

Abstract
Increasing lines of evidence have demonstrated that the development of higher rates of non-diabetic glomerulosclerosis (GS) in African Americans can be attributed to two coding sequence variants (G1 and G2) in the Apolipoprotein L1 (APOL) gene. Recent studies indicate that the gene products of these APOL1 risk variants have augmented toxicity to kidney cells. However, the biological characteristics of APOL1 and its risk variants are not well elucidated. The APOL1 protein can be divided into several functional domains, including signal peptide (SP), pore forming domain (PFD), membrane address domain (MAD), and SRA-interacting domain. To investigate the relative contribution of each domain to cell injury, we constructed a serial expression vectors to delete or express each domain. These vectors were transfected into the human embryonic kidney cell line 293T, and then compared the cytotoxicity. In addition, we conducted studies in which APOL1 wild type (G0) was co-transfected in combination with G1 or G2 to see whether G0 could counteract the toxicity of the risk variants. The results showed that deleting the SP did not abolish the toxicity of APOL1, though deletion of 26 amino acid residues of the mature peptide at the N-terminal partially decreased the toxicity. Deleting PFD or MAD or SRA-interacting domain abolished toxicity, while, overexpressing each domain alone could not cause toxicity to the host cells. Deletion of the G2 sites while retaining G1 sites in the risk state resulted in persistent toxicity. Either deletion or exchanging the BH3 domain in the PFD led to complete loss of the toxicity in this experimental platform. Adding G0 to either G1 or G2 did not attenuate the toxicity of the either moiety. These results indicate that the integrity of the mature APOL1 protein is indispensable for its toxicity. Our study not only reveals the contribution of each domain of the APOL1 protein to cell injury, but also highlights some potential suggested targets for drug design to prevent or treat APOL1-associated nephropathy.
Keywords: APOL1, risk variants, domain, integrity, toxicity
1. Introduction
Cumulative evidence over many years have demonstrated that African Americans (AAs) develop 4–5 fold higher rates of diverse forms of progressive kidney disease, including focal segmental glomerulosclerosis (FSGS), HIVAN, and hypertension-attributed end stage kidney disease (ESKD), compared with European Americans (EAs) (Tzur et al., 2010; Kopp et al., 2011; Quaggin et al., 2011; Genovese et al., 2013; Kasembeli et al., 2015). In several forms of nephropathy, this disparity reaches a greater than 10-fold difference. This overwhelming population health disparity is mainly attributed to two coding sequence variants (G1 and G2) in APOL1 gene (Genovese et al., 2010; Friedman et al., 2011; Foster et al., 2013); however, detailed underlying mechanisms are still poorly understood.
In previous studies, we observed that the APOL1 risk variants (Vs) G1 and G2 confer cytotoxicity to kidney cells such as podocytes (Lan et al., 2014, 2015). Consistent with these observations, Park et al (2014)., reported that podocyte specific expression of APOL1 in G1 and G2 transgenic mice developed severe albuminuria and glomerulosclerosis, while wild type mice expressing G0 APOL1 showed minimal renal abnormalities. Similarly, Olabisi et al (2014)., and Heneghan et al (2014 0., observed that APOL1Vs caused toxicity to Zebrafish and Xenopus Laevis oocytes (). The forgoing studies demonstrate that APOL1Vs confer toxicity to the kidney and its functional cells. However, knowledge of the biological function of APOL1 proteins, especially its risk variants, is not clearly understood.
According to the presumed function, the full length APOL1 wild type protein G0 can be divided into 4 domains: signal peptide (SP, 1-27 AA), pore forming domain (PFD, 60-237 AA), membrane address domain (MAD, 238-303 AA), and SRA-interacting domain (339-398) (Pérez-Morga et al., 2005; Lecordier et al., 2009). Therefore, we sought to advance our understanding of the potential mechanisms of cytotoxicity by determining the relative contribution of these domains to cytotoxicity.
2. Materials and Methods
2.1. Cell Culture
The 293T cell line was purchased from ATCC, and was cultured at 37°C in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1 X penicillin-streptomycin, and 1 mM L-glutamine.
2.2. Trypan blue staining
Cells were rinsed with PBS to remove the floating cells and cell debris, detached with Accutase (Innovative Cell Technologies, San Diego, CA), and then labeled with 0.2% Trypan blue and counted under a light microscope. The unstained cells (white cells) were counted as live cells, while the stained (blue) cells were counted as dead cells.
2.3. Propidium iodide (PI) and Hoechst staining
PI and Hoechst staining were performed as previously described (Vashistha et al., 2009; Lan et al., 2014). Briefly, after each treatment condition of interest, the culture medium was removed from the cells and fresh medium containing Hoechst 33342 (10 μg/ml) was added. Cells were subsequently incubated for 10 min at 37°C. Then, PI solution was added and culture dishes were kept on ice for 7 min. Cell images were recorded with a ZEISS microscope (Carl Zeiss Micro Imaging GmbH, Jena, Germany) equipped with a digital imaging system.
2.4. Plasmid preparation and transfection
Lentiviral vector LG12 was used as the backbone for all the expression vectors in this study, and CMV promoter was used to drive the expression of targeting gene. Plasmid was amplified in E. coli strain DH5α, and was extracted by using QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA), following the manufacturer’s instructions. Host cell 293T was prepared on 35 mm culture dishes, and was transfected with plasmid (400 ng/dish) by using Effectene Transfection Reagent (Qiagen), following the manufacturer’s instructions.
2.5. Statistical analyses
Data are presented as means ± standard deviation (SD) unless otherwise noted. All experiments were conducted and repeated at least three times, either in duplicate or triplicate for each assay. All data were evaluated statistically by analysis of variance (ANOVA), followed by Newman-Keuls multiple comparison tests using Prism 4.0, GraphPad software. In the case of single mean comparison, data were analyzed by student t-test. P values < 0.05 were regarded as statistically significant.
3. Results and Discussion
3.1. Different characters of APOL1 G0 and Vs
In our previous report, we demonstrated that APOL1Vs displayed stronger cytotoxicity to podocytes when compared with APOL1G0 (Lan et al., 2014, 2015). A similar differential cytotoxicity was reported for HEK293 cells (Khatua et al., 2015; Nichols et al., 2015). In the current study, we also used HEK293T cells. To better understand the effect of G1 and G2, we combined both variants into a novel artificial variant G1G2. Our results showed that after 24 h, 293T cells transfected with G0 or Vs all showed a higher percentage of cells with a swollen phenotype resulting in detachment, compared to vector alone (Figure 1A and C). The cell injury phenotype was overt in G1 and G2 expressing cells, and still more pronounced for the G1G2 expressing cells.
Figure 1. Effect of APOL1G0 and variants on 293T cells.


Kidney embryonic cell line 293T was transfected with LG12-APOL1G0 or Vs for 24 h. (A) Cells were stained with Hoechst33342, and then were observed under phase contrast microscopy. Swollen cells were indicated with red arrows. Scale bar, 100 μm. (B) Cells were subjected to PI staining, and the PI positive cells (white arrows) were counted under a fluorescent microscope. (C) Statistical calculation of the swollen cells in (A). (D) Statistical calculation of the PI positive cells in (B). (E) Cells were detached with accutase and then subjected to Trypan Blue staining, and the cell viabilities were calculated. * p < 0.05 when compared with vector; # p < 0.05 when compared with APOL1G0.
We then performed PI staining to determine the percentage of damaged cells. APOL1G0 expressing cells displayed increased numbers of PI positive cells when compared to vector expressing cells. However, APOL1Vs expressing cells demonstrated greater numbers of PI positive cells when compared to APOL1G0 cells (Figure 1B and D). Among these, the double mutant G1G2 cells displayed the highest number of PI positive cells.
To determine the viability of cells, Trypan blue staining of different APOL1 expressing cells was carried out. Results were consistent with other injury phenotypes including swollen cells and PI positive cells amongst different groups: APOL1G1G2 induced the highest cell death ratio, followed by and APOL1G2 and G1, whereas, lowest cell death ratio was displayed by APOL1G0 (Figure 1E).
These results clearly demonstrate that compared with APOL1G0, APOL1Vs confer more severe cell injury to 293T cells. Since APOL1G1G2 exhibits the same properties with G1 and G2, but displayed higher greater toxicity to the host cells, we used these cells to evaluate the role of different domains in the induction of cellular injury.
3.2. Effect of different functional domains on the cytotoxicity
To determine the role of each domain in APOL1 protein, we first cloned the CDS of full-length or truncated APOL1G0 or APOL1Vs into Lentiviral vector LG12 to generate serial expression vectors (Figure 2A). We then transfected these vectors into 293T cells, and evaluated the cell injury after 24 h by using PI staining (Figure 2B).
Figure 2. Effect of APOL1 different domains on 293T cells.


(A) Schematic figure of the expression vector for the APOL1 gene domains.
(B) 293T cells were transfected with the vectors in (A) for 24 h, followed by PI staining. PI positive cells were counted under a fluorescent microscope. * p < 0.05 when compared with full-length vector #1.
APOL1 is produced in liver, pancreas, brain, and kidney, and circulates in the plasma (Duchateau et al., 1997, 2000, 2001). This demonstrates that APOL1 is a secretory protein, with a signal peptide to direct the secretion. Interestingly, only APOL1 has a signal peptide in the APOL1-6 family. Although our previous study showed that endocytosed exogenous APOL1Vs could cause podocyte injury (Lan et al., 2014, 2015), it is not clear whether endogenous APOL1G0 and Vs contribute to toxicity of host cells before secretion. To address this question, we constructed a vector to encode the mature APOL1G0/G1G2 (#2 in Figure 2), and overexpressed these mature proteins in 293T cells with or without deletion of signal peptide. No different effect of the absence or presence of the signal peptide was observed in APOL1G0 or G1G2-induced cellular injury (Figure 2B), indicating that the signal peptide is dispensable for its toxicity.
Besides the full length mature APOL1, a shorter splice variant product with a deletion of 26 amino acid residues at the N-terminal exists in the liver (Duchateau-1997). The molecular weight of the full length APOL1 is 42 kDa, while this truncated version has a MW of 39 kDa. We compared the toxicity of these two peptides in the G0 allele state, and found that there is no significant difference (data not shown). In contrast, in the G1G2 state, the 39kDa variant exhibited lower toxicity to the host cells (#3 in Figure 2). These results indicate that deletion of the N-terminal could partially decrease the toxicity of APOLVs, but that the 39 kDa APOL1 is also toxic to kidney cells.
Since the pore forming domain (PFD) is supposed to be the main domain for lysis of trypanosomes (Pérez-Morga et al., 2005; Lecordier et al., 2009), we overexpressed only this domain in 293T cells (#8 in Figure 2). After 24 h, we did not observe any alterations in number of swollen cells or PI positive cells when compared with vector alone (Figure 2B). Similarly, disruption of the PFD abolished the increased cell injury to the host cells (#4 in Figure 2). These results clearly demonstrated that the PFD is required but not sufficient for APOL1 mediated toxicity.
The membrane-addressing domain (MAD) is located in the middle of APOL1 protein. A newly discovered novel variant G3 was found in some American Africans, but does not show association with kidney disease (Palmer et al., 2015). To investigate the importance of MAD for the toxicity, we truncated this domain, and expressed the remaining part in 293T cells. As shown in Figure 2, deletion of MAD abolished the toxicity of G1G2 (#12 in Figure 2), indicating that integrity of this domain contributes to maintain the cell injury activity of APOL1 protein. However, overexpression of MAD alone did not cause 293T cell injury (#7 in Figure 2).
The Trypanosoma brucei subspecies rhodesiense resists the lytic activity of APOL1G0 by developing a Serum Resistance-Associated (SRA) protein which interacts with the C-terminal helix of APOL1 (Gibson et al., 2005; Shiflett et al., 2007; Uzureau et al., 2013). To test the role of the SRA-interacting domain for the cytotoxicity, we deleted this domain and expressed the remaining part of APOL1 protein in 293T cells (#9 in Figure 2). As shown in Figure 2B, deletion of this domain completely abolished the toxicity, indicating its critical requirement for the toxicity function of APOL1 protein. To further refine this observation, we deleted a shorter sequence of the C-terminus, deleting only the parts harboring the mutation/deletion sites for G1 and G2 (#10 in Figure 2). The resulting truncated APOL1 also lost its toxicity. These findings are consistent with those previously reported by another group (Nichols et al., 2014).
Since all the differences between G0 and the risk variants are located at C-terminus, we wondered whether the C-terminus is the “toxic domain”. To test this hypothesis, we expressed the SRA-binding domain alone or together with MAD in 293T cells (#5 and #6 in Figure 2). To our surprise, none of these constructs showed toxicity to the host cells. These results exclude the hypothesis that the C-terminus is sufficient for toxicity. To further test the role of the C-terminus, we deleted 11 amino acid residues of the C-terminus which removes the G2 site but retains the G1 sites (#11 in Figure 2), and then transfected this truncated construct into 293T cells. Results showed that the toxicity is similar with G1 alone and less than G1G2.
In the middle of pore forming domain, there is a BH3 domain consisting of 9 amino acid residues. It has been reported that this BH3 domain is indispensable for the autophagic cell death of colorectal cancer cell (Wan et al., 2008; Zhaorigetu et al., 2008). However, a recent study showed that replacing the BH3 domain with 9 alanine residues (9A) does not reduce its toxicity to oocytes (Heneghan et al., 2014). To confirm the role of the BH3 domain, we first deleted this domain and transfected the modified APOL1 construct into 293T cells (#13 in Figure 2). Results showed that the BH3 deficient APOL1 could not cause obvious injury to the host cells. We then replaced this domain with 9A (#14 in Figure 2), and found that it also abolished cytotoxicity. These results clearly demonstrated that the BH3 domain is indispensable for the toxicity of APOL1 in the experimental platform examined.
3.3. APOL1G0 cannot attenuate the toxicity of variants
Clinical studies have demonstrated that APOL1- associations are overwhelmingly more pronounced when the patients have two risk alleles (G1/G1 or G1/G2 or G2/G2), while only one risk allele confers little or no kidney disease risk (Reeves-Daniel et al., 2011; Atta et al., 2012; Foster et al., 2013). This phenomenon suggests that APOL1 G0 might have the potential to suppress the toxicity of G1 or G2. To test this hypothesis, we overexpressed both APOL1G0 together with G1 or G2 in 293T cells in 35 mm dishes, and then examined the changes of the cytotoxicity through PI staining after 24 h. Results showed that the PI positive cell ratios of 300 ng APOL1G0, G1, and G2 alone were 0.65%, 2.02%, and 2.19%, respectively; while 150 ng APOL1G0 plus 150 ng APOL1G1 or G2 caused 1.90% and 2.08% PI positive cells, respectively, and none was significantly decreased when compared with G1 or G2 alone (Figure 3). However, 300 ng APOL1G0 plus 300 ng APOL1G1 or G2 dramatically increased the PI positive cell ratios, reaching 4.12% and 4.49%, respectively. In addition, we also counted the swollen cells caused by these plasmids alone or combination, and found the same trend (data not shown). These results indicate that APOL1G0 does not directly itself counteract the toxicity of G1 or G2 as a mechanism for the two-risk allele required.
Figure 3. Effect of APOL1G0 on the toxicity of APOL1G1 and G2.
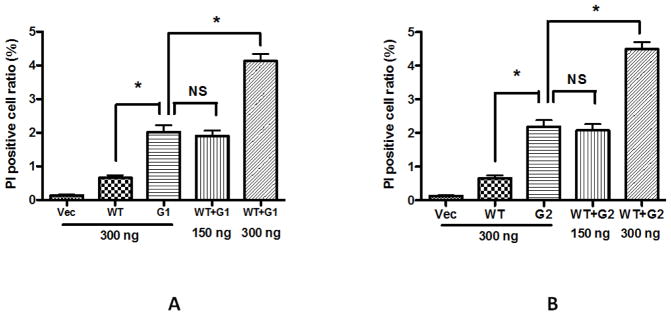
293 cells were transfected with LG12-APOL1G0, G1, G2 alone, or a combination of G0 and G1, or a combination of G0 and G2. After 24 h, cells were stained with PI, and the positive cells were counted. * p < 0.05 when compared with G1 or G2 alone; NS, not statistically different.
APOL1 gene is expressed in only in primates (Monajemi et al., 2002; Poelvoorde et al., 2004), thus suggesting that this gene is dispensable in majority of species. Since its mutation makes a specific population vulnerable not only for development but also for progression of kidney diseases, it suggests that APOL1G0 must be serving some protecting function. However, in the present study, presence of G0 in G1 or G2 expressing kidney cells did not provide any protection. Nonetheless, it is possible that the degree of injury conferred by overexpressing G1 and G2 cells was overwhelming and was beyond the protective potential of APOL1G0. Alternatively, APOL1G0 may have potential to provide protection only to specific type of injuries. This notion is supported by epidemiologic studies in HIVAN, which is almost nonexistent in Caucasians (Yalavarthy et al., 2008).
Several in vitro studies including the present one, demonstrated that even over expression of APOL1G0 carries toxic effects (Wan et al., 2008; Zhaorigetu et al., 2008; Lan et al., 2014). These studies have also demonstrated that interferon-γ enhances APOL1 expression, thus, it is likely that conditions associated with high interferon-γ states such as viral infections would predispose podocytes to injury. Interferon-γ has been known to induce podocyte injury both in vitro and in vivo (Coers et al., 1995; Petrovic-Djergovic et al, 2015). However, APOL1 expression was not evaluated in these studies. It will be interesting to evaluate the role of APOL1 in interferon-γ mediated podocyte injury in future studies.
Although epidemiologic studies indicate lower rates of cardiovascular morbidities in APOL1G1/G2 patients (Langefeld et al., 2015), smooth muscle cells in kidneys displayed higher expression of APOL1 (Madhavan et al., 2011). Thus, it appears that higher APOL1 expression in certain cells may be a marker of their well-being in adverse milieu. Alternatively, mutated APOL1 may be stable in some cells, such as smooth muscle cells, but unstable in other cells, such as podocytes. The mutated APOL1 may be degraded faster and thus resulting in low expression of APOL1.
In vitro studies, higher expression of APOL1 by smooth muscle cells in HIV milieu was associated with secretion of APOL1 (Lan et al., 2015). In our studies, uptake of APOL1 by podocytes was also associated with podocyte injury (Lan et al., 2014, 2015). Therefore, at present it is not clear whether higher expression of APOL1 inflicts de novo podocyte injury or the secretory component of the overexpressing APOL1 cells are contributing to podocyte injury. However, in killing of Tryoanosome Cruzi, secretory component is playing a predominant role (Gibson, 2005).
In summary, we found that the signal peptide is dispensable for the toxicity of both APOL1 wild type and variants; while any of PFD, MAD, and SRA-interacting domain is indispensable for the toxicity. Comparatively, deletion of N-terminal causes lesser loss of the toxicity.
Highlights.
APOLl variants play a role in the development of kidney diseases in African Americans.
APOL1 protein is composed several functional domains.
Deleting signal peptide did not abolish toxicity of APOL1
Deleting pore-forming or membrane address domain or SRA-interacting domain abolished the toxicity of APOL1.
Over-expressing of any domain did not contribute to APOL1 toxicity
Either deletion or exchanging the BH3 domain in the pore forming domain led to complete loss of the toxicity.
Integrity of the mature APOL1 protein is indispensable for its toxicity.
Acknowledgments
This work was supported by grants RO1DK 098074, RO1DK084910, RO1 DK083931 (PCS) from National Institutes of Health, Bethesda, MD. KS was supported by grants from the Ernest and Bonnie Beutler Grant Program at Rambam Medical Center, the Binational Science Foundation, and the Israel Science Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deok Park AS, Kopp JB, Winkler CA, Pullman JA, Miner JH, Susztak K. Podocyte Specific Expression of Mutant APOL1 Induces Albuminuria and Global Sclerosis in Mice. Abstract of 2014 American Society of Nephropathy Annual Conference; 2014. p. 92A. [Google Scholar]
- 2.Atta MG, Estrella MM, Kuperman M, Foy MC, Fine DM, Racusen LC, Lucas GM, Nelson GW, Warner AC, Winkler CA, Kopp JB. HIV-associated nephropathy patients with and without apolipoprotein L1 gene variants have similar clinical and pathological characteristics. Kidney Int. 2012 Aug;82(3):338–43. doi: 10.1038/ki.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coers W, Vos JT, Van der Meide PH, Van der Horst ML, Huitema S, Weening JJ. Interferon-gamma (IFN-gamma) and IL-4 expressed during mercury-induced membranous nephropathy are toxic for cultured podocytes. Clin Exp Immunol. 1995 Nov;102(2):297–307. doi: 10.1111/j.1365-2249.1995.tb03781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duchateau PN, Pullinger CR, Orellana RE, Kunitake ST, Naya-Vigne J, O’Connor PM, Malloy MJ, Kane JP. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J Biol Chem. 1997 Oct 10;272(41):25576–82. doi: 10.1074/jbc.272.41.25576. [DOI] [PubMed] [Google Scholar]
- 5.Duchateau PN, Movsesyan I, Yamashita S, Sakai N, Hirano K, Schoenhaus SA, O’Connor-Kearns PM, Spencer SJ, Jaffe RB, Redberg RF, Ishida BY, Matsuzawa Y, Kane JP, Malloy MJ. Plasma apolipoprotein L concentrations correlate with plasma triglycerides and cholesterol levels in normolipidemic, hyperlipidemic, and diabetic subjects. J Lipid Res. 2000 Aug;41(8):1231–6. [PubMed] [Google Scholar]
- 6.Duchateau PN, Pullinger CR, Cho MH, Eng C, Kane JP. Apolipoprotein L gene family: tissue-specific expression, splicing, promoter regions; discovery of a new gene. J Lipid Res. 2001 Apr;42(4):620–30. [PubMed] [Google Scholar]
- 7.Foster MC, Coresh J, Fornage M, Astor BC, Grams M, Franceschini N, Kao WL. APOL1 Variants Associate with Increased Risk of CKD among African Americans. J Am Soc Nephrol. 2013;24:1484–1491. doi: 10.1681/ASN.2013010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR. Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol. 2011;22:2098–2105. doi: 10.1681/ASN.2011050519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genovese G, Friedman DJ, Pollak MR. APOL1 variants and kidney disease in people of recent African ancestry. Nat Rev Nephrol. 2013;9:240–244. doi: 10.1038/nrneph.2013.34. [DOI] [PubMed] [Google Scholar]
- 10.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR. Association of Trypanolytic ApoL1 Variants with Kidney Disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibson WC. The SRA gene: the key to understanding the nature of Trypanosoma brucei rhodesiense. Parasitology. 2005;131(Pt 2):143–150. doi: 10.1017/s0031182005007560. [DOI] [PubMed] [Google Scholar]
- 12.Heneghan JH, Alper SL, Pollak MA. BH3 Domain-Independent Apolipoprotein L1 Toxicity Rescued by Bcl2 Pro-Survival Proteins. Abstract of 2014 American Society of Nephropathy Annual Conference; 2014. p. 451A. [Google Scholar]
- 13.Kasembeli AN, Duarte R, Ramsay M, Mosiane P, Dickens C, Dix-Peek T, Limou S, Sezgin E, Nelson GW, Fogo AB, Goetsch S, Kopp JB, Winkler CA, Naicker S. APOL1 Risk Variants Are Strongly Associated with HIV-Associated Nephropathy in Black South Africans. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2014050469. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khatua AK, Cheatham AM, Kruzel ED, Singhal PC, Skorecki K, Popik W. Exon 4 Encoded Sequence is a Major Determinant of Cytotoxicity of Apolipoprotein L1. Am J Physiol Cell Physiol. 2015 Apr 29; doi: 10.1152/ajpcell.00384.2014. ajpcell.00384.2014. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, Mokrzycki MH, Kimmel PL, Limou S, Ahuja TS, Berns JS, Fryc J, Simon EE, Smith MC, Trachtman H, Michel DM, Schelling JR, Vlahov D, Pollak M, Winkler CA. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22:2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lan X, Jhaveri A, Cheng K, Wen H, Saleem MA, Mathieson PW, Mikulak J, Aviram S, Malhotra A, Skorecki K, Singhal PC. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol. 2014;307:F326–36. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lan X, Wen H, Saleem MA, Mikulak J, Malhotra A, Skorecki K, Singhal PC. Vascular smooth muscle cells contribute to APOL1-induced podocyte injury in HIV milieu. Exp Mol Pathol. 2015 doi: 10.1016/j.yexmp.2015.03.020. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langefeld CD, Divers J, Pajewski NM, Hawfield AT, Reboussin DM, Bild DE, Kaysen GA, Kimmel PL, Raj DS, Ricardo AC, Wright JT, Jr, Sedor JR, Rocco MV, Freedman BI Systolic Blood Pressure Intervention Trial (SPRINT) Apolipoprotein L1 gene variants associate with prevalent kidney but not prevalent cardiovascular disease in the Systolic Blood Pressure Intervention Trial. Kidney Int. 2015 Jan;87(1):169–75. doi: 10.1038/ki.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lecordier L, Vanhollebeke B, Poelvoorde P, Tebabi P, Paturiaux-Hanocq F, Andris F, Lins L, Pays E. C-terminal mutants of apolipoprotein L-I efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathog. 2009;5(12):e1000685. doi: 10.1371/journal.ppat.1000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol. 2011;22:2119–2128. doi: 10.1681/ASN.2011010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monajemi H, Fontijn RD, Pannekoek H, Horrevoets AJ. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics. 2002 Apr;79(4):539–46. doi: 10.1006/geno.2002.6729. [DOI] [PubMed] [Google Scholar]
- 22.Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D’Agati V, Markowitz G, Kopp JB, Alper SL, Pollak MR, Friedman DJ. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2015 Feb;87(2):332–42. doi: 10.1038/ki.2014.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olabisi OA, Al-Romaih K, Henderson, Ritu Tomar R, Drummond IA, MacRae C, Pollak MA. Studying the Mechanism of Apolipoprotein L1 Nephropathy through Transgenic Zebrafish. Abstract of 2014 American Society of Nephropathy Annual Conference; 2014. p. 451A. [Google Scholar]
- 24.Palmer ND, Ng MC, Langefeld CD, Divers J, Lea JP, Okusa MD, Kimberly RP, Bowden DW, Freedman BI. Lack of Association of the APOL1 G3 Haplotype in African Americans with ESRD. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2014050444. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pérez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homblé F, Vanhamme L, Tebabi P, Pays A, Poelvoorde P, Jacquet A, Brasseur R, Pays E. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science. 2005;309(5733):469–472. doi: 10.1126/science.1114566. [DOI] [PubMed] [Google Scholar]
- 26.Petrovic-Djergovic D, Popovic M, Chittiprol S, Cortado H, Ransom RF, Partida-Sánchez S. CXCL10 induces the recruitment of monocyte-derived macrophages into kidney, which aggravate puromycin aminonucleoside nephrosis. Clin Exp Immunol. 2015 May;180(2):305–15. doi: 10.1111/cei.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poelvoorde P, Vanhamme L, Van Den Abbeele J, Switzer WM, Pays E. Distribution of apolipoprotein L-I and trypanosome lytic activity among primate sera. Mol Biochem Parasitol. 2004 Mar;134(1):155–7. doi: 10.1016/j.molbiopara.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Quaggin SE, George AL. Apolipoprotein l1 and the genetic basis for racial disparity in chronic kidney disease. J Am Soc Nephrol. 2011;22:1955–1958. doi: 10.1681/ASN.2011090932. [DOI] [PubMed] [Google Scholar]
- 29.Reeves-Daniel AM, DePalma JA, Bleyer AJ, Rocco MV, Murea M, Adams PL, Langefeld CD, Bowden DW, Hicks PJ, Stratta RJ, Lin JJ, Kiger DF, Gautreaux MD, Divers J, Freedman BI. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant. 2011 May;11(5):1025–30. doi: 10.1111/j.1600-6143.2011.03513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiflett AM, Faulkner SD, Cotlin LF, Widener J, Stephens N, Hajduk SL. African trypanosomes: intracellular trafficking of host defense molecules. J Eukaryot Microbiol. 2007;54(1):18–21. doi: 10.1111/j.1550-7408.2006.00228.x. [DOI] [PubMed] [Google Scholar]
- 31.Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, Skorecki K. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Human genetics. 2010;128:345–350. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uzureau P, Uzureau S, Lecordier L, Fontaine F, Tebabi P, Homblé F, Grélard A, Zhendre V, Nolan DP, Lins L, Crowet JM, Pays A, Felu C, Poelvoorde P, Vanhollebeke B, Moestrup SK, Lyngsø J, Pedersen JS, Mottram JC, Dufourc EJ, Pérez-Morga D, Pays E. Mechanism of Trypanosoma brucei gambiense resistance to human serum. Nature. 2013;501:430–434. doi: 10.1038/nature12516. [DOI] [PubMed] [Google Scholar]
- 33.Vashistha H, Husain M, Kumar D, Singhal PC. Tubular cell HIV-1 gp120 expression induces caspase 8 activation and apoptosis. Ren Fail. 2009;31:303–312. doi: 10.1080/08860220902780101. [DOI] [PubMed] [Google Scholar]
- 34.Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008;283(31):21540–21549. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yalavarthy R, Smith ML, Edelstein CL. HIV-associated nephropathy in Caucasians: case report and review of literature. Int J STD AIDS. 2008 Nov;19(11):789–90. doi: 10.1258/ijsa.2008.008091. [DOI] [PubMed] [Google Scholar]
- 36.Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;4(8):1079–1082. doi: 10.4161/auto.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]