

Abstract
Polycomb group (PcG) complexes PRC1 and PRC2 are well known for silencing specific developmental genes. PRC2 is a methyltransferase targeting histone H3K27 and producing H3K27me3, essential for stable silencing. Less well known but quantitatively much more important is the genome-wide role of PRC2 that dimethylates ∼70% of total H3K27. We show that H3K27me2 occurs in inverse proportion to transcriptional activity in most non-PcG target genes and intergenic regions and is governed by opposing roaming activities of PRC2 and complexes containing the H3K27 demethylase UTX. Surprisingly, loss of H3K27me2 results in global transcriptional derepression proportionally greatest in silent or weakly transcribed intergenic and genic regions and accompanied by an increase of H3K27ac and H3K4me1. H3K27me2 therefore sets a threshold that prevents random, unscheduled transcription all over the genome and even limits the activity of highly transcribed genes. PRC1-type complexes also have global roles. Unexpectedly, we find a pervasive distribution of histone H2A ubiquitylated at lysine 118 (H2AK118ub) outside of canonical PcG target regions, dependent on the RING/Sce subunit of PRC1-type complexes. We show, however, that H2AK118ub does not mediate the global PRC2 activity or the global repression and is predominantly produced by a new complex involving L(3)73Ah, a homolog of mammalian PCGF3.
Polycomb group (PcG) repressive mechanisms generally involve two kinds of complexes. Polycomb repressive complex 2 (PRC2) methylates histone H3 lysine 27 (H3K27), while PRC1 or its variants can ubiquitylate histone H2A lysine 119 (lysine 118 in Drosophila). Some PRC1 complexes contain a chromodomain component, Polycomb (Pc) in Drosophila or one of several CBX proteins in mammals, that recognizes and binds to trimethylated H3K27. PRC1 and PRC2 complexes are generally bound at PcG target genes, where they establish a repressive chromatin state (for review, see Schwartz and Pirrotta 2013). The repressive action has been variously attributed to chromatin condensation, inhibition of transcription elongation by H2Aub, interference with nucleosome remodeling or transcription initiation, blocking of H3K27 acetylation, or any combination of these, but it is not well understood in detail. Targeted silencing requires PRC1 and PRC2 complexes to be stably bound to the target genes, which are usually marked by both H2AK118/119ub and H3K27me3. In Drosophila, the binding occurs specifically at DNA sequences called Polycomb response elements (PREs), where both PRC1 and PRC2 are thought to be recruited by the combinatorial action of DNA-binding proteins (Kassis and Brown 2013). In mammals, recent work has shown that most PcG binding occurs at CpG islands provided they are not DNA-methylated and not actively involved in transcription (Mendenhall et al. 2010; Lynch et al. 2012; Riising et al. 2014). Recruitment at CpG islands is mediated by KDM2B, a component of a variant PRC1 complex (Farcas et al. 2012; He et al. 2013; Wu et al. 2013). Recent work has shown that the H2AK118/119ub produced by this variant PRC1 can in turn recruit PRC2 complexes (Blackledge et al. 2014; Cooper et al. 2014) through their associated AEBP2 and JARID2 proteins (Kalb et al. 2014).
In addition to producing H3K27me3 at specific target genes, the PRC2 complex is also responsible for H3K27 mono- and dimethylation, whose function is not well understood. In particular, PRC2 produces a widespread and remarkably high level of H3K27me2 in flies and mammals that accounts for 45%–70% of all histone H3, while H3K27me3 constitutes 5%–10% (Peters et al. 2003; Ebert et al. 2004; Jung et al. 2010; Voigt et al. 2012; Ferrari et al. 2014). Quantitatively, H3K27 dimethylation is therefore the major function of the PRC2 complex, but its effects have gone largely unexplored.
Previous studies of the global effects of PRC2 loss of function in embryos lacking the maternal or zygotic esc gene encoding an essential core component of PRC2 (but possessing its close functional relative escl) found that thousands of non-PcG target genes acquired paused RNA Pol II bound in their promoter region (Chopra et al. 2011). The investigators did not consider the presence of H3K27me2 and concluded that the increased RNA Pol II binding was an indirect effect. Several studies in mammalian cells found that loss of PRC2 function affects more genes than those directly targeted by PcG complexes and H3K27me3 and attributed the broader effects to indirect consequences (Boyer et al. 2006; Bracken et al. 2006). Recently, Ferrari et al. (2014) confirmed the abundance and broad distribution of H3K27me2 in mouse embryonic stem cells and found H3K27me1 specifically in transcribed genes. They attributed a role to H3K27me2 in preventing inappropriate enhancer activation and a positive role to H3K27me1 in productive transcription.
Here we characterize the distribution and evaluate the effects of pervasive H3K27 methylation in the Drosophila genome. H3K27me2 is found ubiquitously except in actively transcribed regions or in PcG target regions, which contain H3K27me3. We find that loss of PRC2 function results in global transcriptional activation in both genic and intergenic regions to a degree proportional to the prior level of H3K27me2. We also examine the possible role of PRC1 and related complexes in mediating this activity and in producing global distributions of histone H2A ubiquitylation. We propose a model in which antagonistic genome-wide H3K27 methylation, demethylation, and acetylation control pervasive transcription by regulating the access of transcription factors and RNA Pol II to DNA.
Results
Widespread distribution of H3K27 methylation
All three levels of H3K27 methylation in the Drosophila genome were mapped as part of the modENCODE Project in ML-DmBG3-c2 (hereafter BG3) tissue culture cells (Kharchenko et al. 2011), and we obtained similar results for Sg4 cells (Fig. 1A; Supplemental Fig. 1). These distributions show that H3K27me3 is strongly enriched at specific regions associated with stable PRC2 and PRC1 binding at PREs. The distribution of H3K27me2 is far broader and nearly ubiquitous with the exception of sites enriched for H3K27me3 and regions associated with transcriptional activity as indicated by the presence of RNA Pol II, H3K4me3, and H3K36me3. H3K27me2 levels at non-PcG target genes are inversely correlated with transcriptional activity (Fig. 1B; Supplemental Fig. 1D), and H3K27me2-enriched regions largely correspond to the BLACK chromatin of Filion et al. (2010) (Supplemental Fig. 1A).
Figure 1.

Widespread distribution of H3K27 methylation. (A) Representative genome region showing the distribution of mono- (blue), di- (gray), and trimethylated H3K27 (green). Pc (red), and Pol II (purple) mapped by ChIP-chip in Bg3 cells (the modENCODE Project) or Sg4 cells (this study; Supplemental Fig. 1A). The H3K27me2 track shows the enrichment threshold based on the signal distribution (see Supplemental Fig. 1B–C). (B,C) Meta-gene profiles of H3K27me2 and H3K27me1 for PcG target genes and five subgroups of non-PcG target genes with different expression levels (e.g., NP-Q1 indicates the top 20 percentile active non-PcG target genes) in Bg3 cells (RNA tiling array data from the modENCODE Project). The x-axis represents the positions relative to the transcription start site (TSS; 0.0) and transcription end site (TES; 1.0). ChIP-chip signal values are scaled to the distribution with a mean of zero and a standard deviation of one. The same results normalized to the H3 distribution are shown in Supplemental Figure 1. (D–F) Meta-gene profiles of UTX, Pol II, and H3K27me2 for three subgroups of genes with different UTX binding levels in Sg4 cells.
As in mammalian cells (Ferrari et al. 2014), actively transcribed regions are instead enriched in H3K27me1 (Fig. 1C; Supplemental Fig. 1E). The loss of H3K27me2 might be ascribed to faster nucleosome turnover or to the active demethylation required for effective transcription. The appearance of H3K27me1 could be accounted for in either case by partial remethylation of new nucleosomes or by incomplete demethylation. ChIP shows that UTX, the sole Drosophila H3K27 demethylase, is enriched in active transcription units (Fig. 1D; Supplemental Fig. 1A). The more UTX, the higher the level of RNA Pol II and the greater the depletion of H3K27me2 in the transcription unit (Fig. 1E,F).
Loss of PRC2 function
To study the effects of pervasive H3K27 methylation, we used cultured cell lines carrying homozygous mutations in PcG genes, created by the procedure of Simcox et al. (2008) (see Methods). Line EZ2-2 is homozygous for E(z)61, a temperature-sensitive mutation that loses a large fraction of E(z) function and is embryonic lethal at 29°C (Jones and Gelbart 1990). At 31°C, EZ2-2 cells stop proliferating within 4 d (Fig. 2A), but they recover when returned to 25°C. Control Ras3 cells lacking the E(z)61 mutation continue to grow at 31°C. Figure 2B shows that both H3K27me2 and H3K27me3 decrease progressively and level off at ∼25% the normal value after 8 d at 31°C. Since the cells are not growing, this value represents the balance between demethylation, nucleosome turnover, and remethylation activity. Residual activity likely still occurs because the decrease is more drastic at 33°C. However, massive cell death occurs at this temperature, and the cells do not recover when returned to permissive temperature.
Figure 2.

Global changes of H3K27me2/3 levels after E(z) inactivation. (A) Growth curves of EZ2-2 and Ras3 (w.t.) cells after shift to 31°C. (B) Global H3K27me2/3 levels in EZ2-2 cells at 25°C and 31°C assayed by Western blot. Threefold serial dilutions of nuclear extract were loaded. Total H3 is the loading control. (C,D) H3K27me3 and H3K27me2 levels at PcG target and non-PcG target regions from EZ2-2 cells at 25°C and 31°C. H3K27me2/3 levels were measured by ChIP-qPCR from at least two independent experiments. The relative enrichment values were calculated based on the qPCR values normalized to input DNA. Error bars, SE. The four intergenic regions are identified by the distance in kb from the TSS (e.g., u12k). (E,F) The levels of Pc and E(z) at PcG target regions and two non-PcG target regions assayed by ChIP-qPCR from EZ2-2 cells grown at 25°C and 31°C. (G) H3K27me2 levels in highly expressed (HighExp 10%), intermediate (MidExp 10%), weakly expressed (LowExp 10%), and silent (no RNA-seq reads) non-PcG target genes and PcG target genes were analyzed from EZ2-2 H3K27me2 ChIP-seq data at 25°C and 31°C. (H) H3K27me2 levels in transcriptionally active intergenic non-PcG target (Top 10%) and silent regions (no RNA-seq reads) were analyzed from EZ2-2 H3K27me2 ChIP-seq data at 25°C and 31°C.
Chromatin immunoprecipitation (ChIP) qPCR and genomic ChIP-seq show that at 31°C the levels of H3K27me3 at PcG target genes drop dramatically (Fig. 2C; Supplemental Fig. 2B). While the level of Pc protein is not affected and that of E(z) slightly reduced (Supplemental Fig. 2A), the binding of Pc at PREs decreases to ∼50% but that of E(z) drops rapidly to background levels, indicating that the mutant protein does not bind at 31°C (Fig. 2E,F). Surprisingly, both qChIP and ChIP-seq results show that the level of H3K27me2 at PcG target genes increases significantly (Fig. 2D,G). Since E(z) no longer binds, this is most likely due to partial demethylation and, possibly, to de novo dimethylation.
ChIP-qPCR analysis of several silent intergenic regions and silent genes that are not PcG targets confirmed that H3K27me2 is enriched at 25°C and is depleted at 31°C (Fig. 2C,D). Notably, such regions also contain a low but significant level of H3K27me3. This is not due to antibody cross-reactivity because the ChIP signals for the two modifications are not correlated in non-PcG target regions and because quantitative ChIP shows that the level of H3K27me3 decreases over 10-fold at 31°C, while H3K27me2 decreases two- to fourfold. This suggests that a significant fraction of the total H3K27me3 exists outside of PcG target genes.
Loss of H3K27 methylation affects global transcription
As expected, shifting the EZ2-2 cells to 31°C caused strong derepression of PcG target genes (Fig. 3A). More surprisingly, a considerable increase in transcriptional activity occurred also at all tested non-PcG target regions (Fig. 3B,C). Even genes that are essentially silent at 25°C become significantly active at 31°C. Intergenic regions generally showed a strong relative increase in the levels of steady-state transcript produced. In contrast, intergenic expression levels in a control wild-type Ras3 cell line at 31°C show no significant increase relative to 25°C.
Figure 3.

Global transcriptional increase after E(z) inactivation. (A,B) Expression levels of PcG target genes and silent non-PcG target genes in EZ2-2 cells at 25°C and 31°C were assayed by RT-qPCR and shown relative to RpL32. Since the expression of RpL32 increases approximately twofold at 31°C (Supplemental Fig. 5A), this normalization is conservative. (C) Expression levels of a panel of silent intergenic non-PcG target regions in EZ2-2 cells at 25°C and 31°C. The Ras3 panel shows expression levels in a control wild-type cell line (Ras3) at the same temperatures. (D) Global derepression in coding regions after E(z) inactivation. The log ratio of the expression levels at 31°C and 25°C was calculated for non-PcG targets with different expression levels and for PcG targets. For silent genes, one expression tag read was added to the number of RNA-seq tags to estimate the approximate fold-differences. The bottom and top of each box represent the first and third quartiles, and the mark inside the box is the median value. Whiskers are extended to 1.5× interquartile range. (E) Preferential transcriptional increase at H3K27me2-enriched genes. Expression fold-change between 31°C and 25°C was calculated for the non-PcG target genes with the lowest and the highest levels of H3K27me2.
These effects of PRC2 inactivation are not specific for the EZ2-2 cell line. In a Su(z)12− cultured cell line carrying the loss-of-function Su(z)124 mutation [Su(z)12-2613], the cells lack functional Su(z)12 protein and are totally devoid of H3K27me2 and H3K27me3. RT-PCR analysis shows similar transcriptional derepression in Su(z)124 cells, compared with the wild-type Ras line, or in wild-type S2 cells after RNAi against E(z) (Supplemental Fig. 3). In all cases, the loss of H3K27me2/3 is accompanied by an increase in the level and distribution of transcriptional activity and, except in the case of the Su(z)12− cells, a decrease or arrest of cell proliferation.
To examine the broader transcriptome changes, we compared paired-end RNA-seq reads from total nonribosomal RNA of EZ2-2 cells at 25°C and 31°C, after appropriate normalization (see Methods) (Supplemental Fig. 5). The RNA-seq results confirm the general increase in steady-state transcript accumulation and reveal three major features: (1) The increase is genome-wide and affects intergenic regions as well as most annotated genes (Table 1; Fig. 3D,E; Supplemental Fig. 4). The proportion of intergenic regions showing detectable transcription increased nearly three times (from 15.5% to 44.5%) at 31°C (Table 1; Supplemental Fig. 4), and 79% (1264 genes) of the 1603 silent non-PcG target genes that had no RNA-seq reads at 25°C acquired RNA-seq reads at 31°C. (2) The increase is proportionally highest in the least transcriptionally active regions, but two- to fourfold increases occur even in very active genes (Fig. 3D). (3) The proportional increase is highly correlated to the level of H3K27me2 at 25°C (Fig. 2G,H; Supplemental Fig. 4C) except in the case of PcG target genes, which have high H3K27me3 but low H3K27me2 and become strongly derepressed. The effects in intergenic regions or genes that are silent at 25°C are likely to be underestimated due to insufficient sequencing depth, as shown by the fact that in many such regions transcription could be easily detected by qPCR at 31°C, but no reads were found by RNA-seq (Supplemental Fig. 6). Silent or weakly transcribed regions are enriched in H3K27me2/3 (Figs. 1B, 2G), and, in general, the higher the level of H3K27me2/3 at 25°C, the greater the proportional increase in transcription at 31°C (Fig. 3E).
Table 1.
Genome fraction of transcribing regions in EZ2-2 cells
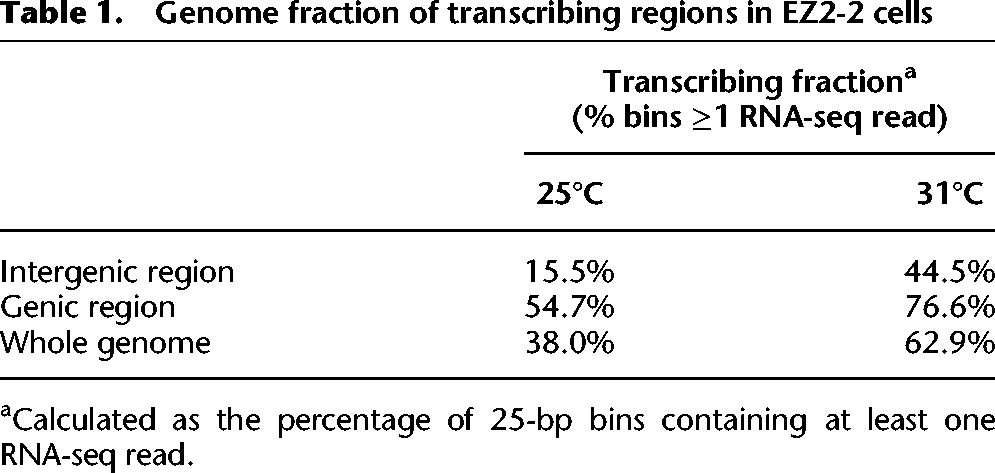
These results suggest that H3K27 methylation represses transcription in a graded manner throughout the genome and that it is the cause, not the consequence, of transcriptional inactivity. They also argue against the idea that the increase in transcription is an indirect effect due to derepression of PcG target genes and production of PcG-regulated activators.
Global derepression depends on activities associated with UTX
In EZ2-2 cells, since less than one cell doubling occurs after 12 d at 31°C, a significant loss of methylation requires active demethylation. To determine the role of H3K27 demethylation in the derepression of intergenic regions at 31°C, we knocked down UTX in these cells and observed significant attenuation of the derepression (Fig. 4A; Supplemental Fig. 6B). All tested intergenic regions showed more than a twofold decrease in transcript accumulation. This result indicates that the derepression upon E(z) inactivation is dependent on UTX function, which appears to continuously remove H3K27me2/3 throughout the genome but especially in transcribed regions. As noted above and also in mammalian cells (Ferrari et al. 2014), the genomic distributions of H3K27me2 and H3K27me1 are inversely related, and H3K27me1 increases in direct proportion to transcriptional activity. We suppose that UTX demethylates H3K27me2 in active genes, producing H3K27me1, which is less efficiently demethylated (Lan et al. 2007; Smith et al. 2008).
Figure 4.

Attenuation of intergenic derepression by depletion of Trithorax group proteins. Expression levels in a panel of intergenic regions were measured by RT-qPCR analysis with total RNA from EZ2-2 cells at 25°C and 31°C incubated with dsRNA targeting LacZ (mock treatment), Utx (A), trr (C), or CBP (E) for 12 d. The expression levels are normalized by the DNA amount in input cells. (B,D,F) The efficiency of the knockdown experiments was monitored by the mRNA levels of the targets.
E(z) inactivation increases transcription of most genes, including the UTX gene itself, whose transcript levels increase fivefold at 31°C. RNAi knockdown of Utx at 31°C reduces Utx RNA to the level present at 25°C (Fig. 4B). Our results show, therefore, that the global derepression observed at 31°C depends on the autocatalytic increase in UTX: Derepression increases UTX, which demethylates more and causes more derepression.
Global increase of H3K27ac and H3K4me1
H3K27 acetylation and H3K4 monomethylation are generally associated with enhancer activation and transcription, and H3K27ac levels are well correlated with the expression levels and are negatively correlated with H3K27me2/3 (Fig. 5C). In EZ2-2 cells shifted to 31°C, both H3K27ac and H3K4me1 increase, as determined at a panel of selected sites by quantitative ChIP-PCR (Fig. 5A,B) and genome-wide by ChIP-seq (Fig. 5C–E). The increase pattern is well correlated with that of transcription: The lower the level at 25°C, the stronger the proportional increase at 31°C (Fig. 5D,E). Higher H3K27ac was found also at intergenic regions, where the level of transcription is very low, although proportionally greatly increased. While we cannot determine whether acetylation is the cause or the consequence of the transcriptional increase, the inverse relationship between H3K27 methylation and acetylation at 25°C and 31°C suggests that the two are in a dynamic antagonism: Loss of methylation provides the opportunity for acetylation.
Figure 5.

H3K27ac and H3K4me1 levels increase upon E(z) inactivation. (A,B) H3K27ac and H3K4me1 in intergenic regions were assayed by ChIP-qPCR in EZ2-2 cells at 25°C and 31°C. (C) Box plots of H3K27ac levels in non-PcG targets with different expression levels and PcG targets at 25°C and 31°C. (D,E) The increase in H3K27ac and H3K4me1 levels after E(z) inactivation is negatively correlated with those levels at 25°C. The ratio of levels at 31°C and 25°C was calculated for non-PcG target genes with different levels at 25°C.
CBP/Nej, a versatile acetyl transferase, is responsible for the acetylation of H3K27 in Drosophila (Tie et al. 2009). Recent studies have attributed H3K4me1 to TRR (Ardehali et al. 2011; Herz et al. 2012), although it has also been assigned to Trithorax (Tie et al. 2014). Significantly, CBP and TRR each form a complex containing UTX (Mohan et al. 2011; Tie et al. 2012), suggesting that these complexes may work in tandem demethylating H3K27 and writing H3K27ac and H3K4me1. To test whether CBP and TRR are involved in intergenic derepression, we knocked each down at 31°C and found that the derepression levels were reduced (Fig. 4C–F; Supplemental Fig. 6). The effects vary: Some intergenic regions were strongly affected by CBP RNAi but were weakly affected by TRR RNAi or vice versa, indicating that derepression in some regions is more dependent on CBP and in others on TRR.
PRC1 involvement in intergenic repression
The chromodomain of the Pc protein in the canonical PRC1 complex binds preferentially H3K27me3 but also H3K27me2 at fivefold lower affinity (Fischle et al. 2003). Given the high levels of H3K27me2 and low but appreciable levels of H3K27me3 broadly distributed in the genome, could these attract the transient presence of PRC1, which might then be responsible for the suppression of pervasive transcription? To test this, we examined the expression level of a panel of non-PcG target regions after RNAi knockdown of PRC1 components in S2 cells (Fig. 6F; Supplemental Fig. 7A–D). Depletion of Pc or RING/Sce results in significantly higher levels of intergenic transcription. A similar increase was observed in a Pc− mutant cell line and a Psc/Su(z)2− mutant cell line compared with wild-type Ras3 cells (Supplemental Fig. 7E,F). These results suggest that PRC1 also plays a role in intergenic repression. The RING/Sce component of PRC1 is a E3 ubiquitin ligase responsible for H2AK118 mono-ubiquitination. No RING/Sce is detected by ChIP in the intergenic regions tested (the modENCODE Project), but a transient presence might produce H2A ubiquitylation, which has been reported to suppress transcriptional activity (Zhou et al. 2008). In mammals, H2AK119ub has been found to recruit PRC2 (Blackledge et al. 2014; Cooper et al. 2014; Kalb et al. 2014), raising the possibility that the ubiquitous activity of a PRC1-type complex might transiently recruit PRC2 and produce ubiquitous H3K27me2.
Figure 6.

PRC1 involvement in intergenic repression. (A) Western blot analysis shows the loss of total H2AK118ub after 12 d of RING/Sce RNAi on S2 cells. LacZ dsRNA was used as control; twofold serial dilutions of nuclear lysate were loaded; and total histones stained with Coomassie blue served as loading controls. (B) H2AK118ub levels in PcG and intergenic non-PcG target regions were assayed by ChIP-qPCR after treatment of S2 cells with dsRNA for LacZ (control) or RING/Sce. H2AK118ub levels in three PcG target regions, one active gene region, and five intergenic non-PcG target regions were measured by ChIP-qPCR. (C) The H2Aub1 levels of intergenic non-PcG (transcriptionally active or silent) and PcG target regions in EZ2-2 cells were determined from the 25°C and 31°C ChIP-seq data. (D) H3K27me2 levels after RING/Sce knockdown in S2 cells were assayed by ChIP-qPCR compared with control treatment with LacZ dsRNA. (E) Decrease of global H2AK118ub level after RNAi depletion of l(3)73Ah in S2 cells. (F) Expression levels in intergenic and genic non-PcG target regions were measured by RT-qPCR with total RNA from mock-treated and Pc, E(z), or L(3)73Ah-depleted S2 cells. Changes in mRNA levels of l(3)73Ah, Pc and E(z) after knockdown are shown in Supplemental Figures 7G, 7B, and 3B, respectively. (G) Distribution of H2AK118ub (blue) in the genomic region surrounding the bithorax complex as determined by ChIP-seq. For comparison, the distributions of Pc (red), H3K27me3 (green), and H3K27me2 (gray) are also shown, indicating the absence of H2AK118ub at the entire BX-C (from Ubx to Abd-B) but presence in other regions both with and without PC or H3K27me2/3.
To determine the genomic distribution of H2AK118ub, we validated a new commercial anti-H2AK119ub antibody (Cell Signaling D27C4) by Western blotting and ChIP upon RING/Sce depletion and found that it is highly specific for Drosophila H2AK118ub (Fig. 6A,B). ChIP-qPCR results confirmed that H2AK118ub is significantly high in intergenic regions and is strongly depleted when RING/Sce is knocked down (Fig. 6B) in S2 cells.
It has been argued that canonical PRC1 is not very active as a H2A ubiquityl transferase and that most of this activity is instead attributable, in flies as in mammals, to alternative RING/Sce-containing complexes that lack a chromodomain component (Lagarou et al. 2008; Gao et al. 2012). We tested first whether H2AK118ub is reduced upon loss of H3K27 methylation in EZ2-2 cells. Western blot analysis shows that at 31°C, H2AK118ub decreases by ∼25% but is not affected in E(z)+ Ras3 cells (Supplemental Fig. 8A,B). Further analysis in EZ2-2 cells by ChIP-qPCR and ChIP-seq showed that the decrease in H2AK118ub at 31°C is largely attributable to a drastic drop at most PcG target genes, while other genomic regions are only mildly affected (Fig. 6C; Supplemental Fig. 8C). These results argue that if canonical PRC1 is recruited by ubiquitous H3K27me2/3, it accounts for only a small fraction of global H2AK118ub, most of which must be attributed instead to variant RING/Sce-containing complexes.
RING ubiquityl transferase activity requires a heterodimer of RING and a RING domain–containing partner such as Psc or Su(z)2 in flies or PCGF proteins in mammals. However, a Pc− cell line (PC1) and a Psc/Su(z)2− cell line (Psc4-1) (Kahn et al. 2014) both retain >60% of the wild-type H2AK118ub level (Supplemental Fig. 8D,E), which is therefore not attributable to either canonical PRC1 or dRAF/KDM2 complexes, both of which contain Psc or Su(z)2. If deletion of Psc and Su(z)2 genes leaves 60% of the H2AK118ub, what might be the alternative RING/Sce partner? The Drosophila genome contains l(3)73Ah, a gene homologous to the RING domain region of Psc and Su(z)2 (Irminger-Finger and Nöthiger 1995) but with the strongest homology to mammalian PCGF3. Knockdown of l(3)73Ah in S2 cells shows that it is responsible for ∼70% of the total H2AK118ub (Fig. 6E; Supplemental Fig. 8F) and therefore that complexes containing this protein are major contributors to H2AK118 ubiquitylation.
Genomic distribution of H2AK118ub
The genomic distribution of H2AK118ub underscores that at least in Drosophila, the role of this modification is far from clear. H2AK118ub is surprisingly widespread, often in correspondence with peaks of H3K27me2/3 with or without PRC1 components (Fig. 6G; Supplemental Fig. 9). In addition, although many PcG target genes contain important peaks of H2AK118ub corresponding to binding of both PRC1 and PRC2, others, including some of the best-known PcG targets such as the entire bithorax complex and the Antp gene, lack appreciable H2AK118ub. We conclude that while H2AK118ub may contribute to PRC2 recruitment, as was recently proposed (Blackledge et al. 2014; Cooper et al. 2014; Kalb et al. 2014), it is neither essential nor sufficient. In particular, RING/Sce knockdown experiments show that while H2AK118ub is efficiently depleted, the level of H3K27me2 is only slightly decreased (Fig. 6B,D), indicating that RING/Sce complexes are not directly involved in the pervasive H3K27 dimethylation.
Discussion
In Drosophila, as in mammalian cells, H3K27 methylation is highly abundant and nearly ubiquitous. While H3K27me2 is the predominant and broadly distributed form, our results show that in most regions, it is accompanied by a low level of H3K27me3 (Figs. 2C, 6G) that makes a significant contribution to the total H3K27me3, unrelated to classical PcG target genes. H3K27me2 is enriched wherever transcriptional activity is low, both in genes and in intergenic regions, but as mass spectrometry/SILAC studies show (Zee et al. 2012; Alabert et al. 2015), it is converted to H3K27me3 at PcG target sites. In transcribed regions, loss of H3K27me2/3 may be caused in part by nucleosome eviction but is, in large part, attributable to the action of the UTX demethylase, which accumulates as a function of transcriptional activity.
Loss of H3K27 methylation at 31°C in EZ2-2 cells or by E(z) RNAi in wild-type cells slows down or arrests cell proliferation, but this was not seen in the Su(z)12− cell line. A proliferation defect in the absence of E(z) function in Drosophila was also reported by Beuchle et al. (2001), and PRC2 function is required for growth in differentiated mammalian cells (Bracken et al. 2003; Pasini et al. 2004) but not in undifferentiated or embryonic stem cells (Montgomery et al. 2005; Pasini et al. 2007). In the EZ2-2 cells, the proliferation deficit upon loss of PRC2 function means that significant changes in the methylation pattern can only be achieved by demethylation or by nucleosome turnover. Since loss of methylation occurs also in regions where transcription remains very low in absolute terms, we conclude that it is due to active demethylation, and knockdown of Utx shows that it is a key factor in derepression, affecting both genic and intergenic transcription. The fact that demethylation occurs in regions that have little or no transcription implies that UTX normally scans the genome in constant antagonism with PRC2, e.g., by a genome-wide hit-and-run mechanism.
Loss of H3K27 methylation causes global transcriptional derepression
Loss of H3K27 methylation, whether by temperature shift in the EZ2-2 cell line, by E(z) RNAi in wild-type cells, or by a null mutation in the Su(z)124 cell line, causes a genome-wide increase in transcriptional activity not only at PcG target genes but also in most other genes and intergenic regions, whether previously active or silent. Overall, the increase in transcription is proportional to the normal levels of H3K27me2/3, which themselves are inversely related to normal transcription levels. The changes are therefore proportionally greatest in weakly transcribed or silent regions, although even very active genes increase in transcription two- to fourfold. Much of this transcription appears to be opportunistic, arising at any site transiently accessible to RNA polymerase. This activity is likely to be underestimated since such unscheduled transcripts are targets for degradation by surveillance mechanisms and nonsense-mediated decay (Isken and Maquat 2007).
One obvious way in which H3K27 methylation could inhibit transcriptional activity is by preventing H3K27 acetylation, which, together with H3K4me1, is associated with enhancer activation and active promoter regions. A dynamic antagonism between H3K27 methylation and H3K27 acetylation and H3K4 methylation at the PcG target sites has been proposed by Tie et al. (2009) and Pasini et al. (2010). Our results show that loss of H3K27 methylation is accompanied by a genome-wide increase of H3K27ac and H3K4me1 correlated with the levels of genome-wide derepression (Fig. 5). RNAi knockdown experiments show that CBP and TRR contribute to transcriptional derepression but to various extents, depending on the site tested. How H3K27ac and H3K4me1 are mechanistically connected is still not well understood, but both CBP and TRR complexes are reported to include UTX (Mohan et al. 2011; Tie et al. 2012), which is associated with active transcription (Smith et al. 2008). In vitro, mammalian and fly UTX demethylases act preferentially on H3K27me2/3 (Lan et al. 2007; Smith et al. 2008). In fact, loss of H3K27me2 in active genes is accompanied by gain in H3K27me1 (Fig. 1B,C), but some de novo monomethylation cannot be excluded (Ferrari et al. 2014). We propose that both UTX-containing complexes contribute to global H3K27 demethylation in a constant, ubiquitous, and dynamic antagonism with PRC2-dependent methylation. This evidence strongly suggests that H3K27 methylation interferes with transcriptional activity and must be removed at active genes. We conclude that ubiquitous H3K27 methylation moderates transcription at active genes, helps prevent spurious transcription of inactive genes, and suppresses low but pervasive transcription of intergenic regions.
Involvement of PRC1 complexes in global repression
Increased expression of non-PcG target upon loss of PRC2 function genes has been reported before and attributed to indirect effects (Chopra et al. 2011; Ferrari et al. 2014). There are two possible ways, one indirect and one direct, in which PRC1 complexes might contribute to global H3K27me-dependent transcriptional repression. The indirect argument proposes that, since PcG target genes are enriched for transcription factors, their derepression might produce a flood of activators whose specific and nonspecific effects might account for the global increase in transcription. Our experiments cannot exclude this interpretation, particularly since we find that loss of PRC1 components also produces increases in transcription of intergenic regions. However, we note that total loss of Pc or of Psc/Su(z)2 in the mutant cell lines or RNAi knockdown of Pc or RING/Sce always have weaker effects than PRC2 inactivation. Transcription factors do not account for the strong correlation between derepression and the level of H3K27me2/3. Derepression by activators would be expected to have particularly strong effects at genes that are the normal targets of such activators and much weaker and uneven effects at other sites. Furthermore, this explanation ignores the role of UTX in transcription initiation and elongation.
The second, more direct possible involvement of canonical PRC1 is by transient binding to ubiquitous H3K27me2/3 through the Pc chromodomain. Transient PRC1 complexes might interfere with transcription in some poorly understood way, possibly involving ubiquitylation of histone H2A (Zhou et al. 2008). Our results show that PRC1 components Pc, RING/Sce, and Psc/Su(z)2 do have a general effect that is similar to but weaker than that of H3K27 methylation on intergenic transcription. However, this is unlikely to be mediated by H2AK119 ubiquitylation. The strongest transcriptional derepression was observed in the EZ2-2 line at 31°C, but at this temperature, the H2AK118ub level is only very slightly reduced, except at PcG target genes. Conversely, knockdown of RING/Sce has a drastic effect on H2AK118ub levels everywhere but has very little effect on H3K27me2 levels. That H2AK118ub or transient PRC1 recruitment might be responsible for the transcriptional repression we observe seems unlikely considering that in EZ2-2 cells at 31°C or in Su(z)12− cells, where PRC1 binding and H2AK118ub at PcG target genes are only mildly reduced, these target genes are nevertheless strongly derepressed.
In flies and mammals, canonical PRC1 complexes are reported to have little H2AK119/118 ubiquitylating activity, which in flies is attributed instead to the dRAF/KDM2 complex (Lagarou et al. 2008) and to a range of variant PRC1 complexes lacking a chromodomain in mammals (Gao et al. 2012). RING/Sce and Psc/Su(z)2 are components of both canonical PRC1 and dRAF/KDM2 complexes (Lagarou et al. 2008). We find that loss of Pc or Psc/Su(z)2 decreases total H2AK118ub by 30%–40%, while ∼70% is attributable to L(3)73Ah, a homolog of mammalian PCGF3. H2AK118ub is therefore produced by at least two different kinds of RING/Sce-containing complexes, one including Psc/Su(z)2 and one L(3)73Ah. However, RNAi knockdown of l(3)73Ah has little or no effect on intergenic transcription (Fig. 6F; Supplemental Fig. 7G). In sum, our results suggest that both canonical PRC1 and H3K27 methylation may play a role in ubiquitous repression, whether independently or linked by the recruitment of PRC1 through the Pc chromodomain. Unfortunately, the unresponsiveness of the Su(z)124− and the Psc/Su(z)2− cell lines to RNAi treatments prevented us from assessing the relationship between the two.
H2AK118ub and PRC2 recruitment
The global distribution of H2AK118ub raises many new questions about its role in PcG recruitment and repression that are beyond the scope of this article. Here we note that while many PcG-binding regions and H3K27me3 domains correspond to high peaks of H2AK118ub, some of the most prominent PcG targets, such as the Antp gene and the bithorax complex, are devoid of H2AK118ub. The Calypso deubiquitylase is likely responsible for removing H2AK118ub, which in some way interferes with effective repression at these loci (Scheuermann et al. 2010). At these genes, at least, PRC2 recruitment does not require H2AK118ub. In contrast, H2AK118ub is widely distributed in the genome in silent genes and intergenic regions, a pattern similar to that of H3K27me2, but is not accompanied by stable binding of either RING/Sce or E(z). Recent work in mammalian cells has suggested that H2AK119ub is responsible for the recruitment of PRC2 to CpG islands. Our results show that, at least in Drosophila, H2AK118ub does not lead to stable binding of PRC2, although it may help in attracting it transiently. RING/Sce knockdown, while very efficiently depleting H2AK118ub, has only a slight effect on H3K27me2 (Fig. 6D), indicating that it is not the main driver of global H3K27 dimethylation. However, in the fly, L(3)73Ah function is essential for viability, probably because, together with H3K27me2, it provides the initial substrate for the recruitment of PcG repression at specific target genes.
Methods
Cell lines and RNAi
Cultured cell lines bearing E(z)61, Pc3, Def(2R)Su(z)2-1.b8, or Su(z)124 PcG mutations were established by the procedure of Simcox et al. (2008) from embryos carrying those mutations in the presence of a transgene expressing a constitutively active form of Ras, RasV12. These cell lines, as well as Schneider S2 cells and its clonal derivative Sg4, were grown at 25°C. For the EZ2-2 temperature-sensitive cell line, E(z) activity was inactivated by shifting to 31°C for 12 or 24 d. RNAi treatments were done according to the method described by Schwartz et al. (2010) with three consecutive treatments with corresponding dsRNA with growth for 4 d between treatments. For the knockdown experiments in the EZ2-2 cell line, one pretreatment with dsRNA was done 4 d before the temperature shift.
Chromatin immunoprecipitation
ChIP was carried out according to the method described by Schwartz et al. (2006). Crosslinking was done with 1.8% formaldehyde for 10 min, stopped with glycine to a final concentration of 0.125 M, and washed with PBS and with ChIP wash buffer. Sonication was done in TE buffer (10 mM Tris-HCl; 1 mM EDTA at pH 8.0) with 0.1% SDS in a bioruptor (Diagenode) at high power 30 sec on/off 5 × 5 min in ice water. The lysate was adjusted to RIPA buffer, and immunoprecipitation was done overnight at 4°C with the antibodies listed in the Supplemental Methods. The complexes were recovered with Protein A–Sepharose beads (Sigma) for 3 h at 4°C; washed five times with RIPA, once with 1 ml LiCl buffer (250 mM LiCl, 10 mM Tris-HCl at pH 8.0, 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate), and twice with TE; and treated with RNase A 50 mg/mL. DNA eluted from the beads was analyzed by qPCR, hybridization to Drosophila Tiling 1.0R microarrays (Affymetrix), or high-throughput sequencing (Illumina HiSeq). The genomic data on BG3 cells were taken from the modENCODE Project (Kharchenko et al. 2011).
Transcriptome analysis
Total RNA and DNA were isolated from the same cell samples. The DNA was used to normalize the relative cell number in different samples by qPCR. Reverse transcription was performed with 1 µg of total RNA using the first-strand cDNA synthesis kit with random hexamers (0.2 µg per reaction) according to the manufacturer's instructions (GE Healthcare). All primer sequences used are given in Supplemental Table S1. For whole-transcriptome analysis, total RNA was depleted of ribosomal sequences with the RiboZero kit (Epicentre), and sequencing libraries were generated with the ovation RNA-seq system V2 kit or encore complete RNA-seq library system (NuGEN) and paired-end sequencing with Illumina HiSeq. For more details, see the Supplemental Methods.
Sequence data processing and analysis
ChIP-seq reads were mapped to the Drosophila genome (FlyBase 5.22) using Bowtie (Langmead et al. 2009), and RNA-seq reads were mapped by GeneSifter (www.genesifter.net). For comparisons between samples and experiments, data were normalized according to ChIP-qPCR or RT-qPCR assays using at least five regions. The RNA-seq data were also normalized using four spike-in standards (single-stranded RNAs made from lambda DNA) added before library construction. The two sets of normalization factors were in good agreement (Supplemental Fig. 5). Sequence data processing is described in detail in the Supplemental Methods.
Data access
The microarray and sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE61280.
Supplementary Material
Acknowledgments
We thank Katsuhito Ohno for purifying the anti-UTX antibody and Dale Dorsett for making us aware of the H2AK119ub antibody from Cell Signaling. This work was supported in part by grants from the Swedish Research Council, Carl Tryggers Stiftelse, and European Network of Excellence EpiGeneSys to Y.B.S. and from the US National Institutes of Health to V.P.
Author contributions: H.-G.L., Y.B.S., and V.P. designed the experiments, analyzed the data, and wrote the manuscript. H.-G.L., Y.B.S., and T.G.K. did the experiments. A.S., H.-G.L., Y.B.S., and T.G.K. generated new cell lines.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.188920.114.
References
- Alabert C, Barth TK, Reverón-Gómez N, Sidoli S, Schmidt A, Jensen ON, Imhof A, Groth A. 2015. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev 29: 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardehali MB, Mei A, Zobeck KL, Caron M, Lis JT, Kusch T. 2011. Drosophila Set1 is the major histone H3 lysine 4 trimethyltransferase with role in transcription. EMBO J 30: 2817–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuchle D, Struhl G, Müller J. 2001. Polycomb group proteins and heritable silencing of Drosophila Hox genes. Development 128: 993–1004. [DOI] [PubMed] [Google Scholar]
- Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LLP, Ito S, Cooper S, Kondo K, Koseki Y, et al. 2014. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and Polycomb domain formation. Cell 157: 1445–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349–353. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini M, Colli E, Helin K. 2003. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22: 5323–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. 2006. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 20: 1123–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra VS, Hendrix DA, Core LJ, Tsui C, Lis JT, Levine M. 2011. The polycomb group mutant esc leads to augmented levels of paused Pol II in the Drosophila embryo. Mol Cell 42: 837–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Dienstbier M, Hassan R, Schermelleh L, Sharif J, Blackledge NP, DeMarco V, Elderkin S, Koseki H, Klose R, et al. 2014. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep 7: 1456–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert A, Schotta G, Lein S, Kubicek S, Krauss V, Jenuwein T, Reuter G. 2004. Su(var) genes regulate the balance between euchromatin and heterochromatin in Drosophila. Genes Dev 18: 2973–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farcas AM, Blackledge NP, Sudbery I, Long HK, McGouran JF, Rose NR, Lee S, Sims D, Cerase A, Sheahan TW, et al. 2012. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. Elife 1: e00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stützer A, Fischle W, Bonaldi T, Pasini D. 2014. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell 53: 49–62. [DOI] [PubMed] [Google Scholar]
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, et al. 2010. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 143: 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. 2003. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17: 1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, Reinberg D. 2012. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell 45: 344–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Shen L, Wan M, Taranova O, Wu H, Zhang Y. 2013. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat Cell Biol 15: 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz H-M, Mohan M, Garruss AS, Liang K, Takahashi Y-h, Mickey K, Voets O, Verrijzer CP, Shilatifard A. 2012. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev 26: 2604–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irminger-Finger I, Nöthiger R. 1995. The Drosophila melanogaster gene lethal(3)73Ah encodes a ring finger protein homologous to the oncoproteins MEL-18 and BMI-1. Gene 163: 203–208. [DOI] [PubMed] [Google Scholar]
- Isken O, Maquat LE. 2007. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mENA function. Genes Dev 21: 1833–1856. [DOI] [PubMed] [Google Scholar]
- Jones RS, Gelbart WM. 1990. Genetic analysis of the enhancer of zeste locus and its role in gene regulation in Drosophila melanogaster. Genetics 126: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HR, Pasini D, Helin K, Jensen ON. 2010. Quantitative mass spectrometry of histones H3.2 and H3.3 in Suz12-deficient mouse embryonic stem cells reveals distinct, dynamic post-translational modifications at Lys-27 and Lys-36. Mol Cell Proteomics 9: 838–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn TG, Stenberg P, Pirrotta V, Schwartz YB. 2014. Combinatorial interactions are required for the efficient recruitment of pho repressive complex (PhoRC) to polycomb response elements. PLoS Genet 10: e1004495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb R, Latwiel S, Baymaz HI, Jansen PWTC, Müller CW, Vermeulen M, Müller J. 2014. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol 21: 569–571. [DOI] [PubMed] [Google Scholar]
- Kassis JA, Brown JL. 2013. Polycomb group response elements in Drosophila and vertebrates. Adv Genet 81: 83–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. 2011. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarou A, Mohd-Sarip A, Moshkin YM, Chalkley GE, Bezstarosti K, Demmers JAA, Verrijzer CP. 2008. dKDM2 couples histone H2A ubiquitylation to histone H3 demethylation during Polycomb group silencing. Genes Dev 22: 2799–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, et al. 2007. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449: 689–694. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MD, Smith AJH, De Gobbi M, Flenley M, Hughes JR, Vernimmen D, Ayyub H, Sharpe JA, Sloane-Stanley JA, Sutherland L, et al. 2012. An interspecies analysis reveals a key role for unmethylated CpG dinucleotides in vertebrate Polycomb complex recruitment. EMBO J 31: 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, Bernstein BE. 2010. GC-Rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet 6: e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan M, Herz H-M, Smith ER, Zhang Y, Jackson J, Washburn MP, Florens L, Eissenberg JC, Shilatifard A. 2011. The COMPASS family of H3K4 methylases in Drosophila. Mol Cell Biol 31: 4310–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery ND, Yee D, Chen A, Kalantry S, Chamberlain SJ, Otte AP, Magnuson T. 2005. The murine Polycomb Group protein Eed is required for global histone H3 lysine-27 methylation. Curr Biol 15: 942–947. [DOI] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Jensen MR, Denchi EL, Helin K. 2004. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23: 4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. 2007. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol Cell Biol 27: 3769–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L, Skotte J, Wutz A, Porse B, Jensen ON, et al. 2010. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res 38: 4958–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AHFM, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AAHA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, et al. 2003. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12: 1577–1589. [DOI] [PubMed] [Google Scholar]
- Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, Helin K. 2014. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell 55: 347–360. [DOI] [PubMed] [Google Scholar]
- Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW, Muller J. 2010. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465: 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz YB, Pirrotta V. 2013. A new world of Polycombs: unexpected partnerships and emerging functions. Nat Rev Genet 14: 853–864. [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Kahn TG, Nix DA, Li X-Y, Bourgon R, Biggin M, Pirrotta V. 2006. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat Genet 38: 700–705. [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Kahn TG, Stenberg P, Ohno K, Bourgon R, Pirrotta V. 2010. Alternative epigenetic chromatin states of polycomb target genes. PLoS Genet 6: e1000805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcox A, Mitra S, Truesdell S, Paul L, Chen T, Butchar JP, Justiniano S. 2008. Efficient genetic method for establishing Drosophila cell lines unlocks the potential to create lines of specific genotypes. PLoS Genet 4: e1000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Lee MG, Winter B, Droz NM, Eissenberg JC, Shiekhattar R, Shilatifard A. 2008. Drosophila UTX is a histone H3 Lys27 demethylase that colocalizes with the elongating form of RNA polymerase II. Mol Cell Biol 28: 1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, Diaz MO, Scacheri PC, Harte PJ. 2009. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136: 3131–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie F, Banerjee R, Conrad PA, Scacheri PC, Harte PJ. 2012. Histone demethylase UTX and chromatin remodeler BRM bind directly to CBP and modulate acetylation of histone H3 Lysine 27. Mol Cell Biol 32: 2323–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie F, Banerjee R, Saiakhova AR, Howard B, Monteith KE, Scacheri PC, Cosgrove MS, Harte PJ. 2014. Trithorax monomethylates histone H3K4 and interacts directly with CBP to promote H3K27 acetylation and antagonize Polycomb silencing. Development 141: 1129–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P, LeRoy G, Drury WJ III, Zee BM, Son J, Beck DB, Young NL, Garcia BA, Reinberg D. 2012. Asymmetrically modified nucleosomes. Cell 151: 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Johansen JV, Helin K. 2013. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol Cell 49: 1134–1146. [DOI] [PubMed] [Google Scholar]
- Zee BM, Britton L-MP, Wolle D, Haberman DM, Garcia BA. 2012. Origins and formation of histone methylation across the human cell cycle. Mol Cell Biol 32: 2503–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Zhu P, Wang JK, Pascual G, Ohgi KA, Lozach J, Glass CK, Rosenfeld MG. 2008. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell 29: 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.