane bound in order to interact with exchange factors and complete nucleotide exchange necessary for myrArf-1 function33,50,51,53,54. Overall, myrArf-1 provided good validation that changes in protein conformation upon membrane association could be measured and localized within the protein using our Langmuir monolayer HX MS system.
Conclusions
We have developed a new method for analyzing conformational features of membrane-associated peptides and peripheral membrane proteins by combining HX MS and Langmuir monolayers. We believe this to be the first description of using HX MS and Langmuir monolayers to study conformation during membrane association. A Langmuir trough setup originally designed for neutron reflection studies was modified to rapidly introduce deuterated buffer under a monolayer where peptide/protein had associated. The strength of the interaction of the peptide/protein with the monolayer was sufficient enough that peptide/protein was not washed away during exchange of the subphase, giving us confidence that non-specific or casual binding events were likely not responsible for the HX differences that were observed. The method had deuterium recovery equivalent to that of solution HX experiments; the additional steps associated with the monolayer did not accelerate back-exchange. With this method, conformational changes could be monitored in peptides (melittin) and proteins (myrArf-1). The reported lipid induced structural changes observed in these systems are consistent with what is predicted or known from other published data. An important advantage of using the Langmuir monolayer system is that the lipid packing density can be controlled and reproduced from monolayer to monolayer. For proteins that undergo conformational changes as a result of lipid packing density, this is a very valuable feature. We show in an accompanying paper32 how when packing density is altered, different conformations of the myrNef protein result.
There is of course the fact that monolayers are not membranes, so the question remains as to how well results obtained for monolayer interaction of proteins that interact with membranes in cells represent reality. An answer may depend on the particular protein. At one extreme, transmembrane proteins are likely not suited for analysis in this Langmuir monolayer system. At the other extreme, those peripheral membrane proteins that enter only one monolayer of a membrane may behave highly similarly in Langmuir monolayers as they do in the presence of a lipid bilayer. The behavior of (and validity of Langmuir monolayers for) proteins falling between the extremes is less certain and more measurements are needed. It may be possible to perform HX with tethered lipid bilayers, rather than just monolayers, thereby allowing a much wider range of inserted or membrane-associated proteins to be investigated.
The aspiration step to remove sample from the trough requires some practice to reliably obtain only 300 μLs and capture mostly monolayer and peptide/protein with minimal excess of subphase buffer. With practice, one can become quite good at the entire protocol, including aspiration, as demonstrated by the reproducibility of the measurements. During the course of the work reported here and in the accompanying paper32, 48 independent Langmuir monolayers were prepared and analyzed [peptide standards (Fig 2) in triplicate; melittin (Fig 3) in triplicate for two time points equaling 6 total; myrArf-1 (Fig 4) in triplicate for 5 time points equaling 15 total; myrNef (accompanying paper) in triplicate, 4 time points at 2 pressures for 24 total]. The deuterium measurement reproducibility from monolayer to monolayer was remarkably high and gave us high confidence in the measurements (triplicate results would not be as meaningful were the three MS measurements of deuterium obtained from one monolayer). As we have advocated37,55 replication that recapitulates as much biology (in this case, fresh monolayers and protein binding) as possible results in the most meaningful results. One downside to the method is that 100 mL of 99.9% deuterium buffer were required per analysis (we consumed >5 liters in all the work here); modifications to the trough and associated pump tubing could reduce the required volume (and cost) substantially. While lipids were present in each aspirate and were injected into the LC/MS system with every run, they were mostly captured by the inline traps and did not interfere with the signals of nearly all peptides. The exception to this were highly hydrophobic and eluted very late in the gradient when lipids also were eluting.
Another major advantage of this technique is the opportunity to combine global structural information from other techniques such as neutron or X-ray reflection with local information from HX. Our Langmuir monolayer HX MS method was designed to be integrated with a neutron or X-ray reflection workflow. In such a scheme, protein association with monolayers can be monitored in the same trough described here using neutron or X-ray reflection, the profile of the protein with respect to the monolayer obtained, packing density of the monolayer modulated (if desired) and the impact on conformation monitored. Once an interesting conformation is identified by neutron or X-ray reflection (or at any point, conformation or condition such as packing density), the protein could be labeled with deuterium right at the reflectometer and the sample passed to a UPLC-MS system for HX measurement. We believe there are significant benefits to such a strategy and the presented data provide strong justification for interrogating membrane protein association using both neutron reflection and HX MS which we further describe and exemplify in the accompanying paper32.
Supplementary Material
Acknowledgments
We gratefully acknowledge Mark Weir and Prof. Thomas E. Smithgall for cloning the Arf-1 gene into the pET-Deut vector. We also thank Prof. Thomas E. Wales for helpful discussion and critical insight with the manuscript. This work was supported by NIH grants GM086507 and GM101135, and a research collaboration with the Waters Corporation. Sandia National Laboratories is a multi-program laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-AC04-94AL85000.
Footnotes
Supporting Information: Supporting information available: This material is available free of charge via Internet at http://pubs.acs.org.
References
- 1.Cho W, Stahelin RV. Annu Rev Biophys Biomol Struct. 2005;34:119–151. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 2.Overington JP, Al-Lazikani B, Hopkins AL. Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 3.Rask-Andersen M, Almen MS, Schioth HB. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 4.White SH. Nature. 2009;459:344–346. doi: 10.1038/nature08142. [DOI] [PubMed] [Google Scholar]
- 5.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou HX, Cross TA. Annu Rev Biophys. 2013;42:361–392. doi: 10.1146/annurev-biophys-083012-130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hebling CM, Morgan CR, Stafford DW, Jorgenson JW, Rand KD, Engen JR. Anal Chem. 2010;82:5415–5419. doi: 10.1021/ac100962c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan CR, Hebling CM, Rand KD, Stafford DW, Jorgenson JW, Engen JR. Mol Cell Proteomics. 2011;10:M111 010876. doi: 10.1074/mcp.M111.010876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan CR, Miglionico BV, Engen JR. Biochemistry. 2011;50:3394–3403. doi: 10.1021/bi200197e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim M, Sun ZY, Rand KD, Shi X, Song L, Cheng Y, Fahmy AF, Majumdar S, Ofek G, Yang Y, Kwong PD, Wang JH, Engen JR, Wagner G, Reinherz EL. Nat Struct Mol Biol. 2011;18:1235–1243. doi: 10.1038/nsmb.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nasr ML, Shi X, Bowman AL, Johnson M, Zvonok N, Janero DR, Vemuri VK, Wales TE, Engen JR, Makriyannis A. Protein Sci. 2013;22:774–787. doi: 10.1002/pro.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karageorgos I, Wales TE, Janero DR, Zvonok N, Vemuri VK, Engen JR, Makriyannis A. Biochemistry. 2013;52:5016–5026. doi: 10.1021/bi400430k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker CH, Morgan CR, Rand KD, Engen JR, Jorgenson JW, Stafford DW. Biochemistry. 2014;53:1511–1520. doi: 10.1021/bi401536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barclay LA, Wales TE, Garner TP, Wachter F, Lee S, Guerra RM, Stewart ML, Braun CR, Bird GH, Gavathiotis E, Engen JR, Walensky LD. Mol Cell. 2015;57:873–886. doi: 10.1016/j.molcel.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.West GM, Chien EY, Katritch V, Gatchalian J, Chalmers MJ, Stevens RC, Griffin PR. Structure. 2011;19:1424–1432. doi: 10.1016/j.str.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan Y, Piyadasa H, O'Neil JD, Konermann L. J Mol Biol. 2012;416:400–413. doi: 10.1016/j.jmb.2011.12.052. [DOI] [PubMed] [Google Scholar]
- 17.Mehmood S, Domene C, Forest E, Jault JM. Proc Natl Acad Sci U S A. 2012;109:10832–10836. doi: 10.1073/pnas.1204067109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Y, Brown L, Konermann L. J Am Chem Soc. 2011;133:20237–20244. doi: 10.1021/ja206197h. [DOI] [PubMed] [Google Scholar]
- 19.Koshy SS, Eyles SJ, Weis RM, Thompson LK. Biochemistry. 2013;52:8833–8842. doi: 10.1021/bi401261b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vadas O, Dbouk HA, Shymanets A, Perisic O, Burke JE, Abi Saab WF, Khalil BD, Harteneck C, Bresnick AR, Nurnberg B, Backer JM, Williams RL. Proc Natl Acad Sci U S A. 2013;110:18862–18867. doi: 10.1073/pnas.1304801110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rey M, Mrazek H, Pompach P, Novak P, Pelosi L, Brandolin G, Forest E, Havlicek V, Man P. Anal Chem. 2010;82:5107–5116. doi: 10.1021/ac100171m. [DOI] [PubMed] [Google Scholar]
- 22.Lipid Polymorphism and Membrane Properties. Vol. 44 Academic Press; San Diego, CA: 1997. [Google Scholar]
- 23.The Structure of Biological Membranes. 2nd. CRC Press; Boca Raton, FL: 2005. [Google Scholar]
- 24.Boggs JM. Biochim Biophys Acta. 1987;906:353–404. doi: 10.1016/0304-4157(87)90017-7. [DOI] [PubMed] [Google Scholar]
- 25.Epand RM, Epand RF. Biochim Biophys Acta. 2009;1788:289–294. doi: 10.1016/j.bbamem.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee S, Maxfield FR. Traffic. 2000;1:203–211. doi: 10.1034/j.1600-0854.2000.010302.x. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez SA, Tricerri MA, Ossato G, Gratton E. Biochim Biophys Acta. 2010;1798:1399–1408. doi: 10.1016/j.bbamem.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takatori S, Mesman R, Fujimoto T. Biochemistry. 2014;53:639–653. doi: 10.1021/bi401598v. [DOI] [PubMed] [Google Scholar]
- 29.Akgun B, Satija S, Nanda H, Pirrone GF, Shi X, Engen JR, Kent MS. Structure. 2013;21:1822–1833. doi: 10.1016/j.str.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kent MS, Murton JK, Sasaki DY, Satija S, Akgun B, Nanda H, Curtis JE, Majewski J, Morgan CR, Engen JR. Biophys J. 2010;99:1940–1948. doi: 10.1016/j.bpj.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maget-Dana R. Biochim Biophys Acta. 1999;1462:109–140. doi: 10.1016/s0005-2736(99)00203-5. [DOI] [PubMed] [Google Scholar]
- 32.Pirrone GF, Emert-Sedlak L, Smithgall TE, Kent MS, Engen JR. Anal Chem. 2015 doi: 10.1021/acs.analchem.5b01725. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Kahn RA, Prestegard JH. Structure. 2009;17:79–87. doi: 10.1016/j.str.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wales TE, Fadgen KE, Gerhardt GC, Engen JR. Anal Chem. 2008;80:6815–6820. doi: 10.1021/ac8008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Mol Cell Proteomics. 2006;5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Wales TE, Engen JR. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 37.Engen JR, Wales TE. Annu Rev Anal Chem (Palo Alto Calif) 2015;8 doi: 10.1146/annurev-anchem-062011-143113. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendez MA, Nazemi Z, Uyanik I, Lu Y, Girault HH. Langmuir. 2011;27:13918–13924. doi: 10.1021/la202970g. [DOI] [PubMed] [Google Scholar]
- 39.Raghuraman H, Chattopadhyay A. Biosci Rep. 2007;27:189–223. doi: 10.1007/s10540-006-9030-z. [DOI] [PubMed] [Google Scholar]
- 40.Vogel H. FEBS Lett. 1981;134:37–42. doi: 10.1016/0014-5793(81)80545-5. [DOI] [PubMed] [Google Scholar]
- 41.Dempsey CE, Butler GS. Biochemistry. 1992;31:11973–11977. doi: 10.1021/bi00163a003. [DOI] [PubMed] [Google Scholar]
- 42.Andersson M, Ulmschneider JP, Ulmschneider MB, White SH. Biophys J. 2013;104:L12–14. doi: 10.1016/j.bpj.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klocek G, Schulthess T, Shai Y, Seelig J. Biochemistry. 2009;48:2586–2596. doi: 10.1021/bi802127h. [DOI] [PubMed] [Google Scholar]
- 44.Krueger S, Meuse CW, Majkrzak CF, Dura JA, Berk NF, Tarek M, Plant AL. Langmuir. 2001;17:511–521. [Google Scholar]
- 45.Yang L, Harroun TA, Weiss TM, Ding L, Huang HW. Biophys J. 2001;81:1475–1485. doi: 10.1016/S0006-3495(01)75802-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gimenez D, Sanchez-Munoz OL, Salgado J. Langmuir. 2015;31:3146–3158. doi: 10.1021/la504293q. [DOI] [PubMed] [Google Scholar]
- 47.D'Souza-Schorey C, Chavrier P. Nat Rev Mol Cell Biol. 2006;7:347–358. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- 48.Gillingham AK, Munro S. Annu Rev Cell Dev Biol. 2007;23:579–611. doi: 10.1146/annurev.cellbio.23.090506.123209. [DOI] [PubMed] [Google Scholar]
- 49.Donaldson JG, Jackson CL. Nat Rev Mol Cell Biol. 2011;12:362–375. doi: 10.1038/nrm3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldberg J. Cell. 1998;95:237–248. doi: 10.1016/s0092-8674(00)81754-7. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Kahn RA, Prestegard JH. Nat Struct Mol Biol. 2010;17:876–881. doi: 10.1038/nsmb.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi XE, Wales TE, Elkin C, Kawahata N, Engen JR, Annis DA. Anal Chem. 2013;85:11185–11188. doi: 10.1021/ac403173p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franco M, Chardin P, Chabre M, Paris S. J Biol Chem. 1995;270:1337–1341. doi: 10.1074/jbc.270.3.1337. [DOI] [PubMed] [Google Scholar]
- 54.Franco M, Chardin P, Chabre M, Paris S. J Biol Chem. 1996;271:1573–1578. doi: 10.1074/jbc.271.3.1573. [DOI] [PubMed] [Google Scholar]
- 55.Moroco JA, Engen JR. Bioanalysis. 2015 doi: 10.4155/bio.15.46. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.