

Abstract
We demonstrate a platform that integrates photonic crystal enhanced fluorescence (PCEF) detection of a surface-based microspot fluorescent assay with a microfluidic cartridge to achieve simultaneous goals of high analytic sensitivity (single digit pg/mL), high selectivity, low sample volume, and assay automation. The PC surface, designed to provide optical resonances for the excitation wavelength and emission wavelength of Cyanines 5 (Cy5), was used to amplify the fluorescence signal intensity measured from a multiplexed biomarker microarray. The assay system is comprised of a plastic microfluidic cartridge for holding the PC and an assay automation system that provides a leak-free fluid interface during introduction of a sequence of fluids under computer control. Through the use of the assay automation system and the PC embedded within the microfluidic cartridge, we demonstrate pg/mL-level limits of detection by performing representative biomarker assays for interleukin 3 (IL3) and Tumor Necrosis Factor (TNF-α). The results are consistent with limits of detection achieved without the use of the microfluidic device with the exception that coefficients of variability from spot-to-spot are substantially lower than those obtained by performing assays with manual manipulation of assay liquids. The system’s capabilities are compatible with the goal of diagnostic instruments for point-of-care settings.
1. Introduction
As molecular diagnostics are increasingly utilized as a minimally invasive tool for determining the health status of patients and for guiding the course of therapy, there is intense interest in transitioning central laboratory-based tests towards the point-of-care through assays that can be performed easily within health clinics. There is also a drive towards gaining a more comprehensive view of a patient’s health status from a single specimen through multiplexed tests that are capable of determining the presence and concentration of many analytes simultaneously. The ability to perform such testing using only a single droplet of serum would aid in the widespread acceptance of such tests, particularly if a sample could be gathered by a finger-stick, rather than drawing blood by a highly skilled phlebotomist. For example, rapid detection of immune-related biomarkers (i.e. specific antibodies) to pathogenic antigens, tumor antigens, or allergenic antigens is an important aspect of allergy characterization and diagnosis of autoimmune disease [1, 2] [3]. Detection of soluble protein biomarkers is used heavily in oncology for early detection of several forms of cancer [4–12], cardiovascular disease [13, 14], neurological disorders and infectious disease, as disease specific biomarker panels continue to be discovered and validated by clinical studies [15]. New types of biomarkers are the subject of intense research interest, including circulating tumor DNA (ctDNA) [16–18], and microRNA (miRNA) biomarkers for cancer [19–21]. The concept of a “liquid biopsy” is gaining acceptance as an approach for assessing many aspects of a patient’s health without expensive medical imaging technology, exposure to ionizing radiation, or extraction of tissue from the patient. When implemented with a sensitive, inexpensive, rapid, and multiplexed detection technology, a liquid biopsy may serve as a primary form of health assessment, early disease detection, and treatment-tracking approach that will complement more invasive and expensive diagnostic modalities.
A key unmet need for soluble biomarker detection for achieving clinical relevance is improved sensitivity. Early diagnostic applications for cancer, for example, require detection of biomarkers that are produced by, or in response to, a small number of cells. The biomarker is diluted throughout the volume of blood within a person, so that when a sample of peripheral blood is drawn, the biomarker concentration can be <1–10 pg/mL. (For reference, 1 pg/mL of a typical cytokine such as IL-6 is equivalent to ~45 fM. A 10 μl sample at this concentration contains 0.45 attomoles of IL-6.) In addition to high sensitivity, clinical diagnostic laboratories require automated platforms that can perform rapid multiplexed biomarker analysis at low cost/assay and that can quantify small changes in concentration. Consensus is emerging that multiple, mutually exclusive biomarkers in an assay will lead to better clinical management of disease, compared to assessment of a single biomarker, and will allow subtle differences in patient populations (gender, race, age) to be understood [22–24].
Among the current methodologies of biomarker detection, sandwich antibody microarrays hold great potential due to their demonstrated capacity to detect analytes at <10 pg/mL concentration in complex media (such as serum) [1, 25] with the use of only 3–10 μL samples, no assay cross-reactivity, and fast binding kinetics. Using a photonic crystal (PC) surface to enhance the fluorescence output from a biomarker microarray, we have previously published the results of studies that successfully achieved high sensitivity for multiplexed cancer biomarker detection [26–29]. More recently, we developed a high sensitivity, compact, and inexpensive detection platform for Photonic Crystal Enhanced Fluorescence (PCEF) that uses a silicon-based PC surface and a compact/inexpensive laser line-scanning detection instrument in an effort to maximize the coupling efficiency of light into the PC structure [27]. Our previous work demonstrates clearly that PCEF surfaces can be inexpensively mass-manufactured from silicon wafers using conventional processes used in integrated circuit manufacturing, and that silicon-based PCs do not introduce background fluorescence (which is also amplified by PCEF), thus enabling detection of biomarkers present at low concentrations.
Our previously-demonstrated application of PCEF for multiplexed, high sensitivity detection of soluble protein and miRNA biomarkers was performed as a proof-of-principle using an assay protocol that required a highly skilled person to introduce all reagents and to perform all assay wash steps manually using a pipette. For this approach, a relatively large (1 × 0.5 in2) silicon-based PC chip was fitted with a silicone rubber gasket with a screw-on metal holding fixture that would enable the operator to define up to ten distinct assay wells upon the PC surface. Microspot fluorescent immunoassays for TNF-α and IL-3 performed on this platform demonstrated that the system can achieve pg/mL-level sensitivity. Although only a 10 uL sample was consumed with the use of 2mm-diameter wells, manual sample handling presented difficulties in injecting liquid and required dedicated time/labor. The performance and implementation characteristics of performing PCEF microarray assays with manual liquid sample handling greatly limits its scalability and accessibility to clinicians, including a disjointed workflow that required manual intervention across multiple steps and used the PC surface area inefficiently. Therefore, the development of a simpler and automatic format is desired to move PCEF-based biomarker assays closer to clinical applications such as point-of-care diagnostics.
In this paper, we present a new platform that combines PCEF and microfluidic sample handling to achieve high analytic sensitivity (1.57 pg/ml), high selectivity, low sample volume, and assay automation. The novel elements of the present work include the design and fabrication of a low-cost PC-integrated microfluidic cartridge that incorporates a stereolithographic 3D printed cartridge body [30–32] a laser-cut fluidic channel [33–35] a low autofluorescent transparent glass viewing window, and a substantially smaller (2 × 8 mm2) PC chip. The function of the cartridge is to hold the PC in place during the assay, to provide a leak-free and simple-to-operate interface to an external assay automation system, and to provide a stable mechanical platform during fluorescence scanning. The assay automation system introduces the correct sequence of fluids through the cartridge from a set of liquid reservoirs (external to the cartridge) under computer control. Here we demonstrate the first use of the cartridge and assay automation system for two representative biomarker assays in the form of a small sandwich antigen microarray. While substantially reducing the operator time and complexity required to perform an assay, we demonstrate that achieved limits of detection are equivalent to those obtained with the more “manual” approach, while the use of microfluidic flow (rather than pipette-based liquid handling) was found to significantly improve the spot-to-spot reproducibility of the assay. We found that the coefficients of variability across spots were decreased most dramatically for the lowest concentration assays.
2. Materials and Methods
2.1 Design and fabrication of a PC-embedded microfluidic device
The PC is comprised of a periodic surface structure fabricated with a low refractive index (RI) silicon dioxide (SiO2, RI=1.46) layer on a silicon substrate [27]. The grating structure is coated with a high RI titanium dioxide (TiO2, RI=2.35) thin film. The PC has a period of 360 nm, a duty cycle of 36%, a grating depth of 40 nm, and a TiO2 thickness of 130 nm. A commercial vendor (Novati Technologies Inc., Austin TX) was contracted for performing photolithography and reactive ion etching (RIE) of the SiO2 grating structure over 8-inch diameter wafers, while TiO2 thin films were deposited upon whole wafers at a second vendor (Intlvac Inc., Niagara Falls NY). Following lithography, etching, and TiO2 deposition, the wafers were diced into 2 × 8 mm2 pieces. An SEM image showing the surface structure of the PC is presented in the inset of Figure 1 (c). As shown in Figure 1(a), the cartridge assembly is comprised of a 3D printed plastic base with inlet/outlet through-holes, a Double-Sided Adhesive (DSA) fluid channel, and a nonfluorescent glass coverslip. The plastic base was designed by three-dimensional (3D) design software (PTC Creo Parametric 2.0) and fabricated by stereolithography of optically clear resin (WaterClear Ultra 10122) using a Viper SLA System. The base layer is 75 mm long, 25 mm wide, 6 mm thick, and incorporates a recessed region (2.2 mm×8.2 mm), into which the PC chip of the same dimension was attached by adhesive. The middle layer is comprised of DSA (3M™ Optically Clear Adhesive 8212) with a ~2 × 12 mm wide and 150 μ m deep channel in the center, aligned with the fluid inlet/outlets and the PC. In order to ensure device cleanness, non-contact laser cutting was used to cut the channel area on the DSA. The bottom layer is a thin transparent coverslip, which allows a video camera to monitor the flow inside the channel during the assay, and subsequently for the laser scanning instrument to illuminate the microarray spots for fluorophore excitation. Shott Glass (#1098576, 24 × 60 × 0.17 mm) was chosen for its tight thickness tolerance, which minimizes spherical aberration, and its low background fluorescence, therefore promoting high quality images of the fluorescence signal during scanning. The three layers were joined by the DSA layer in a custom-built mechanical alignment fixture and pressed by a roller at room temperature to seal them together. Room temperature sealing of the chip is desired, so as to not damage protein-based capture molecules that are printed upon the PC before final cartridge assembly. Figure 1(c) shows a photo of a fabricated microfluidic chip in which red food dye was introduced through the inlet.
Figure 1.

2.2 Computer-controlled microfluidic system
Using components shown in Figure 2(a), the entire assay process was automated with no intervention from the user other than introduction of a droplet of test sample into the inlet reservoir (volume ~10 μL). A 3-to-1 valve (Cole-Parmer Manifold Mixing Solenoid Valve) introduces either compressed nitrogen, buffer solution, or Cy5-labeled detection antibody into the channel. The assay liquids are stored in 2 mL reservoirs, as they are common to all assays that will be performed by the instrument, and are pushed from the reservoir to the cartridge by pneumatic pressure. The fluid selection is determined by a programmable microfluidic controller (FlowTest™ Programmable Microfluidic Controller). The fluid driving force was supplied by compressed nitrogen, which was regulated by a pressure controller (Fluigent MFCS-EZ) that provided pulseless and highly stable (<0.1% CV) flows. A digital camera (Dino-Lite AM3111T) located beneath the fixture monitored the flow through a hole in the fixture. Under computer control, the system regulates the fluid flow rate, while enabling real-time monitoring of the exposure status of the PC by an integrated video camera.
Figure 2.

To ensure leak-proof sealing and zero dead volume, flanged end tubes and a custom-machined metal frame were used in our system [36]. The seal was based on a forced fit between the flanged ends of chemically inert polytetrafluoroethylene (Cole-Parmer PTFE EW-06605-27 1/16″) tubing and the flat outer surface of the microfluidic cartridge. The flange was created by heating the end of the tubing and pressing it against a metal washer [36]. The metal frame maintains alignment between the tubing and the inlet/outlets of the microfluidic chip, while screws between the upper and lower metal holding plates of the fixture apply sufficient pressure for a leak-proof seal. A schematic of the sealing fixture is shown in Figure 2(b).
2.3 Immunoassay procedure using microfluidic sample handling
To demonstrate the capability of the microfluidic system, microspot immunoassays for two cytokines (interleukin 3 (IL-3) and tumor necrosis factor alpha (TNF-α)) were performed using PCs embedded in the cartridge. Before printing spots, the PC surface was cleaned and activated with a vapor-phase epoxysilane process. The epoxysilane chemistry was chosen for its low background fluorescence [37] and high binding capacity for antibody molecules [38]. The devices were first cleaned by sonication in 2″ petri dishes of acetone, isopropanol, and deionozied (DI) water for 2 minutes each. The devices were then dried in a stream of N2 and then treated in an oxygen plasma system (Diener, Pico) for 10 minutes (power of 100W, pressure of 0.75mTorr). The backside of each device was then adhered to the inside of a screw top lid of a 2″ glass container. At the base of the container, 100 uL of (3-Glycidoxypropyl) trimethoxysilane (GPTS, Sigma Aldrich, Saint Louis, MO) was placed and the lid was securely placed over the dish. After securely tightening the lids, each dish with a device adhered to its lid was placed in a vacuum oven for an overnight incubation at a temperature of 80 °C and a pressure of 30 Torr. The devices were then detached from the lids and sonicated in 2″ petri dishes of toluene, methanol, and DI water for 2 minutes each and dried under a stream of N2.
After silanization, the protein arrays were printed onto the PC surface. A PC holds two subarrays, and each subarray contains 4 sets of 4 replicate spots for each protein, resulting in an array with 16 spots. The first row is a fluorescent-tagged protein for array orientation/location (Alexa-Fluor-555 fluorescencent streptavidin conjugates, Life Technologies), the second row is the TNF-α antibody (R&D systems Inc.), the third row is a negative control of Phosphate Buffered Saline (PBS) (pH=7.4), and the last row is the IL-3 antibody (R&D systems Inc.). The antibody microarrays on the PC were printed by a desktop nanofabrication system (Arrayit NanoPrint LM60 Microarrayer) while the chip was in a free format (i.e. not yet attached to the cartridge base). The printing was performed under a controlled environment using an environmental chamber (ambient temperature and 50% relative humidity). Four pins (946MP3 Microarray Printing Pins) were used to print the two antibodies at a concentration of 1 mg/mL, which results in 5 × 109 molecules/mm2. The binding affinity is ~50nM for TNF-alpha antibody while ~ 1nM for IL3 antibody [39]. The positive control was printed at a protein concentration of 10 μg/mL, and the negative control was comprised of printed buffer solution. Printing pins were cleaned between sample pickups with 15 s sonication and 4 cycles of washing (2.5 s) and drying (1 s). Measured spot diameters were 79±2 μm. Row spacing was 149±3 μm and column spacing was 200±0.8 μm. The printed substrates were incubated in a sealed box with a desiccant overnight at ambient temperature. Next, the arrays were blocked with casein blocking buffer (BioRad, Hercules, CA) for 1 h before they were washed three times with 0.02% (v/v) tween 20 in PBS (PBST). The substrates were then dried and put in a sealed box for future use.
Following array printing and surface blocking, a PC substrate with antibody microspots was glued into the recess of the plastic base of the cartridge. Next, the glass cover slip was bonded to the base using the laser-patterned DSA film to complete the fabrication process. Each completed microfluidic cartridge was used to perform one immunoassay for one mixture of different antigen concentrations in casein buffer. One thing to be noted is that 50 chips were printed with microarrays and assembled into a microfluidic channel one time, in order to help users avoid the spotting and assembly steps and directly run the automatic assay every time. Since the antibody microarrays can be stored in freezer at dry environment for more than six months, the quality of the microfluidic chips are not effected by the storage. 10 μL of test sample, comprised of a mixture of TNF-α and IL-3 in PBS was introduced into the microchannel inlet by a pipette. The hydrophilic nature of the inner channel surface facilitates liquid flow into the channel by capillary action, so the droplet of test sample covers the PC array without any intervention from the user or application of external fluid driving force. The cytokine TNF-α (R&D systems Inc.) was assayed at the following seven concentrations: 1 ng/mL, 0.25 ng/mL, 62.50 pg/mL, 15.62 pg/mL, 7.8 pg/mL, 3.9 pg/mL, and 1.9 pg/mL. The concentration of IL-3 (R&D systems Inc.) is 10 times higher than that of TNF-α in all assayed antigen mixtures. After a 3-hour incubation of the test sample with the microarray, the cartridge was loaded into the assay automation system, to perform the washing and labeling steps of the assay. The device was washed by flowing PBST for 1 min with an applied pressure of 25 mBar to remove the unreacted antigens. Next, the 1 ug/mL fluorescent-labeled secondary antibody mixture (Alexa Fluor® 647 TNF-α antibody and Alexa Fluor® 647 IL-3 antibody, at the mixing ratio of 1:1, Novus Biologicals, LLC) was introduced into the microchannel with an applied pressure of 25 mBar for 30 s. After 1 h incubation, the microchannel was washed again by flowing PBST for 1 min to remove the unreacted fluorescent-labeled secondary antibody solution. Finally, an air flow was produced in the microchannel by applying high-pressure nitrogen gas (345 mBar) to the inlet to remove remaining liquid. The PC surface was completely cleaned and dried in 10 s due to its hydrophobicity, as observed by the imaging camera. Following the immunoassay process, the microfluidic chip was removed from the automation system and transferred to the PCEF laser scanning instrument for fluorescent imaging.
2.4 PCEF scanner and image acquisition
We have designed and constructed a PCEF microarray detection instrument that provides collimated illumination and the ability to tune the incident angle to precisely match the resonant coupling condition [40] while focusing the light from a fiber-coupled semiconductor laser to a 8 μm line with a cylindrical lens [41]. The purpose of the custom detection instrument is to optimize coupling of laser illumination to the PC surface. As described in previously published reports [40] [41], the excitation laser is collimated in the plane perpendicular to the grating lines, but focused in the plane parallel to the grating (Figure 3). Collimated light with electric field polarization perpendicular to the grating lines is able to couple most efficiently with the PC resonant mode, but the light need only be collimated with respect to a single axis. Along the orthogonal axis, light can be focused without compromising coupling efficiency to the PC, and thus focus along one axis is used to achieve high illumination intensity. The system is designed for optimal interaction with the PC for both enhanced excitation and enhanced extraction, using design principles discussed, modeled, and demonstrated in [27]. The focal point of the cylindrical lens is located at the back focal plane of the objective. Linear translation of the cylindrical lens by a computer-controlled motion stage results in adjustment of the incident angle to achieve the ‘on resonance’ illumination. A semiconductor laser diode (AlGaAs, 70 mW, λ=637 nm) is expanded to a diameter of 1 mm, and focused to an 8 μm wide line onto the PC surface by a cylindrical lens. A mirror coupled to a computer-controlled linear translation stage enables adjustment of the incident angle from 0–20 degrees with 0.01 degree increments. The grating lines of the PC are oriented perpendicular to the scan line, allowing the laser (polarized output perpendicular to the grating) designed to excite the Transverse Magnetic (TM) mode of the PC.
Figure 3.

Using the scanner described above, a fluorescent image of the PC surface was obtained by adjusting the incident angle of the laser illumination line upon a region of the PC adjacent to the microspots and then translating the PC holding stage in increments of 2 μm past the array region, gathering a fluorescent intensity image of the line for each motion increment. Then, the fluorescent intensities of each line were assembled into a two-dimensional image of fluorescence intensity on the PC surface through a custom-built C# user interface. Spot segmentation and intensity calculations of the constructed fluorescence images were performed using ImageJ. Net spot intensity was calculated as the local background subtracted spot intensity where the local background is an annular region around a given spot. Spot signal-to-noise ratio (SNR) was calculated as the local background subtracted spot intensity divided by the standard deviation of the local background.
3. Results and discussions
3.1 Raw fluorescent intensity
Because all previous PCEF measurements of biomarker microarrays were gathered with PCs not packaged within a cartridge, we first sought to determine whether laser scanning through the glass window would have a detrimental effect on the spatial resolution, intensity, or background levels of the array. To investigate the potential effects of scanning the array through the glass window, we performed the immunoassay for 1.9 pg/mL TNFα using the PC-embedded cartridge, and then scanned the microarrays before and after removing the glass window. The illumination angle (incident angle=4.12 degree) and camera settings (sensitivity gain=25, exposure time=40ms) were the same for both scans. Although we scanned the same area twice, the intensity decay due to photobleaching after the first scan is less than 0.001% [42] and can be safely ignored, as the laser only illuminated the area for 120 ms, given that the exposure time of the camera was 40 ms and oversampling rate was 3. Fluorescence intensities of each microspot and its surrounding background were obtained by quantifying fluorescent images (Figure 4(a)), and subsequently averaged over the four replicates. Interestingly, the spot intensity obtained by scanning through the glass window (the first red bar in Figure 4(b)) is 20% higher than that obtained without the window (the first black bar in Figure 4(b)). The increased intensity was attributed to the fluorescence from a small amount of fluorescently labeled secondary antibody that was absorbed onto the glass during the assay procedure. We can confirm this assumption by examining the average background intensity, which decreased from 2236 counts to 1744 counts by removing the glass (Figure 4(b)). Net intensity was calculated as the background subtracted spot intensity, through which any fluorescence from the glass was removed. In contrast to the increase in the average spot intensity and background intensity, the average net intensity decreased to 93% if spots were scanned through the glass (the third bars in Figure 4(b)). This sma ll signal loss is attributed to partial light reflection at the interface between air and glass. Given that refractive index is n1=1.00 for air and n2=1.50 for glass, the transmittance of the fluorescence after passing the upper surface of glass is 96%, calculated by the equation:
Figure 4.

Ignoring the phenomena of multiple internal reflection in the glass, transmittance of the fluorescence through the glass, adding the signal loss at the second surface, is 92%, which is close to the percentage of the signal we obtained in the experiment. Considering the small thickness and good flatness of the glass, any absorption or scattering can be neglected. Although there was a slight decrease in the net fluorescence signal, the fluorescent spots were still distinguishable from the background, even for the assay with the lowest concentration.
3.2 Fluorescence enhancement of a PC-embedded cartridge
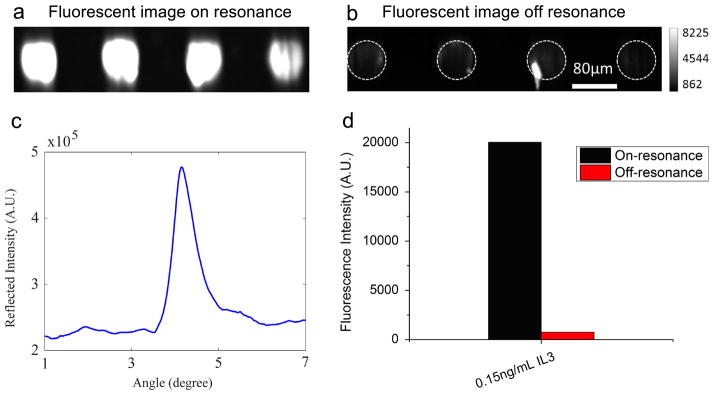
The PC is designed to increase the fluorescence intensity of Cy5 dyes through the enhanced excitation and extraction mechanism described previously. The enhanced extraction effect is always present, regardless of the illumination conditions. In a previous report [26], we demonstrated that enhanced extraction resulted in an approximately 5-fold increase in fluorescence intensity compared to detection on unpatterned glass. The effect of enhanced excitation can be determined by comparing the fluorescence output under the following two conditions: (a) when the incident angle of the excitation laser was adjusted to illuminate the PC at the resonant angle (‘on resonance’), and (b) when the angle of incidence was selected not to coincide with the resonant coupling condition (‘off resonance’). Although fluorescence enhancement by a silicon PC has been demonstrated previously [27], it is still necessary to characterize the enhancement factor and show that the same enhancement can be observed using a PC embedded in a microfluidic device that is covered by a glass window. In the experiment to characterize the enhanced excitation of the PC in the microfluidic device, 0.15 ng/mL of IL-3 was assayed, and the microspots were scanned at both on- and off-resonance conditions. First, the reflection spectrum of the PC (Figure 5(c)) was acquired by illuminating the surface over a range of incident angles at the fixed excitation wavelength λ=637 nm. The on-resonance angle of illumination was 4.12 degrees, as indicated in the spectrum, while the off-resonance angle was chosen to be 3.00 degrees. The fluorescent images shown in Figure 5(a)(b) were obtained at on- and off-resonance conditions, respectively. Because of the low fluorescence at the off-resonance condition, exposure time of the camera was set to be 400 ms, 10 times higher than that of the on-resonance condition. Therefore, the on-resonance value was multiplied by a factor of 10 before direct comparison to the off-resonance value. It can be observed in Figure 5(d) that by scanning the PC at its resonant angle, the fluorescence intensity was enhanced by a factor of 26, which is similar to what was reported previously [26, 42]. In general, we expect a PC embedded in the cartridge to perform identically to a PC in the “free” chip format, as the absorption and reflection of light by the thin glass cover are minimal.
Figure 5.
3.3 Standard curves and limit of detection
The performance of the PC-embedded cartridge was studied in the context of a microspot-based fluorescent sandwich immunoassay. Seven concentrations of antigen mixtures were assayed by the PC microfluidic system as described in the previous sections. No leakage from the flanged tubes was observed for any of the performed experiments. The assay procedure was controlled by a computer with minimal human intervention. Figure 6(a) shows representative fluorescence images of microspots at four sets of assayed concentrations. Compared with previously reported results obtained by manually handling the liquids [27], one obvious improvement realized by using the automatic system is the intensity uniformity within spots of the same cytokine. In order to quantitatively characterize how easily a spot can be distinguished from background noise, we defined signal to noise ratio (SNR) as the net signal divided by the standard deviation. A spot with SNR larger than 3 is regarded as detectable. Figure 6(a) shows that all the cytokine spots were detectable over the 1.9 pg/mL-10 ng/mL range. For example, SNRs of the TNF-α spots and the IL-3 spots at the lowest concentrations are 50.0 and 78.3, respectively.
Figure 6.

The signal intensities from each dilution in the concentration series were used to generate standard curves for both TNF-α (Figure 6(b)) and IL-3 (Figure 6(c)) using Prism. The limit of detection (LOD) is defined as the concentration corresponding to the blank intensity (i.e. the intensity of the negative control spot of PBS buffer) plus 3 standard deviations from all assay spots. Negative controls performed by exposing the capture antibodies to a casein sample resulted in no observable fluorescence signal above the background. Therefore, LOD values for the two cytokines TNF-α and IL-3 are 1.57 pg/mL and 17.96 pg/mL, respectively. While the LOD values are similar, the intensity uniformity within the spots is greatly improved compared to that obtained without the use of the microfluidic system. Coefficients of variance (CVs) were calculated for the IL-3 spots at the lowest four concentrations in order to quantitatively compare the intensity uniformity (Figure 7). It is obvious that CVs decreased dramatically by using the microfluidic system for sample handling. For example, the CV decreased from 44.91% to 10.54% for the microspots that were assayed at the lowest IL3 concentration. The improvement in reproducibility is attributed to three factors. First, the non-contact nature of the microfluidic method compared to manual injection removes the possibility of array damage through physical contact with pipette tips commonly used for injection/removal of assay liquids during sample introduction and washing steps. Second, no air bubbles are present with the automatic microfluidic system. Bubbles, when present over the assay during any step, can greatly decrease the incubation efficiency and result in variance from spot to spot. Because the liquid flow is continuous in the microfluidic system, no bubbles are introduced during the assay process. Using the manual approach, the small volume of the well and the pipette tip often results in generation of bubbles during liquid injection. Third, the wash steps are more thorough within the microfluidic system compared to the manual system. Note that we flowed ~3mL wash buffer for ~30s to remove the unbound molecules using the automatic microfluidic system while, with a manual assay, we only pipette ~15 μL wash buffer three times into the well.
Figure 7.

We directly compare our results to those obtained for the same analytes with commercially available approaches. We are especially concerned with the ability to achieve assay results at the point-of-care that are faster, more sensitive, and less invasive (in terms of sample volume required) than laboratory-based approaches. The traditional microplate ELISA remains one of the most popular assay platforms used for cancer biomarker analysis, which has no multiplexing capability with a detection limit of on the order of 10–50 pg/mL for an overnight sample incubation. Recently a multiplexed, sandwich assay has been developed which is based on a bead-based approach (Luminex). While the sensitivity of this bead-based approach approaches 2–10 pg/mL, it typically uses much larger volumes of serum (>50 μl) than available in a pin prick, requires an expensive non-automated detection instrument, provides limited multiplexing capability, and a more lengthy assay protocol than microarrays. The PC microfluidic approach, however, optimally use 10 μl of sample to detect analytes at 1 pg/ml concentration, lacks assay cross-reactivity, and demonstrates the fastest binding kinetics (<4 hours incubation), while providing equivalent or lower limits of detection than bead-based assays due to the ability to wash away unbound material. Our new platform enables more rapid, high sensitivity detection and a simplified output that will be readily usable by a clinician.
4. Conclusion
In this work, we designed, fabricated, and demonstrated a microfluidic cartridge and assay automation system for performing multiplexed and high sensitivity fluorescent microarray sandwich assays that incorporate photonic crystal fluorescence enhancement. We have demonstrated 80 fM detection limits for two representative biomarkers (TNF- and IL3) from a 10 μl sample volume with small intensity variances across the replicate spots and detection limits that are identical to those obtained with the PC utilized in a highly manual assay protocol. The chip is held in a simple, inexpensive, and room-temperature-assembled cartridge that interfaces with an assay automation instrument. The assay instrument is comprised of a computer-controlled pressure manifold and three-way valve that automates the washing, labeling, and drying steps of the immunoassay. The cartridge interfaces with the assay automation instrument through a simple and leak-free press-fit seal that introduces no liquid dead volume. The described platform integrates many features that are required for a practical point-of-care immunoassay targeted at liquid-biopsy diagnostics: it is automatic and easy-to-use; it can perform multiple assays simultaneously; it requires only a small volume of test sample and reagents (~500 μL); and high analytic sensitivity and high selectivity are achieved. Moreover, the use of the automation system greatly reduces spot-to-spot coefficients of variability compared to manual handling.
We demonstrated 80 fM detection limits for two representative biomarkers (TNF-α and IL3) from a 10 ml sample volume
We designed, fabricated, and demonstrated a microfluidic cartridge and assay automation system
Fluorescence intensity was enhanced 20 times by photonic crystal surface that is embedded in the microfluidic chip.
We achieved low sample consumption and small intensity variances across the replicate spots using the microfluidic system combined with photonic crystal structure.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM086382A, R33CA177446, R01GM108584) and the National Science Foundation (CBET 07-54122). Any opinions, findings, conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Institutes of Health or the National Science Foundation. The authors thank Scott McDonald and David Switzer at machine shop of ECE department for their help making the holding fixture.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, Dean EJ, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nature Medicine. 2002;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- 2.Wong J, Sibani S, Lokko NN, LaBaer J, Anderson KS. Rapid detection of antibodies in sera using multiplexed self-assembling bead arrays. Journal of Immunological Methods. 2009 Oct 31;350:171–182. doi: 10.1016/j.jim.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Debnath M. Springer Science + Business Media, editor. Molecular Diagnostics: Promises and Possibilities. 2010. Segments of Molecular Diagnostics - Market Place; pp. 503–513. [Google Scholar]
- 4.Ward DG, Suggett N, Cheng Y, Wei W, Johnson H, Billingham LJ, et al. Identification of serum biomarkers for colon cancer by proteomic analysis. British Journal of Cancer. 2006 Jun 19;94:1898–1905. doi: 10.1038/sj.bjc.6603188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nam MJ, Madoz-Gurpide J, Wang H, Lescure P, Schmalbach CE, Zhao R, et al. Molecular profiling of the immune response in colon cancer using protein microarrays: Occurrence of autoantibodies to ubiquitin C-terminal hydrolase L3. Proteomics. 2003 Nov;3:2108–2115. doi: 10.1002/pmic.200300594. [DOI] [PubMed] [Google Scholar]
- 6.Tannapfel A, Anhalt K, Hausermann P, Sommerer F, Benicke M, Uhlmann D, et al. Identification of novel proteins associated with hepatocellular carcinomas using protein microarrays. Journal of Pathology. 2003 Oct;201:238–249. doi: 10.1002/path.1420. [DOI] [PubMed] [Google Scholar]
- 7.Patz EF, Campa MJ, Gottlin EB, Kusmartseva I, Guan XR, Herndon JE. Panel of serum biomarkers for the diagnosis of lung cancer. Journal of Clinical Oncology. 2007 Dec 10;25:5578–5583. doi: 10.1200/JCO.2007.13.5392. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Bast RC, Yu YH, Li JN, Sokoll LJ, Rai AJ, et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Research. 2004 Aug 15;64:5882–5890. doi: 10.1158/0008-5472.CAN-04-0746. [DOI] [PubMed] [Google Scholar]
- 9.Luo LY, Katsaros D, Scorilas A, Fracchioli S, Bellino R, van Gramberen M, et al. The serum concentration of human kallikrein 10 represents a novel biomarker for ovarian cancer diagnosis and prognosis. Cancer Research. 2003 Feb 15;63:807–811. [PubMed] [Google Scholar]
- 10.Diamandis EP, Scorilas A, Fracchioli S, van Gramberen M, de Bruijn H, Henrik A, et al. Human kallikrein 6 (hK6): A new potential serum biomarker for diagnosis and prognosis of ovarian carcinoma. Journal of Clinical Oncology. 2003 Mar 15;21:1035–1043. doi: 10.1200/JCO.2003.02.022. [DOI] [PubMed] [Google Scholar]
- 11.Diamandis EP, Borgono CA, Scorilas A, Yousef GM, Harbeck N, Dorn J, et al. Immunofluorometric quantification of human kallikrein 5 expression in ovarian cancer cytosols and its association with unfavorable patient prognosis. Tumor Biology. 2003;24:299–309. doi: 10.1159/000076462. [DOI] [PubMed] [Google Scholar]
- 12.Maurya P, Meleady P, Dowling P, Clynes M. Proteomic approaches for serum biomarker discovery in cancer. Anticancer Research. 2007 May-Jun;27:1247–1255. [PubMed] [Google Scholar]
- 13.Vasan RS. Biomarkers of cardiovascular disease - Molecular basis and practical considerations. Circulation. 2006 May 16;113:2335–2362. doi: 10.1161/CIRCULATIONAHA.104.482570. [DOI] [PubMed] [Google Scholar]
- 14.Wang TJ, Gona P, Larson MG, Tofler GH, Levy D, Newton-Cheh C, et al. Multiple biomarkers for the prediction of first major cardiovascular events and death. New England Journal of Medicine. 2006 Dec 21;355:2631–2639. doi: 10.1056/NEJMoa055373. [DOI] [PubMed] [Google Scholar]
- 15.Jain KK. The Handbook of Biomarkers: Springer Science + Business Media. Humana Press; 2010. [Google Scholar]
- 16.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Science Translational Medicine. 2014 Feb 19;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawson SJ, Tsui DWY, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. New England Journal of Medicine. 2013;368:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 18.Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014 doi: 10.1038/nm.3519. vol. advance online publication, 04/06/online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khoo SK, Petillo D, Kang UJ, Resau JH, Berryhill B, Linder J, et al. Plasma-Based Circulating MicroRNA Biomarkers for Parkinson’s Disease. Journal of Parkinson’s Disease. 2012 Jan 01;2:321–331. doi: 10.3233/JPD-012144. [DOI] [PubMed] [Google Scholar]
- 20.Jeffrey SS. Cancer biomarker profiling with microRNAs. Nat Biotech. 2008;26:400–401. doi: 10.1038/nbt0408-400. 04//print. [DOI] [PubMed] [Google Scholar]
- 21.Sita-Lumsden A, Dart DA, Waxman J, Bevan CL. Circulating microRNAs as potential new biomarkers for prostate cancer. Br J Cancer. 2013;108:1925–1930. doi: 10.1038/bjc.2013.192. 05/28/print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neagu M, Constantin C, Tanase C, Boda D. Patented biomarker panels in early detection of cancer. Recent Patents on Biomarkers. 2011;1:10–24. [Google Scholar]
- 23.Gonzalez RM, Daly DS, Tan R, Marks JR, Zangar RC. Plasma biomarker profiles differ depending on breast cancer subtype but RANTES in consistently increased. Cancer Epidemiology, Biomarkers, and Prevention. 2011 doi: 10.1158/1055-9965.EPI-10-1248. vol. Under Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zangar RC, Varnum SM, Bollinger N. Studying cellular processes and detecting disease with protein microarrays. Drug Metabolism Reviews. 2005;37:473–487. doi: 10.1080/03602530500205309. [DOI] [PubMed] [Google Scholar]
- 25.Fan R, Vermesh O, Srivastava A, Yen BKH, Qin LD, Ahmad H, et al. Integrated barcode chips for rapid, multiplexed analysis of proteins in microliter quantities of blood. Nature Biotechnology. 2008 Dec;26:1373–1378. doi: 10.1038/nbt.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang CS, George S, Lu M, Chaudhery V, Tan R, Zangar RC, et al. Application of Photonic Crystal Enhanced Fluorescence to Cancer Biomarker Microarrays. Analytical Chemistry. 2011 Feb 15;83:1425–1430. doi: 10.1021/ac102989n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.George S, Chaudhery V, Lu M, Takagi M, Amro N, Pokhriyal A, et al. Sensitive detection of protein and miRNA cancer biomarkers using silicon-based photonic crystals and a resonance coupling laser scanning platform. Lab on a Chip. 2013;13:4053–4064. doi: 10.1039/c3lc50579k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ganesh N, Zhang W, Mathias PC, Chow E, Soares JANT, Malyarchuk V, et al. Enhanced fluorescence emission from quantum dots on a photonic crystal surface. Nat Nano. 2:515–520. doi: 10.1038/nnano.2007.216. 08//print 2007. [DOI] [PubMed] [Google Scholar]
- 29.Pokhriyal A, Lu M, Chaudhery V, Huang CS, Schulz S, Cunningham BT. Photonic crystal enhanced fluorescence using a quartz substrate to reduce limits of detection. Optics Express. 2010 Nov 22;18:24793–24808. doi: 10.1364/OE.18.024793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melchels FPW, Feijen J, Grijpma DW. A review on stereolithography and its applications in biomedical engineering. Biomaterials. 2010 Aug;31:6121–6130. doi: 10.1016/j.biomaterials.2010.04.050. [DOI] [PubMed] [Google Scholar]
- 31.Zhang AP, Qu X, Soman P, Hribar KC, Lee JW, Chen S, et al. Rapid Fabrication of Complex 3D Extracellular Microenvironments by Dynamic Optical Projection Stereolithography. Advanced Materials. 2012;24:4266–4270. doi: 10.1002/adma.201202024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Au AK, Lee W, Folch A. Mail-order microfluidics: evaluation of stereolithography for the production of microfluidic devices. Lab on a Chip. 2014;14:1294–1301. doi: 10.1039/c3lc51360b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choudhury IA, Shirley S. Laser cutting of polymeric materials: An experimental investigation. Optics & Laser Technology. 2010 Apr;42:503–508. [Google Scholar]
- 34.Eltawahni HA, Hagino M, Benyounis KY, Inoue T, Olabi AG. Effect of CO2 laser cutting process parameters on edge quality and operating cost of AISI316L. Optics & Laser Technology. 2012 Jun;44:1068–1082. [Google Scholar]
- 35.Nie J, Liang Y, Zhang Y, Le S, Li D, Zhang S. One-step patterning of hollow microstructures in paper by laser cutting to create microfluidic analytical devices. Analyst. 2013;138:671–676. doi: 10.1039/c2an36219h. [DOI] [PubMed] [Google Scholar]
- 36.Wilhelm E, Neumann C, Duttenhofer T, Pires L, Rapp BE. Connecting microfluidic chips using a chemically inert, reversible, multichannel chip-to-world-interface. Lab on a Chip. 2013;13:4343–4351. doi: 10.1039/c3lc50861g. [DOI] [PubMed] [Google Scholar]
- 37.Dorvel B, Reddy B, Block I, Mathias P, Clare SE, Cunningham B, et al. Vapor-Phase Deposition of Monofunctional Alkoxysilanes for Sub-Nanometer-Level Biointerfacing on Silicon Oxide Surfaces. Advanced Functional Materials. 2009;20:87–95. [Google Scholar]
- 38.Zhu H, Klemic JF, Chang S, Bertone P, Casamayor A, Klemic KG, et al. Analysis of yeast protein kinases using protein chips. Nature genetics. 2000;26:283–290. doi: 10.1038/81576. [DOI] [PubMed] [Google Scholar]
- 39.Borras TGL, Tietz J. Generic Approach for the Generation of Stable Humanized Single-chain Fv Fragments from Rabbit Monoclonal Antibodies. Journal of Biological Chemistry. 2010;285:12. doi: 10.1074/jbc.M109.072876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Block ID, Mathias PC, Ganesh N, Jones SI, Dorvel BR, Chaudhery V, et al. A detection instrument for enhanced-fluorescence and label-free imaging on photonic crystal surfaces. Optics Express. 2009 Jul 20;17:13222–13235. doi: 10.1364/oe.17.013222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaudhery V, Lu M, Huang C-S, Polans J, Tan R, Zangar RC, et al. A line-scanning detection instrument for photonics crystal enhanced fluorescence. Optics Letters. 2012 doi: 10.1364/OL.37.002565. vol. In Print. [DOI] [PubMed] [Google Scholar]
- 42.Chaudhery V, Lu M, Huang C, George S, Cunningham B. Photobleaching on Photonic Crystal Enhanced Fluorescence Surfaces. Journal of Fluorescence. 2011 Mar 01;21:707–714. doi: 10.1007/s10895-010-0760-8. [DOI] [PubMed] [Google Scholar]
- 43.http://static-content.springer.com/esm/art%3A10.1007%2Fs00125-015-3535-6/MediaObjects/125_2015_3535_MOESM2_ESM.pdf
- 44.Talvitie M. Evaluating a TNF-α immunoassay using EnSpire AlphaPLUS: ELISA and AlphaLISA technologies. 2010 Available: http://www.perkinelmer.co.jp/Portals/0/resource/tech/tech_ls/protocol_collection/APP_EnSpire_Evaluating_TNFa_Immunoassay.pdf.