

Abstract
The germline mutation rate is an important parameter that affects the amount of genetic variation and the rate of evolution. However, neither the rate of germline mutations in laboratory mice nor the biological significance of the mutation rate in mammalian populations is clear. Here we studied genome-wide mutation rates and the long-term effects of mutation accumulation on phenotype in more than 20 generations of wild-type C57BL/6 mice and mutator mice, which have high DNA replication error rates. We estimated the base-substitution mutation rate to be 5.4 × 10−9 (95% confidence interval = 4.6 × 10−9–6.5 × 10−9) per nucleotide per generation in C57BL/6 laboratory mice, about half the rate reported in humans. The mutation rate in mutator mice was 17 times that in wild-type mice. Abnormal phenotypes were 4.1-fold more frequent in the mutator lines than in the wild-type lines. After several generations, the mutator mice reproduced at substantially lower rates than the controls, exhibiting low pregnancy rates, lower survival rates, and smaller litter sizes, and many of the breeding lines died out. These results provide fundamental information about mouse genetics and reveal the impact of germline mutation rates on phenotypes in a mammalian population.
Germline mutations are the ultimate source of congenital diseases, individual phenotypic variations, and evolutionary phenotypic changes. The per generation de novo mutation rate affects genetic variability and the speed of evolution (Kimura 1983; Drake et al. 1998). Advancements in high-throughput sequencing have made it possible to determine the mutation rates in various organisms. For example, the mean germline base-substitution mutation rate is calculated at 1.2 × 10−8 per nucleotide per generation for humans (Conrad et al. 2011; Kong et al. 2012; Campbell and Eichler 2013), 1.2 × 10−8 for chimpanzees (Venn et al. 2014), 2.8 × 10−9 for Drosophila melanogaster (Keightley et al. 2014), and 2.7 × 10−9 for Caenorhabditis elegans (Denver et al. 2009). The per generation mutation rate varies between species (Lynch 2010a) and within them; for example, the rate in humans varies several-fold between individuals, and is influenced by age, sex, and other genetic or environmental factors (Conrad et al. 2011; Kong et al. 2012; Campbell and Eichler 2013).
More recently, the importance of understanding the risks and effects of germline mutagens, such as environmental chemicals and radiation, on the future health of animal populations, including humans, has become clear (Yauk et al. 2015). However, only limited information about the long-term effects of mutation-rate differences is currently available. For example, we know that an increased mutation rate will increase the frequency of congenital disease. However, the overall phenotypic effects of accumulated mutations on future populations living under higher mutation-rate conditions are largely unknown. Furthermore, the range of germline mutation rates that will permit the long-term survival of mammalian populations is unclear. Thus, here we established a new experimental model for assessing germline mutation rates and their phenotypic effects on future populations living under higher mutation-rate conditions.
For this model, we raised two lines of mice, wild-type C57BL/6 mice (control mice) and homozygous Pold1exo/exo mice (mutator mice), for more than 20 generations with phenotypic inspection, and established a set of mutation accumulation (MA) lines. Pold1exo/exo mice lack the 3′-5′ exonuclease activity of DNA polymerase delta (of which a catalytic subunit is encoded by the Pold1 gene) on a C57BL/6 background (Uchimura et al. 2009) and have a high rate of DNA replication errors. DNA polymerase delta, a major enzyme in DNA replication and genome maintenance, contributes to faithful DNA synthesis, mainly lagging-strand synthesis, through its intrinsic 3′-5′ exonuclease proofreading activity (Burgers 2009; Prindle and Loeb 2012). Disrupting its exonuclease activity increases the spontaneous mutation (mainly base-substitution) rates in yeast (Simon et al. 1991) and mouse somatic cells (Albertson et al. 2009) and increases tumor susceptibility in mice (Goldsby et al. 2002) and humans (Palles et al. 2013). In the current study, ∼30% of the mutator mice died from thymic lymphoma at 3–8 mo of age, but the tumor rate did not change over the generations studied. The mice were bred by one-to-one natural mating between siblings, without artificial selection. At the time of this writing, we had studied up to 24 generations of these breeding lines (Fig. 1).
Figure 1.

Pedigrees of control and mutator mice in a long-term breeding study. All breeding lines that were separated by five or more generations from the original line are shown: blue, wild-type; black, surviving mutator lines; and red, extinct mutator lines. Green arrows indicate whole-genome sequencing was performed on mice from the indicated generations. The first appearance of a conspicuous phenotypic anomaly is shown for each breeding line. Gray shading indicates body weight was analyzed for the generations indicated (see Fig. 3, legend).
For the current study, we performed whole-genome sequencing of the mouse MA lines and estimated the per generation mutation rate in the control and mutator mice. This is the first application of high-throughput sequencing to determine the spontaneous germline mutation rate of wild-type mice and provides the first direct estimate of a genome-wide germline mutation rate in laboratory mice. We also recorded detailed phenotypic data through several generations and found that the different germline mutation rates between the mutator and control mice had a significant impact on the health of their descendants. Our findings indicate that the combination of genome sequencing and MA analyses in mice is a promising tool for understanding the biological significance of mutation rate and de novo mutations in a population at the whole-genome level.
Results
Detection of de novo mutations in breeding lines
To determine the per generation mutation rate and nature of the de novo mutations, we detected de novo mutations in four MA lines (two control lines: conA and conB; two mutator lines: mutC and mutD), where “de novo” mutations are those that arose during our breeding experiment. First, we sequenced the whole genome for seven mice at a >40× average coverage, using an Illumina sequencer, and mapped the data to the C57BL/6J mm10 database as a reference. The seven mice included one male from each of the four MA lines (conA, conB, mutC, and mutD), the original male and female pair (see Fig. 1, Adam/Eve) for the mutC and mutD lines, and one male from the mutator MA line mutE, which was derived from a different original pair of mutator mice than mutC and mutD (shown in Fig. 1). The mutE mouse was used to evaluate the frequency of recurrent genetic alterations, including single-nucleotide variant mutations (SNVs) and small insertion/deletion-type variant mutations (indels), resulting from a nonrandom mutagenesis process in the mutator mice (shown in the Supplemental Information). To obtain credible comparisons between mutator and control mice, we analyzed high-quality sequenced autosomal effective whole-genome coverage (EWC) regions (see Methods) of the two Adam/Eve mice and the four mice from the conA, conB, mutC, and mutD lines. The SNVs and indels were analyzed in two different EWC regions, because conditions required to accurately detect indels are more stringent than for SNVs. These regions covered 61.6% (for SNVs) and 39.1% (for indels) of the total autosomal region and 71.6% (for SNVs) and 70.4% (for indels) of the autosomal coding sequences in the reference genome (details in Supplemental Table S1).
We detected SNVs and indels using SAMtools/BCFtools (Li et al. 2009). Candidate de novo mutations in the mutator lines were selected by comparing variant calls in mutC or mutD with those in the ancestral Adam/Eve pair; this eliminated artifacts caused by misalignment to the reference sequence, as well as unique ancestral variants (details in Supplemental Table S2). We also filtered variants called in the conA and conB lines using those from the mutator Adam and Eve, rather than the ancestral pair of the control lines. The initial variants derived from the control ancestral mice that were not completely removed by this filter were subsequently assessed by Sanger sequencing using the genomes of the original male–female pair (generation 0) for conA and conB.
We found considerably fewer candidate de novo SNVs in the control lines (211 and 235 homozygous and 105 and 103 heterozygous variants in lines conA and conB, respectively, which is expected to include some initial variants from the original mice in these lines) than in the mutator lines (1304 and 1472 homozygous and 1944 and 1633 heterozygous variants in lines mutC and mutD, respectively) within the EWC region, and these were distributed throughout the autosomal chromosomes (Supplemental Fig. S1). To estimate the rate of true de novo mutations other than false positives due to miscalls and the initial variants from the original mice in conA and conB, a total of 165 de novo SNV candidates (to validate 10 homozygous and 30 heterozygous candidates per sequenced individual) were randomly selected for Sanger sequencing (Supplemental Table S3). Although five candidates failed to be sequenced due to poor PCR amplification or sequencing analysis conditions, the other 160 candidates were successfully sequenced and confirmed in the sequenced individuals, indicating that our high-throughput sequencing analysis was very reliable.
In the mutator lines, all of the selected candidates (80 SNVs) were confirmed to be bona fide de novo SNVs, present in the sequenced mouse but not in its ancestors. In the control lines, 40% (8/20) of the homozygous and 93.3% (56/60) of the heterozygous de novo candidates were confirmed to be bona fide de novo SNVs, present in the sequenced mouse but not in the original male–female pair for conA and conB. The remaining 60% (12/20) of homozygous and 6.7% (4/60) of heterozygous candidates were also present in the ancestral male–female pair (generation 0) and confirmed to be the initial variants derived from the original pair. The values corrected for this frequency of true de novo variants were used as the number of de novo mutations in control lines for estimating the mutation rate (Table 1). The theory that successive inbreeding would remove the initial variants from the heterozygous state (the 20-generation inbreeding coefficient is 0.986) was borne out; there were more homozygous than heterozygous candidates at the end of the breeding period.
Table 1.
Estimated per generation mutation rates in mice
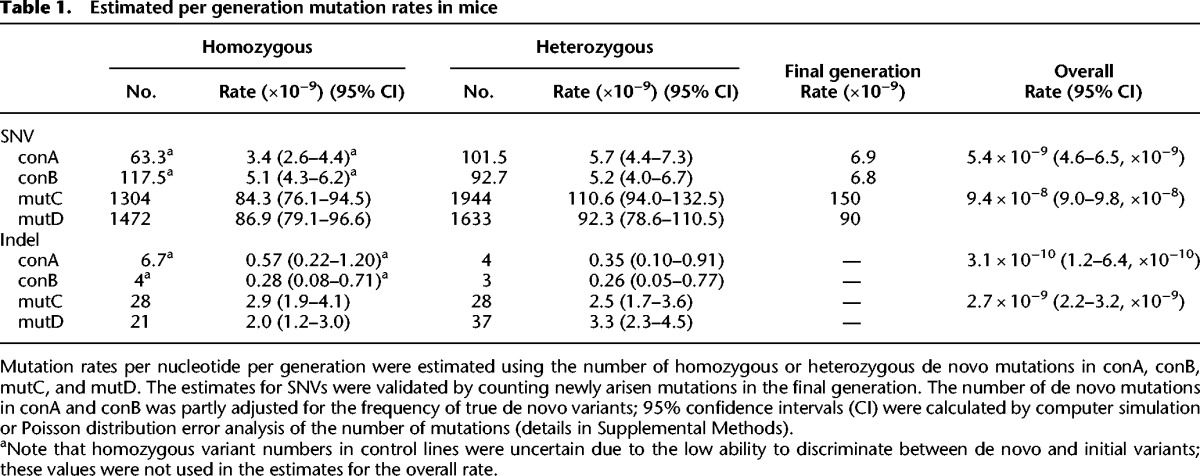
To detect de novo SNVs without missing true mutations, we assessed the false-negative rate by a synthetic point mutation approach, in which mutations were simulated by altering the sequence read data (Keightley et al. 2014). This analysis indicated that the rate of false negatives was very low in our pipeline: Of a total of 4504 homozygous synthetic mutations, none was missing, and of 4504 heterozygous mutations generated by binomial distribution in the EWC region, only two were missing (0.04%; for details, see Supplemental Information). In addition, considering that the frequency distribution of the called de novo mutations (Supplemental Fig. S2) was clearly different between homozygotes and heterozygotes and that the frequency of the heterozygous variants was symmetrical with respect to 50%, similar to the theoretically expected binomial distribution, the miscalling and false-negative rates in our pipeline appeared to be very low.
Our detection of de novo variants with few sequencing errors and few missing variants was highly accurate compared with previous reports (Keane et al. 2011; Simon et al. 2013) in which the whole genome of C57BL/6N mice was sequenced. Our higher accuracy might have been due to our better analysis conditions—100-bp (or 150-bp) paired-end sequencing with greater coverage; an updated reference sequence (mm10 database); the choice of more sequence-amenable regions (EWC regions) for analysis, which were strictly selected; and more efficient filtering out of sequencing errors using the Adam/Eve samples—compared with those used in previous reports (for details, see Supplemental Information).
Similarly, de novo indels were also examined in the EWC region. There were fewer indel than SNV candidates. As with SNVs, fewer candidate variants were called in the control lines (10 and 12 homozygous and five and three heterozygous variants in lines conA and conB, respectively, of which the homozygous variants is expected to include some initial variants from the original mice) than in the mutator lines (28 and 21 homozygous and 28 and 37 heterozygous variants in lines mutC and mutD, respectively). Randomly selected variant validation by Sanger sequencing identified three initial variants in a total of 14 tested variants in conA and conB, as well as only one sequencing error in a total of 26 checked variants (Supplemental Table S3).
Estimation of germline mutation rates
We estimated the per generation mutation rate from the number of accumulated homozygous or heterozygous de novo mutations, using the expected coalescent time for two alleles in a sequenced individual (Supplemental Fig. S3). In the control mice, there was close agreement among the four SNV mutation rate estimates based on the number of heterozygous and homozygous variants in the conA and conB lines, although some variability was observed in the de novo homozygous-derived mutation rate (Table 1). This variability may have been due to the small number of variants checked in the base population. By use of the mean of the two heterozygous-derived mutation rates, we estimated the base-substitution mutation rate to be 5.4 × 10−9 (95% confidence interval [CI] = 4.6 × 10−9–6.5 × 10−9) per nucleotide per generation. For the de novo indel mutations, the estimated per generation indel rate in wild-type mice was 3.1 × 10−10 (95% CI = 1.2 × 10−10–6.4 × 10−10), which was 5.7% of the SNV rate; note that there were differences in the EWC regions used for SNV and indel, and the estimation of the indel rate was based on fewer de novo variants than that of the SNV rate (Table 1).
Our estimated SNV mutation rate in control mice was 15% of the previous estimate for laboratory mice, 3.7 × 10−8, which was obtained using a specific locus test (SLT) and in vivo reporter transgenic mouse approaches (Lynch 2010a). Taking the indel mutation rate into consideration, our estimate was still 16% of the previous one. This rate difference was probably due to the different genomic regions analyzed; the loci targeted by SLT and the transgenic mouse approaches may have upwardly biased the mutation rates compared with the rate for the whole genome. On the other hand, the base-substitution rate was slightly higher than a previously reported phylogenic estimate of ∼3.1 × 10−9 (at non-CpG sites) in rodents (Eory et al. 2010).
Data from the mutator mice yielded four estimates that were in substantial agreement. The overall mutation rate was 9.4 × 10−8 (95% CI = 9.0 × 10−8–9.8 × 10−8) for SNV and 2.7 × 10−9 (95% CI = 2.2 × 10−9–3.2 × 10−9) for indel. These rates were 17.2-fold (for SNV) and 8.6-fold (for indel) higher than the mutation rates in the control mice. These differences were smaller than the 60-fold increased revertant mutation rate reported in cultured mouse Pold1exo/exo fibroblasts and higher than the threefold increased mutant frequency obtained by in vivo cII reporter transgene assay in Pold1exo/exo mouse somatic tissues (Albertson et al. 2009). The per generation mutator mouse SNV mutation rate was intermediate between the rate (5.4 × 10−9) of wild-type mice and the known rate (8.0 × 10−7) of mice treated with the widely used chemical mutagen N-ethyl-N-nitrosourea (ENU) (Table 2; Keays et al. 2006; Gondo et al. 2009).
Table 2.
Mammalian mutation rates and their effects on survival

Our estimates presupposed unbiased chromosomal segregation and the absence of any selective pressure on each mutation during the serial inbreeding term. However, according to our data (see next section), some portion of the mutations was suspected to be subject to purifying selection, which could have caused the mutation rate to be underestimated. To evaluate such effects on our estimated mutation rates, we determined the rates using another rough approach, a final-generation approach, that did not require the above assumptions. In this approach, we counted the number of SNVs that arose during the final generation of the 30 heterozygous de novo variants analyzed in the validation experiment, extrapolated the residual variants, and estimated the per generation mutation rate (Supplemental Table S3). The mutation-rate estimates of wild-type and mutator mice using this approach were in similar ranges as the original estimates (Table 1). Although a somewhat higher mutation rate (1.5 × 10−7) was obtained using the final-generation approach for the mutC heterozygous mutations than the other estimates in mutator mice, it could be explained by the relatively long generation span (264 days) of the parents of the whole-genome-sequenced mutC individual (Supplemental Table S4). The per generation mutation rate is known to increase with parental age, possibly as a result of increased cell division (DNA replication) in the paternal germline (Crow 2000; Kong et al. 2012). In any case, the high consistency of the estimates generated by the two approaches indicated that the mutation rate was nearly constant throughout the breeding term and that most of the de novo mutations were neutral. These findings also indicated that our estimates of the per generation mutation rates in mice were accurate, although we could not rule out some effects of selective pressure and biased chromosomal segregation.
The spectrum of de novo SNVs observed is shown in Supplemental Table S5. In control mice, nucleotide transitions occurred approximately 2.1 times more frequently than transversions. As CpG-containing sites are known mutational hot spots in mammals, due to the oxidative deamination of methylated cytosines, the rate of total CpG-site substitutions was 16.8-times higher than that of non-CpG substitutions. The rate of change from strong (G:C) to weak (A:T) base pairs was 3.9-times higher than in the opposite direction, indicating that the GC content (= 42.4% in the EWC region) might not be in equilibrium in wild-type mice or that GC-biased gene conversion may play an important role in maintaining the equilibrium (Duret and Galtier 2009). These tendencies are similar to those reported for human de novo mutations (Kong et al. 2012). In the mutator mice lacking the proofreading activity of replicational DNA synthesis by DNA polymerase delta, all of the base-substitution types of mutations occurred with greater frequency than in control mice, and the transversion rate and non-CpG-site mutation rate, which are kept low in wild-type mice, were markedly increased. The increased transversion rate was consistent with previous results using Pold1exo/exo and wild-type mouse somatic cells (Albertson et al. 2009). No G:C to A:T bias was found in the mutator mice; instead, there was a slight increase in the opposite direction. For indels, many of the de novo mutations in both the control and mutator MA lines were detected at sites with repetitive sequence elements, and de novo insertions of A:T base pairs were markedly increased in the mutator mice (Supplemental Table S6).
Although the relatively large number of accumulated de novo mutations and the highly accurate detection of them supported our estimates, the actual per generation mutation rate for the entire genome may be higher. This is because the EWC regions are practical sites for whole-genome sequencing analysis, and the rest of the genome includes complex regions that are difficult for DNA polymerases to access and are presumably more mutable. The indel mutation rate in particular may have been underestimated, since more repeat sequences, which are mutable regions for indels, were excluded from our analysis of the EWC region for indels than for SNVs.
Assuming that mutations are inherited in a neutral fashion, the number of accumulated de novo SNVs in a genome is predicted to increase with the number of generations, as shown in Supplemental Fig. S4. In our study, mutator mice bred for 20 generations gave rise to 6335 SNVs (the sum of the numbers of heterozygous and homozygous variants). This is much higher than the estimated number of mutations induced by ENU treatment (2105 SNVs) (Gondo et al. 2009) or the number of de novo SNVs predicted to distinguish the original C57 mouse stock (Russell 1978) from the C57BL/6J strain currently distributed by Jackson Laboratory (3292 SNVs, calculated using our estimated mutation rate and the known generation number, F226). Considering that our comparison of the variant data between the mutC/mutD lines and the mutE line indicated that the frequency of recurrent mutations in the mutator mice was low (details in Supplemental Information), the long-term breeding of mutator mice is a promising method for enhancing mutagenesis across the entire genome.
Long-term phenotypic effects of mutation rates
The difference in mutation rates between the wild-type and mutator mice had several long-term effects on phenotypes. First, there were 4.1 times more visible abnormalities in the mutator lines (11.0%, n = 6229) than in the control lines (2.7%, n = 1649; Fisher's exact test; P = 9.2 × 10−32). Phenotypic variations included mutants with human-audible vocalizations (Supplemental Movie S1), shortened limbs and tail, or diluted coat color (Fig. 1; Supplemental Fig. S5; Supplemental Table S7). The frequency of anomalies in the mutator line (particularly hydrocephaly and minor color-variation phenotypes) increased at a rate of 0.68% per generation (95% CI = 0.55%–0.81%; P < 1.1 × 10−16) (Fig. 2), suggesting some relationship between increased anomalies and the accumulation of mutations (Supplemental Fig. S4). The frequency of abnormal phenotypes varied between independent mutator breeding lines, suggesting that breeding line–specific mutations were present (Supplemental Table S8). While these abnormal phenotypes did not always exhibit clear Mendelian-like inheritance, we observed several inheritance patterns. For example, we identified a causative recessive base-substitution mutation in the human-audible vocalizer mutant mouse (data not shown). Mutations causing coat-color dilution and syndactylism both exhibited recessive trait inheritance. Although mutations causing priapism and a shortened tail and limbs appeared only in closely related populations, these traits did not follow simple Mendelian-like inheritance; these may be multifactorial or low-penetrance mutations. Since mice with abnormal phenotypes often reproduced at a low rate, some de novo mutations may have been removed by purifying selection during the breeding process.
Figure 2.
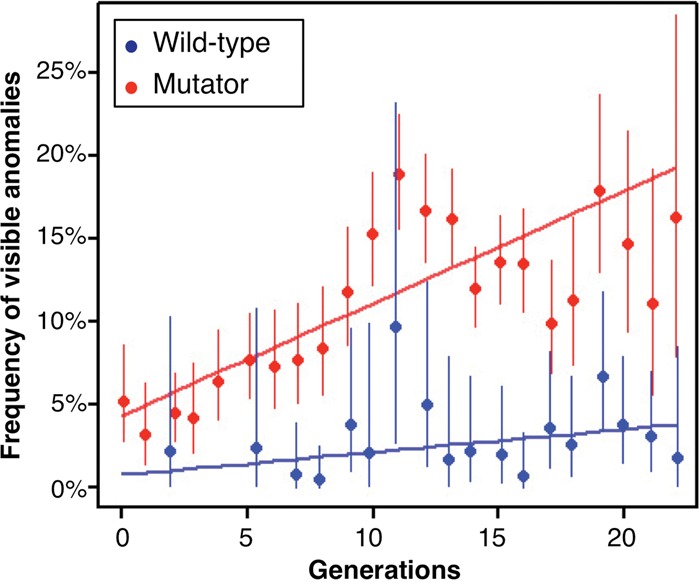
Frequency of visible phenotypic anomalies in breeding lines. Frequency of visible anomalies in each successive generation. Circles indicate observed frequencies with 90% CI, determined by Fisher's exact test. Since fewer than 20 mice were screened in the early-generation (fewer than seven generations) populations of control mice, mean phenotypic frequencies are shown for generations 0–3 and 4–6. Solid lines show the fit with a binomial linear model.
After several generations of breeding, mutation rates can be reflected in body weight, which is a polygenic trait. Although there was no specific artificial selection for body weight in the breeding, we found several statistically significant differences in the average weight (8-wk-old mice) between the breeding lines (Fig. 3A, Supplemental Fig. S6A). The mean body weight of all the measured mutator mice tended to decrease as the number of generations rose (P = 1.1 × 10−14 in males, and P = 4.1 × 10−18 in females) (Fig. 3B; Supplemental Fig. S6B), with an estimated loss of 0.115 g in males (95% CI = 0.086–0.144) and 0.090 g in females (95% CI = 0.070–0.110) per generation. In contrast, the weight was fairly stable from generation to generation in the control mice (P = 0.14, per generation weight loss = 0.032 g [95% CI = −0.010–0.075] in males, and P = 0.092, per generation weight loss = 0.028 g [95% CI = −0.004–0.061] in females; note that the investigated generation number was smaller than that of the mutator lines). Related to this weight-decreasing tendency, many developmental delays were observed among the mutator mice, particularly in lines with high numbers of generations (Fig. 3C).
Figure 3.
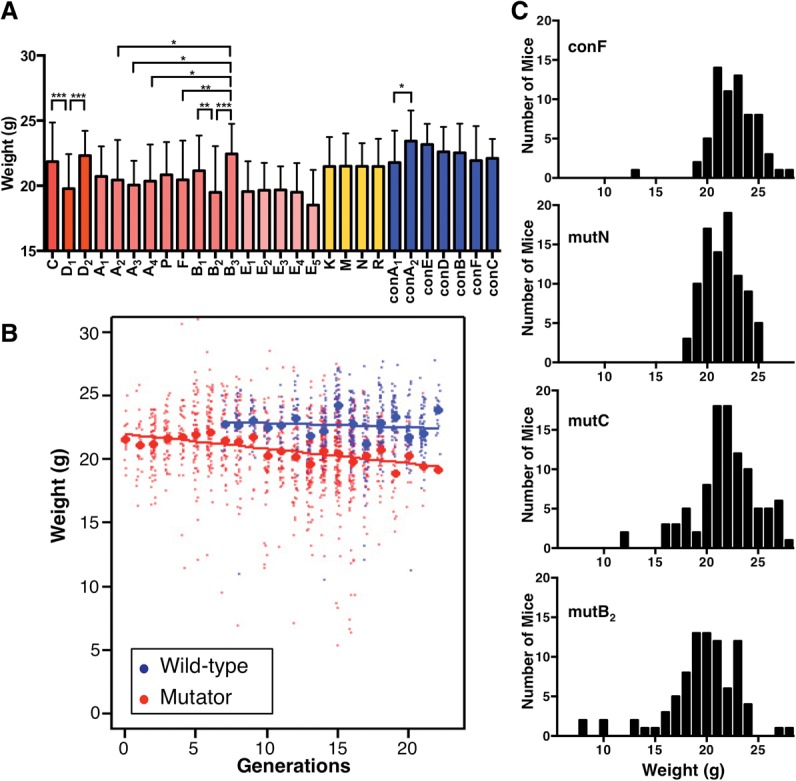
Body-weight changes in breeding lines. (A) Body weights of 8-wk-old males: blue, control; yellow or red, mutator lines. Error bars, SDs. Each data point represents the mean weight in the gray-shaded generations in Figure 1; breeding-line names correspond to the sublines in Figure 1. (*) Statistically significant differences between sublines belonging to the same subgroup ([C∼D2], [A1∼B3], [E1∼E5], [K∼R], and [conA1∼conC]), by Tukey's multiple comparison test; (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. (B) Body weights of 8-wk-old males, plotted against the number of generations: red, mutator; blue, control. Weights of individual mice (dots) and means (circles) are shown for each generation. Solid lines show the fit for simple linear regression. (C) Histograms of the male weights in representative breeding lines.
The most striking phenotypic difference between mutator and control mice was in their reproductive capacity. Although the ability of control mice to reproduce remained almost constant through the generations (P = 0.84, with a linear fit), the ability of mutator mice decreased markedly with generation number (P = 4.3 × 10−4, with a linear fit), with the average number of offspring per mating decreasing by 0.042 (95% CI = 0.014–0.071) per generation (Fig. 4A). Notably, this tendency was nonlinear: It was most evident in the first few breeding generations, indicating the importance of the effects of recessive mutations. We tested one of the simplest models, the recessive lethal mutation model proposed by Dr. Lyon (Lyon 1959), which considers only the effect of completely lethal de novo recessive mutations. This model fit our data better (Akaike information criterion [AIC] = 3790.3) (Akaike 1974) than a simple linear model (AIC = 3803.3) and suggested that 1.98 lethal mutations occurred per generation per diploid genome (95% CI = 1.14–2.81; P = 5.1 × 10−7) in the mutator lines. Although there are many possible causes besides the effect of lethal mutations, the results suggested that the reduced reproductivity was caused by deleterious de novo mutations that were mostly recessive. These results also indicated that after the first several generations of mutator mouse breeding, an equilibrium between the occurrence of deleterious de novo mutations and a purifying selection against such mutations was established.
Figure 4.
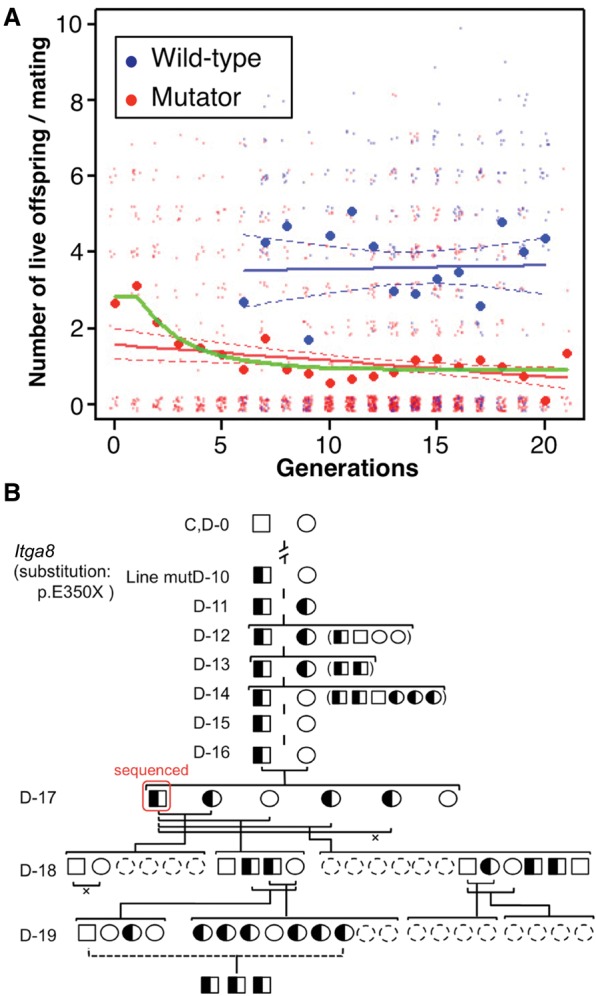
Reduced mutator mouse reproduction. (A) Live offspring per mating, plotted against the number of generations; red, mutator mice; blue, control; dots, individual mice; circles, mean for the generation. Solid lines show linear fit and dashed lines show the 95% CI, assuming a negative binomial distribution; green line, regression curve using the recessive lethal mutation model for the mutator lines. (B) The Itga8-mutant genotype in mutD; squares, males; circles, females; filled, homozygotes (but not present here); half-filled, heterozygotes; empty, no Itga8 mutation; and dashed-line circles, post-natal deaths. (×) Failure to reproduce. In 26 tested offspring obtained from the heterozygous intercross, there were no homozygotes (6.5 homozygotes would be expected for a neutral mutation, χ2-test; P < 0.01), indicating that this mutation had a recessive lethal effect on the phenotype.
Reproductive ability was decreased in the mutator lines due to lower birth rates per mating, smaller litter sizes at post-natal day 0 (P0), and a higher rate of post-natal death (Supplemental Table S9). Consistent with these findings, we observed many cases of oligozoospermia and pre- and post-natal death (including pups neglected by their mother) in the mutator breeding lines (data not shown). These findings suggested that the effects of deleterious mutations that accumulated in the mutator MA lines included not only the pre- and post-natal lethality of individuals but also sterility and impaired rearing ability in the parental mice. As with the abnormal phenotypes and body weight, the extent of the reproductive phenotype varied among the mutator breeding lines (Supplemental Table S10).
Accumulated mutations and phenotypes in the breeding lines
In the MA lines, whole-genome sequencing analysis showed many functionally suspected de novo mutations that accumulated in coding regions, noncoding RNAs, and untranslated regions (Supplemental Table S11). Of these mutations, all of the de novo nonsynonymous mutations observed in the EWC region are shown in Supplemental Table S12. About 80% of them occurred at sites that are conserved among species (80% [64/80] in mutator lines, 78% [7/9] in control lines). Protein variation effect analyzer (PROVEAN) predictions (Choi et al. 2012) indicated that ∼60% of these mutations had deleterious effects on protein function (54% [43/80] in mutator lines, 67% [6/9] in control lines). Notably, we found more than 10 suspected deleterious amino acid substitutions in genes whose knockouts are associated with embryonic or post-natal lethality, sterility, or developmental retardation, accumulated in a single mouse from one of the mutator MA lines.
Among the variants, we focused on three significant mutations observed as heterozygous SNVs in the mutD line, and investigated their inheritance history: two stop-gain SNVs, one in the integrin alpha 8 (Itga8) gene and the other in the T-cell activation inhibitor mitochondrial (Tcaim) gene, and one SNV abolishing the start codon in the parathyroid hormone 2 (Pth2) gene (resulting in the deletion of five amino acids from Pth2) (Fig. 4B; Supplemental Fig. S7). In the descendants of the sequenced individual in line mutD, those with homozygous mutations in Tcaim and Pth2 appeared normal and were fertile, indicating that such mutations did not have serious deleterious effects. In contrast, the mutation in Itga8 may have been partially responsible for the mutD line's poor ability to reproduce (Supplemental Table S10), since no homozygotes of this mutation were observed among 26 offspring obtained from the heterozygous intercross (Fig. 4B). The lack of homozygous Itga8 individuals is consistent with the post-natal lethal phenotype of Itga8 KO mice (Muller et al. 1997), and this example suggests that certain recessive lethal mutations are associated with reduced reproductive ability.
In addition to the de novo mutations examined here, other types of mutations or environmental effects may have contributed to the phenotypes observed in the MA lines. Although we could not show them clearly due to the lack of a validation experiment, many candidate de novo SNVs and indels were also called in chromosomal regions, including sex chromosomes, outside of the EWC region. A single candidate de novo heteroplasmic SNV was also identified in the mitochondrial genome in the mutC line. We used BreakDancer software (Chen et al. 2009) to look for de novo deletion-type structural variants, and found two such deletions (204 bp and 1013 bp) in the mutD line (Supplemental Table S13). Karyotype analyses did not reveal any chromosomal abnormalities in either the control or mutator MA lines (Supplemental Fig. S8). Although we could not completely rule out the possibility that phenotypic changes in the mutator mice resulted from genetic variations in the base populations, it is more likely that most of the phenotypic changes were due to a higher mutation rate (Supplemental Information).
Mice in the mutator lines often did not care well for their pups, and this behavior was associated with a decrease in the pups’ survival rate (the percentage of pups reaching 8 wk of age) (Supplemental Table S9). The major phenotypic changes in body weight and reproductive capacity observed in mutator mice are known to be affected by environmental factors, especially maternal effects and litter size (Falconer and Mackay 1996). Thus, many of the phenotypic changes described here may have resulted from complex interactions between accumulated genetic variations and environmental factors.
Discussion
The germline mutation rate is a fundamental parameter in genetics and evolutionary biology studies. However, information is still lacking about this rate and the resultant phenotypic effects of MA on individuals in future populations living under higher mutation-rate conditions. Here, we determined the spontaneous germline mutation rate and its spectrum in wild-type laboratory (C57BL/6) and Pold1exo/exo mutator mice by the whole-genome sequencing of MA lines, and we examined the phenotypes of these mice over several generations.
To date, whole-genome sequencing in mammals has been used only to determine the germline base-substitution mutation rates for humans (Conrad et al. 2011; Kong et al. 2012; Campbell and Eichler 2013) and chimpanzees (Venn et al. 2014). The mutation spectra exhibit similar features (e.g., transition type substitutions occur dominantly over transversion ones, CpG sites are mutable, G:C-to-A:T mutation bias is present) in humans, chimpanzees, and the mice presented here, indicating that the molecular mechanisms of germline mutagenesis are conserved among mammals. However, the mutation rate in mice was lower than in the other two species (Table 2). This lower rate may be partly explained by the shorter generation times and/or smaller number of germ cell divisions per generation in rodents versus apes (Drost and Lee 1995) or by the greater effect of selective pressure on the mutation rate of rodents living in a natural environment, due to their larger effective population size compared with that of apes (Lynch 2010a).
Our findings that the mutator MA lines showed a 17.2-fold increase in the SNV rate and 8.6-fold increase in the indel rate compared with the control lines illuminated the significance of the spontaneous germline mutation rate in wild-type mice. Although a higher per generation mutation rate is expected to generate more frequent phenotypic anomalies and a greater mutational load (Lyon 1959), as well as quantitative phenotypic variances (including body weight and bone length) (Casellas 2011), we actually found that in the Pold1exo/exo mutator MA lines (1) abnormal phenotypes appeared 4.1 times more frequently than in control lines, (2) quantitative traits changed on a larger scale between and within each line, and (3) the reproductive capacity was considerably reduced. The decrease of the reproductive ability made it difficult to maintain the inbred strains. The mean number of offspring in the mutator MA lines after several generations fell to 36% of that of the starting population (generations 0–2) (Supplemental Table S9). Furthermore, only a small fraction (18% on average) of the sister-brother matings in the MA lines could produce littermates including both sexes. In addition, since mice with early death phenotypes (hydrocephaly, tumors, or other pathological phenotypes, which often caused death after 8 wk of age) were excluded from our analysis of reproductive capability, the condition of the mutator breeding colony as a whole was worse than the above description. Indeed, this low reproductive capacity caused many of the lines, including the mutC line used for sequencing analysis, to become extinct (Fig. 1), and we had limited success in maintaining the mutator MA lines for 24 or more generations, at the time of this writing. These findings suggest that the mutation rate of the mutator mice was close to the upper limit for the practical maintenance of an inbred laboratory mouse population by natural full-sib mating. This value is supported by the finding that TOY-KO mice, which lack three genes involved in repairing oxidative DNA damage and preventing mutations, have a base-substitution mutation rate that is approximately twice that of the Pold1exo/exo mice (Table 2) and become extinct within eight generations due to progressively decreasing fitness (Ohno et al. 2014). On the other hand, cell population analysis suggested that a 10,000-fold increase in the mutation rate is the upper limit for cell proliferation in diploid yeast and, presumably, in mouse cells (Herr et al. 2011, 2014). Our findings suggest that a much lower mutation rate is the permissible maximum value for maintaining an animal population compared with a cell population.
The tolerable range of mutation rates for human populations is a matter of ongoing discussion. The mutational load theory (Muller 1950; Kimura and Maruyama 1966) proposed that each de novo deleterious allele would ultimately be extinguished from populations as one “genetic death,” irrespective of the mutation's fitness effects, if the human population were to be sustainable. According to this theory, recent estimates of the per generation mutation rate (1.2 × 10−8) and of the frequency of deleterious alleles in human mutations mean that an individual would have a reproduction failure rate of at least 88% due to “genetic death” (Table 2), which seems inconsistent with the low rate of human reproduction (Kondrashov and Crow 1993; Eyre-Walker and Keightley 1999). Several theoretical models—such as selection prior to birth (including neonatal death and prezygotic selection) (Reed and Aquadro 2006); synergistic epistasis (Kimura and Maruyama 1966); and truncation selection, a more efficient form of directional selection (Crow and Kimura 1979)—have been proposed as mechanisms for removing deleterious mutations. In addition, a recent study proposed that the relative fitness model resolves such a mutational load problem in humans (Lesecque et al. 2012). However, since we still have limited knowledge about the distribution of the fitness effects of each de novo mutation and how natural selection against deleterious mutations actually works in our population, it is difficult to foresee the net phenotypic consequence of high mutation rates in humans (Lynch 2010b). In our presented study, the extremely small population size (n = 2) for inbred laboratory mice means that natural selection is largely inoperative, which also limits meaningful comparisons between humans and mice. However, our observation that a mutation rate eight times that of the mean human rate caused deleterious effects in a laboratory mouse population after several generations highlights the importance of assessing the risk of germline mutagenesis in human populations.
Our estimated mutation rates from highly accurate whole-genome sequencing provide useful information for further studies on germline mutagenesis and on mammalian genetics. The baseline spontaneous mutation rate in laboratory mice is important for maintaining genetically stable inbred mouse strains in genetic studies and for assessing the mutagenic effects of environmental chemicals and irradiation. On the other hand, our established mutator mouse MA lines, in addition to revealing the capacity of laboratory mice to accumulate mutations, provide a new mutant mouse resource. Our results also showed that whole-genome sequencing analysis combined with mouse MA line studies, including mutator MA lines, is an effective new tool with which to study the physiological functions and maintenance mechanisms of de novo mutations in a population (Supplemental Fig. S9). These studies may help elucidate the future risk of mutagens in mammalian germline and the mechanisms of evolutionary processes in mammals.
Methods
Animals
C57BL/6J mice (JAX mice from Charles River) were used as wild-type control mice and as the background for generating the mutator Pold1exo/exo line (details in Supplemental Information) (Uchimura et al. 2009). The mice were maintained at a constant temperature (22 ± 1°C) in a 12-h:12-h light/dark cycle, with ad libitum food and water access. The housing environment and diet were constant throughout the experiment. All experiments were approved by the Animal Committee of Osaka University and were performed in accordance with the 1996 National Institutes of Health guidelines.
Mouse breeding and initial phenotypic screening
At 4 wk of age, pups were weaned and assessed for any visible phenotypic anomalies. At 8 wk of age, pups were weighed and assessed again for phenotypic anomalies, and tail tips were collected and preserved at −80°C for genomic DNA analysis. All capable females were bred in a one-to-one natural mating with a male sibling over a 1-wk period, and mating was repeated as necessary to obtain the required number of mice. Sibling males were selected arbitrarily. Mice with prostration (possibly due to thymic lymphoma or hydrocephaly) were not used. Details about phenotypic analysis, treating abnormalities, body weight, and reproductive ability are shown in the Supplemental Methods.
In each mutator line, the first homozygous Pold1exo/exo mating pair was defined as the 0th generation. Lines that were derived from different origins, or separated from the previous breeding line by more than 10 generations, were defined as distinct breeding lines. To maintain the mutE line, we performed a one-time exceptional breeding between the fourth and sixth generations (its resultant populations were treated as the sixth generation, as shown in Fig. 1) because we were missing a male–female mating pair within the littermates in August 2009.
Whole-genome sequencing analysis
Sequencing reactions, mapping, and variant calling
DNA for whole-genome sequencing was extracted from tail tissue using the Qiagen DNeasy blood and tissue kit. Paired-end libraries were prepared using the Illumina TruSeq sample prep kit according to the Illumina user manual. The Adam, Eve, and mutC sequencing libraries were amplified by PCR (eight cycles), and the mutD, mutE, conA, and conB libraries were constructed without PCR amplification. The libraries were applied to an Illumina HiSeq 2000 sequencer with a 100-bp read length or to a HiSeq 2500 with a 150-bp read length.
Sequence reads were mapped to the mouse reference genome (UCSC mm10) by BWA v0.6.2 (Li and Durbin 2009) using the default parameters. Although sex chromosomes were not analyzed in the present study, all of the chromosomes were compared to the reference genome to avoid misalignment. PCR or optical duplicates were removed using Picard v1.77, and base quality recalibration and indel realignment were performed by GATK v2.3.5 (McKenna et al. 2010; DePristo et al. 2011). We recalibrated the base quality using dbSNP build 132 on mm10.
EWC regions
To estimate the de novo mutation rate with high confidence, we defined the EWC regions as follows: For SNVs, (1) the lower bound coverage was set to 50% and the upper bound coverage to 3× the peak coverage of the MQ60 reads; (2) the minimum ratio of MQ60 reads to all mapped reads on each site was set to 80%; and (3) mutation calls required both forward and reverse strand coverage. For indels, in addition to the criteria for SNVs, (4) repeat sequences detected by RepeatMasker and Tandem Repeats Finder (derived from UCSC) were removed, as were (5) homopolymers (≥8 bp) and di-/tri-/tetra nucleotide repeats (≥10 bp). The minimum base quality for a base to be considered was 13. The EWC region was defined for each sample (mutC, mutD, conA, conB, Adam, and Eve), and then the common EWC region was used for analysis. Supplemental Table S1 shows a breakdown of the common EWC regions.
Variant calls and de novo SNVs/indels
Variants were called with SAMtools/BCFtools v0.1.18 (Li et al. 2009) using highly reliable reads with mapping qualities ≥60. The parameters “mpileup -B -F 0.25 -m 3” and “view -vcg” were used for SAMtools and BCFtools, respectively. The raw variants were then subjected to the following custom filters: (1) The lower bound of the alternate allele frequency was set to 25%, and (2) alternative allele identification required both forward- and reverse-strand coverage. In both filters, the minimum base quality for a base to be considered was 13.
To identify de novo variants in mutC and mutD, we focused on variants with an alternative allele frequency that was present in <10% of the original population (i.e., Adam/Eve). This threshold was determined in part by validation by Sanger sequencing. When investigating mutations in conA and conB, we also focused on variants with an alternative allele frequency of <10% in Adam and Eve; this removed sequencing artifacts and some initial variants present in the C57BL/6J mating pair but did not completely remove the initial variants present in the base conA and conB breeding pairs. A validation experiment, in which randomly selected candidate de novo variants in conA, conB, and the ancestral mating pair were subjected to Sanger sequencing, partially compensated for the imperfect filtering. We modified the de novo variant numbers based on the validation results, and used these values to estimate the mutation rates.
For the validation experiment, randomly selected candidate de novo variants were subjected to Sanger sequencing. For the PCR amplifications and subsequent Sanger sequencing, we tried several primers (up to three primers were tested for each forward and reverse site) and PCR conditions (including a buffer for GC-rich regions and a nested PCR assay) to obtain good results. The details for the PCR and Sanger sequencing analyses are provided in the Supplemental Material (Excel file).
Per generation germline mutation rates
The per generation per nucleotide mutation rate (μ) is estimated by the following formula,
where M is the number of accumulated mutations, G base pairs is the analyzed genome size, and L is the number of generations in which de novo mutations occurred. We calculated M based on the validation results by the Sanger sequencing (Supplemental Table S3). We also calculated M by analyzing 30 validated heterozygous de novo SNVs to determine the number of mutations occurring in the last generation (where L = 1, G = 2× the size of the EWC region). For estimations using the accumulated de novo heterozygous or homozygous variant number, the size of the EWC region was used as G, and L was calculated from the number of generations of inbreeding and the probability of coalescence in each generation, as shown below (and in Supplemental Fig. S3):
Pk represents the probability that two autosomal alleles in an individual in the preceding k generation will coalesce under full-sibling mating conditions.
Ln represents the expected number of generations, from the 0th generation to an investigated individual (at the nth generation), in which the accumulated de novo heterozygous or homozygous mutations occur.
Detailed information about the estimates of overall mutation rates and CIs for μ, including conservative CIs, is presented in the Supplemental Methods. To predict the accumulated number of mutations (assumed to be inherited in a neutral fashion) at a given generation number with a specific mutation rate, we used the formula M = μ × G × L. We used 2.73 × 109 bp as the mouse genome size (G), and estimated the number of accumulated mutations at each breeding generation (Supplemental Fig. S4).
Data access
The nucleotide sequence data have been submitted to the DDBJ Sequence Read Archive (DRA; http://trace.ddbj.nig.ac.jp/dra/index_e.shtml) under accession number DRA002606.
Supplementary Material
Acknowledgments
We thank M. Furusawa for fundamental ideas for our breeding experiment; N. Honda, Y. Hidaka, and M. Kumagai for their help with the experiments; and C. Choji and A. Kinjo for mouse care. This work was financially supported by Neo-Morgan Laboratory, the GCOE program of Osaka University FBS, and a Grant-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (21650099, 23650234, 25640047 to T.Y.; 21657062, 24650235 to A.U.; and 221S0002 for Innovative Areas “Genome Science”).
Author contributions: A.U. and T.Y. designed the research. A.U., J.N., and T.Y. wrote the manuscript. A.U. and M.H. performed the breeding experiment and analyzed the data. Y.M., A.T., and A.F. were involved in the whole-genome sequencing and detection of genetic variants. A.U. and J.N. contributed to the theoretical analysis, particularly in the estimation of mutation rates, and to the regression analyses of phenotypic data. M.O. performed the cytogenetic analysis. I.M. and S.W. performed a part of the inheritance test and supervised the screening of abnormal phenotypes.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.186148.114.
References
- Akaike H. 1974. New look at statistical-model identification. IEEE Trans Automat Contr 19: 716–723. [Google Scholar]
- Albertson TM, Ogawa M, Bugni JM, Hays LE, Chen Y, Wang Y, Treuting PM, Heddle JA, Goldsby RE, Preston BD. 2009. DNA polymerase ε and δ proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci 106: 17101–17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers PM. 2009. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem 284: 4041–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CD, Eichler EE. 2013. Properties and rates of germline mutations in humans. Trends Genet 29: 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas J. 2011. Inbred mouse strains and genetic stability: a review. Animal 5: 1–7. [DOI] [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. 2009. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. 2012. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7: e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF. 2000. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 1: 40–47. [DOI] [PubMed] [Google Scholar]
- Crow JF, Kimura M. 1979. Efficiency of truncation selection. Proc Natl Acad Sci 76: 396–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Dolan PC, Wilhelm LJ, Sung W, Lucas-Lledo JI, Howe DK, Lewis SC, Okamoto K, Thomas WK, Lynch M, et al. 2009. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci 106: 16310–16314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, Crow JF. 1998. Rates of spontaneous mutation. Genetics 148: 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost JB, Lee WR. 1995. Biological basis of germline mutation: comparisons of spontaneous germline mutation rates among Drosophila, mouse, and human. Environ Mol Mutagen 25 Suppl 26: 48–64. [DOI] [PubMed] [Google Scholar]
- Duret L, Galtier N. 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet 10: 285–311. [DOI] [PubMed] [Google Scholar]
- Eory L, Halligan DL, Keightley PD. 2010. Distributions of selectively constrained sites and deleterious mutation rates in the hominid and murid genomes. Mol Biol Evol 27: 177–192. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD. 1999. High genomic deleterious mutation rates in hominids. Nature 397: 344–347. [DOI] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. 1996. Introduction to quantitative genetics, 4th ed Longman, Essex, England. [Google Scholar]
- Goldsby RE, Hays LE, Chen X, Olmsted EA, Slayton WB, Spangrude GJ, Preston BD. 2002. High incidence of epithelial cancers in mice deficient for DNA polymerase δ proofreading. Proc Natl Acad Sci 99: 15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondo Y, Fukumura R, Murata T, Makino S. 2009. Next-generation gene targeting in the mouse for functional genomics. BMB Rep 42: 315–323. [DOI] [PubMed] [Google Scholar]
- Herr AJ, Ogawa M, Lawrence NA, Williams LN, Eggington JM, Singh M, Smith RA, Preston BD. 2011. Mutator suppression and escape from replication error-induced extinction in yeast. PLoS Genet 7: e1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr AJ, Kennedy SR, Knowels GM, Schultz EM, Preston BD. 2014. DNA replication error-induced extinction of diploid yeast. Genetics 196: 677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. 2011. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477: 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keays DA, Clark TG, Flint J. 2006. Estimating the number of coding mutations in genotypic- and phenotypic-driven N-ethyl-N-nitrosourea (ENU) screens. Mamm Genome 17: 230–238. [DOI] [PubMed] [Google Scholar]
- Keightley PD, Ness RW, Halligan DL, Haddrill PR. 2014. Estimation of the spontaneous mutation rate per nucleotide site in a Drosophila melanogaster full-sib family. Genetics 196: 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1983. The neutral theory of molecular evolution. Cambridge University Press, Cambridge. [Google Scholar]
- Kimura M, Maruyama T. 1966. The mutational load with epistatic gene interactions in fitness. Genetics 54: 1337–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov AS, Crow JF. 1993. A molecular approach to estimating the human deleterious mutation rate. Hum Mutat 2: 229–234. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesecque Y, Keightley PD, Eyre-Walker A. 2012. A resolution of the mutation load paradox in humans. Genetics 191: 1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010a. Evolution of the mutation rate. Trends Genet 26: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010b. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci 107: 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon FM. 1959. Some evidence concerning the “mutational load” in inbred strains of mice. Heredity 13: 341–352. [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium, Waterston RH Lindblad-Toh K Birney E Rogers J Abril JF, Agarwal P, Agarwala R Ainscough R Alexandersson M, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
- Muller HJ. 1950. Our load of mutations. Am J Hum Genet 2: 111–176. [PMC free article] [PubMed] [Google Scholar]
- Muller U, Wang D, Denda S, Meneses JJ, Pedersen RA, Reichardt LF. 1997. Integrin α8β1 is critically important for epithelial–mesenchymal interactions during kidney morphogenesis. Cell 88: 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Sakumi K, Fukumura R, Furuichi M, Iwasaki Y, Hokama M, Ikemura T, Tsuzuki T, Gondo Y, Nakabeppu Y. 2014. 8-Oxoguanine causes spontaneous de novo germline mutations in mice. Sci Rep 4: 4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, et al. 2013. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prindle MJ, Loeb LA. 2012. DNA polymerase δ in DNA replication and genome maintenance. Environ Mol Mutagen 53: 666–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed FA, Aquadro CF. 2006. Mutation, selection and the future of human evolution. Trends Genet 22: 479–484. [DOI] [PubMed] [Google Scholar]
- Russell ES. 1978. Origins and history of mouse inbred strains: contributions of Clarence Cook Little. In Origins of inbred mice (ed. Morse HC), pp. 33–43. Academic Press, New York. [Google Scholar]
- Simon M, Giot L, Faye G. 1991. The 3′ to 5′ exonuclease activity located in the DNA polymerase δ subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J 10: 2165–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MM, Greenaway S, White JK, Fuchs H, Gailus-Durner V, Wells S, Sorg T, Wong K, Bedu E, Cartwright EJ, et al. 2013. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol 14: R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura A, Hidaka Y, Hirabayashi T, Hirabayashi M, Yagi T. 2009. DNA polymerase δ is required for early mammalian embryogenesis. PLoS One 4: e4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venn O, Turner I, Mathieson I, de Groot N, Bontrop R, McVean G. 2014. Nonhuman genetics. Strong male bias drives germline mutation in chimpanzees. Science 344: 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Fujiyama A, Hattori M, Taylor TD, Toyoda A, Kuroki Y, Noguchi H, BenKahla A, Lehrach H, Sudbrak R, et al. 2004. DNA sequence and comparative analysis of chimpanzee chromosome 22. Nature 429: 382–388. [DOI] [PubMed] [Google Scholar]
- Yauk CL, Aardema MJ, Benthem JV, Bishop JB, Dearfield KL, DeMarini DM, Dubrova YE, Honma M, Lupski JR, Marchetti F, et al. 2015. Approaches for identifying germ cell mutagens: report of the 2013 IWGT workshop on germ cell assays. Mutat Res Genet Toxicol Environ Mutagen 783: 36–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.