

Abstract
Chromium in its toxic Cr(VI) valence state is a common contaminant particularly associated with alkaline environments. A well-publicized case of this occurred in Glasgow, United Kingdom, where poorly controlled disposal of a cementitious industrial by-product, chromite ore processing residue (COPR), has resulted in extensive contamination by Cr(VI)-contaminated alkaline leachates. In the search for viable bioremediation treatments for Cr(VI), a variety of bacteria that are capable of reduction of the toxic and highly soluble Cr(VI) to the relatively nontoxic and less mobile Cr(III) oxidation state, predominantly under circumneutral pH conditions, have been isolated. Recently, however, alkaliphilic bacteria that have the potential to reduce Cr(VI) under alkaline conditions have been identified. This study focuses on the application of a metal-reducing bacterium to the remediation of alkaline Cr(VI)-contaminated leachates from COPR. This bacterium, belonging to the Halomonas genus, was found to exhibit growth concomitant to Cr(VI) reduction under alkaline conditions (pH 10). Bacterial cells were able to rapidly remove high concentrations of aqueous Cr(VI) (2.5 mM) under anaerobic conditions, up to a starting pH of 11. Cr(VI) reduction rates were controlled by pH, with slower removal observed at pH 11, compared to pH 10, while no removal was observed at pH 12. The reduction of aqueous Cr(VI) resulted in the precipitation of Cr(III) biominerals, which were characterized using transmission electron microscopy and energy-dispersive X-ray analysis (TEM-EDX) and X-ray photoelectron spectroscopy (XPS). The effectiveness of this haloalkaliphilic bacterium for Cr(VI) reduction at high pH suggests potential for its use as an in situ treatment of COPR and other alkaline Cr(VI)-contaminated environments.
INTRODUCTION
Chromium (Cr) is a significant component of contaminated soil and groundwater through a variety of environmental exposures from its widespread use in metallurgy and industrial processes (1–3). Under most environmental conditions, it is stable as the Cr(VI) and Cr(III) valence states (4). The Cr(III) state dominates under reducing conditions, forming largely insoluble Cr(III) hydroxide phases (5, 6), which are widely considered nontoxic (7). In contrast, the Cr(VI) species dominates under oxidizing conditions, forming the toxic, carcinogenic, and highly soluble oxyanions HCrO4−, CrO42−, and Cr2O42− (8, 9). Due to the greater stability and mobility of Cr(VI) at high pH, it is particularly associated with contaminated alkaline environments (10). A well-known example of alkaline Cr(VI) contamination relates to the poorly controlled disposal of waste from the “high-lime” chromite ore (FeCr2O4) processing technique, chromite ore processing residue (COPR) (11, 12). Roasting the chromite ore with lime causes oxidation of Cr(III) to Cr(VI), enabling leaching with water (12). However, due to inefficiencies in the process, COPR contains significant concentrations of Cr, typically 3 to 7% by mass, of which 1 to 30% is typically in the Cr(VI) state (13, 14), while the addition of lime produces typically high pH values of 11 to 13 (15). The Cr(VI) forms part of a complex mineralogy, which upon saturation with groundwater readily yields alkaline leachate with high concentrations of aqueous Cr(VI) (15–17). COPR-related contamination is a global issue, with significant cases reported in the United Kingdom, United States, Eastern Europe, India, Pakistan, and China (11, 12, 18). For example, in Glasgow, United Kingdom, the poorly controlled disposal of >2 million metric tons of COPR has resulted in widespread contamination with highly alkaline Cr(VI) leachate, with groundwater and surface water containing up to 100 mg liter−1 (16, 19–21). These values are far in excess of the World Health Organization's upper limit for Cr(VI) in drinking water, 0.05 mg liter−1 (22).
A potential treatment of Cr(VI) contamination involves harnessing the microbial metabolism of bacteria that are capable of enzymatic metal reduction, reducing Cr(VI) to the relatively insoluble Cr(III) (23, 24). The ability to enzymatically reduce Cr(VI) has been observed among a diverse range of bacteria (25, 26), primarily among the facultative anaerobes (27). Microbial Cr(VI) reduction is often attributed to enzymes that have alternative metabolic functions (28), while a restricted range of bacteria are capable of using Cr(VI) as the terminal electron acceptor for growth (29, 30). Most previous studies have been carried out at near-neutral conditions, and pH extremes have proved a major limiting factor to enzymatic reduction (31). As Cr(VI) contamination is primarily associated with alkaline environments (4), several studies have sought to culture alkaliphilic bacteria capable of Cr(VI) reduction at high pH (32–37). Alkaliphiles exhibit optimum growth under alkaline conditions (pH 9 to 12) (38), while a number of these, the haloalkaliphiles, also require salinity for optimum growth (39). Haloalkaliphiles of the Halomonas genus are especially well represented in high-pH and high-salt environments (40, 41), and a number of studies have found these organisms to be capable of Cr(VI) reduction (34, 36). Halomonas species have also been reported for other remedial reactions, such as the reduction of nitrate (42), while the isolate used in this current study was also able to reduce the nuclear contaminant Tc(VII) to Tc(IV) (43).
Despite the identification of a small number of alkaliphilic bacteria capable of reducing Cr(VI) in model laboratory solutions, there remains a need to test these bacterial systems against environmental Cr(VI) contamination. The aim of this study is to determine whether the microbial metabolism of alkaliphilic bacteria can be harnessed for the reductive precipitation of Cr(VI) in high-pH leachates of COPR, modified with haloalkaliphilic medium and bacteria. This was explored by using a haloalkaliphilic soda lake isolate which has previously been reported to be effective at high-pH reduction of Tc(VII) (43). The findings of this study would therefore represent the first investigative results of the direct treatment of COPR leachates using an alkaliphilic Cr(VI)-reducing bacterium.
MATERIALS AND METHODS
Organism and culture conditions.
The facultative anaerobic haloalkaliphilic bacterium was originally isolated from Mono Lake (California, USA), herein referred to as the Mono Lake isolate, by N. N. Lyalikova (Institute of Microbiology, Russian Academy of Sciences, Moscow, Russia). The isolate was cultured using sterile anaerobic growth medium (pH 10) consisting of a basal medium of 13 g liter−1 Na2CO3, 4 g liter−1 NaHCO3, 50 g liter−1 NaCl, and 0.5 g liter−1 K2HPO4, and the following additional growth nutrients: 0.1 g liter−1 MgSO4·7H2O, 0.1 g liter−1 NH4Cl, 2 g liter−1 sodium acetate (Na acetate), 2 g liter−1 yeast extract, and 2 ml of mineral elixir (2.14 g liter−1 nitrilotriacetic acid, 0.1 g liter−1 MnCl2·4H2O, 0.3 g liter−1 FeSO4·7H2O, 0.17 g liter−1 CoCl2·6H2O, 0.2 g liter−1 ZnSO4·7H2O, 0.03 g liter−1 CuCl2·H2O, 0.005 g liter−1 AlKSO4·12H2O, 0.005 g liter−1 H3BO3, 0.09 g liter−1 Na2MoO4, 0.11 g liter−1 NiSO4·6H2O, and 0.02 g liter−1 Na2WO4·2H2O).
COPR sample collection and preparation of a COPR extracted medium.
A sample of COPR was obtained from a borehole at a site in southeastern Glasgow, United Kingdom, transferred to a sterile plastic container, and stored in the dark for approximately 2 years at 10°C until use. The COPR has been extensively characterized in a previous study and found to be composed of a cementitous mineralogy, with considerable leachable Cr(VI) content (44, 45), consistent with previous studies (15, 17). To prepare the COPR extract medium, a subsample of field moist COPR was added to the basal medium at 5:100 (wt/vol) for growing cell experiments and 10:100 (wt/vol) for resting cell experiments, under aerobic conditions in the absence of any growth nutrients. The COPR medium slurry was homogenized by shaking and left in the dark at 20°C for 24 h to equilibrate. The resulting slurry was then filter sterilized using a 0.22-μm filter.
Growing cell experiments.
Experiments using a growing culture of the Mono Lake isolate were conducted by inoculation of the previously stated 5:100 (wt/vol) COPR–basal medium extract in the presence of added growth nutrients. Growing cell cultures were prepared by mixing 11 ml of the filter-sterilized 5:100 COPR extract with 3.5 ml of sterile growth medium in sterile 20-ml serum bottles, which were then sealed using butyl rubber stoppers and aluminum crimps, in equilibrium with air. The serum bottles were then inoculated with a 1-ml aliquot of a growing anaerobic culture, maintained at 20°C, of the Mono Lake isolate. The bottles were then incubated at 30°C in the dark for the duration of the experiment. Samples (1 ml) were removed using a N2 degassed syringe and centrifuged (Sigma 1-14 Microfuge) at 13,000 × g for 5 min, and the supernatant was then removed for Cr(VI) analysis. The remaining solids were resuspended in 1% NaCl and again centrifuged (Sigma 1-14 Microfuge) at 13,000 × g for 5 min. The supernatant was then discarded and replaced with 100 μl of 1% NaCl, and the solution was homogenized for protein analysis.
Resting cell experiments.
The isolate was also used in a series of anaerobic resting cell experiments in the presence of the 10:100 (wt/vol) COPR–basal medium extract as detailed in the medium preparation section. Under aerobic conditions, growth medium was inoculated with a culture of the Mono Lake bacterium and incubated in a sterile Erlenmeyer flask on a shaking incubator at 30°C. The culture was then harvested in late log phase (approximately 24 h) using a centrifuge (Sigma 6k15) at 5,000 × g for 20 min and washed three times using basal medium under an N2 atmosphere. The washed cells were then used to inoculate resting cell experiments to a final protein concentration of 81.5 μg ml−1 (equivalent to an optical density at 600 nm of 0.45). The resting cell experiments were composed of 20 ml of the COPR extract medium supplemented with 2 g liter−1 Na acetate and 2 g liter−1 yeast extract. All cultures were contained in sterile serum bottles sealed using butyl rubber stoppers and aluminum crimps and degassed using N2 gas passed through a 0.22-μm filter. Resting cell experiments were established at pH 10, 11, and 12 in triplicate, alongside noninoculated abiotic controls. The starting pH of the medium was 12, and the pH was subsequently adjusted to pH 11 and 10 in the corresponding cultures using sterile 3 M HCl. Aqueous samples were removed to monitor the geochemical parameters of the experiment using an N2 degassed syringe and centrifuged (Sigma 1-14 Microfuge) at 13,000 × g for 5 min, and a subsample of the supernatant was then analyzed for aqueous Cr(VI) concentration.
Calculation of Cr(VI) removal reaction rates.
The Cr(VI) concentration data were fitted to a pseudo-first-order reaction rate model. The reaction rate is proportional to the concentration of aqueous Cr(VI), while the cell concentration is assumed to remain in far excess and constant throughout the experiment:
where [Cr(VI)] is the concentration of aqueous Cr(VI), t is time, and kobs is the observed first-order reaction rate constant. The first-order reaction rate model was applied only when significant aqueous Cr(VI) removal (>50%) was observed.
Aqueous-phase analysis.
The pH of the samples was measured using a meter (Denver Instrument UB-10 meter) and a probe (Cole-Parmer 5990-45 CCP), calibrated using relevant pH buffers. The aqueous Cr(VI) concentration was determined by the 1,5-diphenylcarbazide (DPC) UV-visible light (UV-vis) spectrophotometric method and compared to K2CrO4 standards of known Cr(VI) concentration (46).
Protein assay.
Protein concentrations were determined using a bicinchoninic acid (BCA) and Cu(II)SO4 spectrophotometric assay (47), quantified by comparison to bovine serum albumin (BSA) standards. All UV-vis measurements were recorded on a Jenway 6715 UV-vis spectrophotometer.
Solid-phase analysis.
At the end of the resting cell experiment, the replicates that exhibited Cr(VI) removal were sampled for solid-phase analysis. The aqueous slurry was centrifuged (Sigma 1-14 Microfuge) at 13,000 × g for 5 min, the supernatant was removed and replaced with 18.2 MΩ water, and the resulting solution was homogenized. This was repeated three times, and the resulting pellet was dried in an anoxic glove box prior to analysis.
For transmission electron microscope (TEM) imaging, the pellet was resuspended in ethanol, and droplets were placed on an Agar Scientific holey carbon film grid and allowed to dry. The TEM analysis was performed on a Philips CM200 FEG TEM equipped with a field emission gun (FEG) and an energy-dispersive X-ray analyzer (EDX), Oxford Instruments X-Max 80-mm2 silicon drift detector (SDD) INCA EDX.
X-ray photoelectron spectroscopy (XPS) was performed on a Kratos Axis Ultra spectrophotometer with a monochromated Al Kα X-ray source. Analysis was carried out with an analyzer pass energy of 80 eV (wide scans) and 20 eV (narrow scans) with a total energy resolution of 1.2 and 0.6 eV, respectively, at a base pressure of 5 × 10−8 Pa. All spectra were fit with a Shirley background model (48) and had their photoelectron binding energies (BE) referenced to the C 1s adventitious carbon peak (285 eV BE). Fitting of the Cr 2p region was conducted using 70% Lorentzian and 30% Gaussian curves.
DNA extraction, PCR amplification, and 16S rRNA gene sequencing.
The DNA of the isolate was extracted using a PowerSoil DNA isolation kit (MO BIO Laboratories, Inc., Carlsbad, CA, USA). The DNA was amplified by PCR using several broad-specificity 16S rRNA gene primers to obtain overlapping amplified 16S rRNA fragments, including 8F (49), 530F (50), 519R (51), 943R (50), and 1492R (51).
The dideoxynucleotide method was used to determine the nucleotide sequences (52), using an ABI Prism BigDye terminator cycle sequencing kit in combination with an ABI Prism 877 integrated thermal cycler and ABI Prism 377 DNA sequencer (PerkinElmer Applied Biosystems, Warrington, United Kingdom). A contig was generated from the sequences (typically 900 bp in length), using DNA Dragon v1.6 (SequentiX Digital DNA Processing, Klein Raden, Germany). The consensus sequence (1,446 bp in length) was analyzed against the NCBI (US) database using BLAST program packages and matched to known 16S rRNA gene sequences.
The phylogenetic tree was constructed using MEGA5 (53). The evolutionary history was inferred by using the neighbor-joining method (54), and the evolutionary distances were computed using the maximum composite likelihood method (55).
RESULTS AND DISCUSSION
Phylogenetic characterization.
Sequencing of the 16S rRNA gene of the Mono Lake isolate showed that the organism belongs to the Halomonas genus of the Gammaproteobacteria (Fig. 1). The isolate occupies a clade with Halomonas mongoliensis and shares 16S rRNA gene sequence similarity to a variety of Halomonas species (Fig. 1).
FIG 1.

Phylogenetic tree based on 16S rRNA gene sequences of the Mono Lake isolate and other members of the Halomonas genus. GenBank nucleotide sequence accession numbers are in parentheses. The bar represents 1% divergence in the 16S rRNA gene sequence.
Halomonas species are often highly represented in isolates from hypersaline and alkaline environments such as soda lakes (40, 41). These obligate heterotrophs have gained attention for other potentially useful behavior, such as their ability to degrade aromatic compounds (56–58) and produce alkaline enzymes with possible biotechnological applications (59, 60). In addition to this, a number of Halomonas species, including H. mongoliensis (61), the closest phylogenetic match to the Mono Lake isolate, have been found to be capable of anaerobic nitrate reduction under alkaline (pH 10) and high-salt conditions (4 M Na+) (36). Several closely related Halomonas strains have also been found to be capable of alkaline Cr(VI) reduction under anaerobic conditions (34, 36). As the Mono Lake isolate studied here has also been shown to reduce Tc(VII) to Tc(IV) (43), it may potentially reduce a variety of redox-active metals.
Cr(VI) reduction during growth.
Upon inoculation of aerobic COPR extract on growth medium with the Mono Lake Halomonas species, the aqueous Cr(VI) concentration decreased to below detection limits within 170 h (Fig. 2), while the pH was maintained at 10 throughout the experiment. Concurrent to this removal of Cr(VI), protein concentrations increased steadily over the reaction period reaching more than 100 μg ml−1 after 168 h of incubation. This concurrent increase in protein levels, while Cr(VI) decreased, is clear evidence for growth of the isolate in the presence of significant Cr(VI) concentrations. As these cultures were initially incubated under oxic conditions, where the facultative anaerobic Halomonas species likely consumes O2 as an initial electron acceptor, it is unclear whether growth is directly coupled to Cr(VI) reduction.
FIG 2.

Aqueous Cr(VI) concentration and protein concentration after inoculation of the COPR leachate growth media with the Mono Lake Halomonas species. Error bars represent the standard deviations of duplicate experiments.
Cr(VI) reduction by resting cells.
To identify the impact of pH on the kinetics of Cr(VI) reduction, a series of resting cell experiments were conducted at pH 10, 11, and 12 (Fig. 3). The Cr(VI) concentrations of cell-free abiotic control experiments showed little change over the duration of the experiment at all pH values, remaining at ∼2,500 μM throughout. However, rapid Cr(VI) removal was noted in the presence of cells of the Mono Lake Halomonas isolate in the cultures at pH 10 and 11, along with reductive precipitation of Cr(III), confirmed by XPS analysis of the resulting precipitates (Fig. 4 and Table 1). Thus, Cr(VI) removal was due to direct, anaerobic, enzymatic Cr(VI) reduction by the Mono Lake Halomonas isolate.
FIG 3.

Aqueous Cr(VI) concentration (a) and pH (b) of the COPR leachate in sterilized controls (open symbols) and when inoculated with a late-log-phase culture of a Halomonas isolate from Mono Lake (solid symbols). The black arrows indicate sampling times for solid-phase analysis. Error bars represent the standard deviations of triplicates. (c) Pseudo-first-order Cr(VI) removal rate kinetics, plotted as ln [Cr(VI)] against time, calculated from the averages of triplicate Cr(VI) values of experiments containing resting cells of Halomonas sp. These experiments were performed at a starting pH of 10 and 11.
FIG 4.
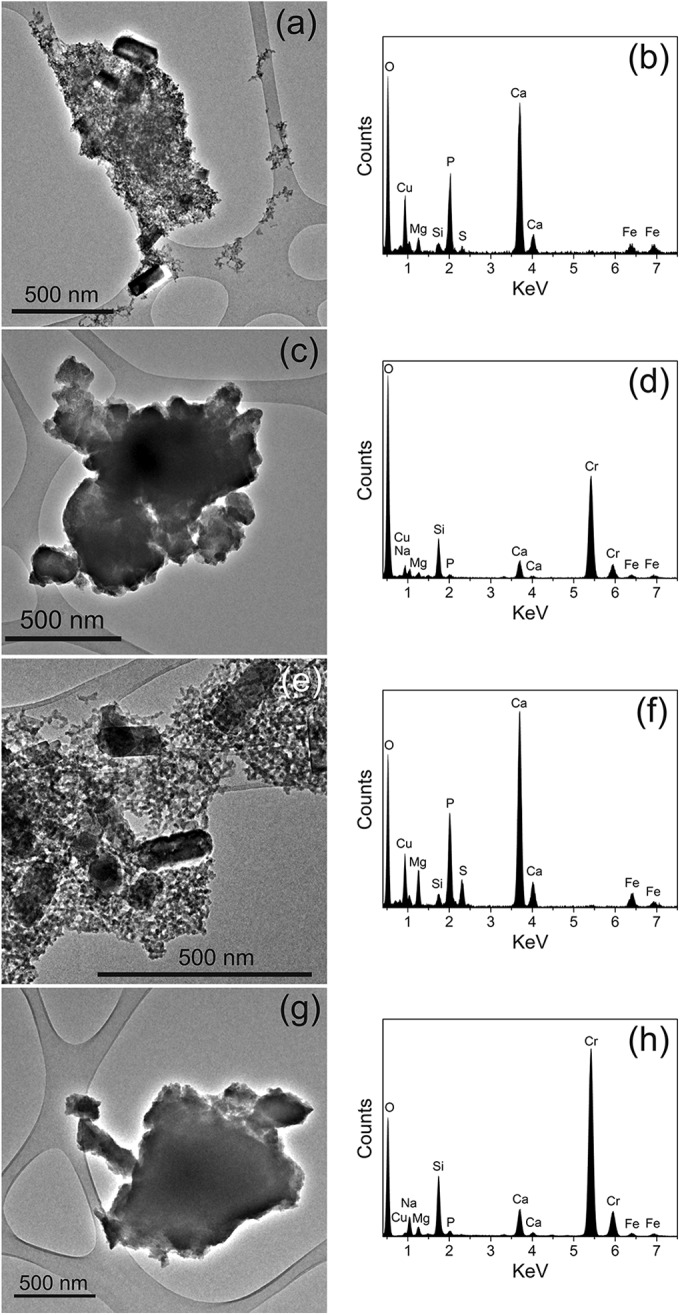
TEM images and their corresponding EDX spectra of Cr-containing solid phases from resting cell experiments at pH 10 (a to d) and pH 11 (e to h). The Cu component in the EDX spectra is due to its presence in the carbon-coated copper grids.
TABLE 1.
Summary of XPS data obtained from precipitates formed by reduction of Cr(VI) by resting cell cultures of a Halomonas species from Mono Lake
| Starting pH | XPS elemental composition (atomic %) |
XPS valence state [Cr(III)/Cr(VI)] | |||||||
|---|---|---|---|---|---|---|---|---|---|
| C | O | Cr | Na | Si | Ca | N | P | ||
| 10 | 67.8 | 20.3 | 0.4 | 0.4 | 5.4 | 0.2 | 6.0 | 0.3 | 100:0 |
| 11 | 73.0 | 21.7 | 0.6 | 0.2 | 3.3 | 0.5 | 0.8 | 74:26 | |
The solution pH was found to have a strong control over enzymatic Cr(VI) reduction, with no appreciable Cr(VI) removal observed in the pH 12 replicate, suggesting a loss of metabolic activity under extremely alkaline conditions. The rapid removal of Cr(VI) noted at pH 10 and 11 translated to kobs values that were considerably higher at pH 10 (0.0409 h−1; R2 = 0.96) compared to the pH 11 replicate (0.0126 h−1; R2 = 0.98) (Fig. 3c). As the starting pH of 11 evidently buffers down to 10.5 during incubation by the addition of the bacterial isolate, it is this level that should be assumed to be the upper limit of sustained Cr(VI) removal observed in this study. Reduction of Cr(VI) at this highly alkaline pH is in line with the upper limits of enzymatic Cr(VI) reduction reported previously (34, 36, 62, 63). These values are also consistent with previously reported optimum growth conditions of pH 9 to 10 for closely related Halomonas species (42, 61). The pH values of the COPR leachate and contaminated groundwater are typically within the range of 9 to 12.5 (16, 64). Therefore, the observed removal of Cr(VI) at alkaline values up to 10.5 indicates that a bioremediation approach using the Halomonas species may represent a possible treatment of a proportion of COPR leachates, without the need for pH amendment prior to inoculation, while treatment of higher pH COPR leachates would require some degree of buffering to a lower pH, albeit to a lesser extent than required for bioremediation using neutrophilic bacteria.
Characterization of the Cr precipitates.
Upon visual inspection of the resting cell incubations at the end of the experiments, a purple precipitate was observed in the pH 10 cultures and a green precipitate was observed in the pH 11 cultures. The observed color differences in the precipitates would indicate the presence of differing precipitate phases where Cr(III) minerals can occur as green or purple minerals (65, 66). The fate of the Cr removed from solution in the resting cell experiments was assessed using TEM-EDX alongside XPS analysis.
The TEM images of the resulting precipitates and their corresponding EDX spectra are presented in Fig. 4, for the pH 10 (Fig. 4a to d) and pH 11 (Fig. 4e to h) incubations. The XPS wide-scan and Cr 2p region spectra are presented in Fig. 5, and their elemental composition and Cr valence states are presented in Table 1. TEM analysis shows that the precipitates formed at both pH 10 and pH 11 appear to possess a similar morphology, both containing a finer granular component (10- to 30-nm diameter) along with larger cubic structures (100 to 250 nm in length), visible in Fig. 4a and e. Both the precipitates formed at pH 10 and pH 11 also contained larger, micron-sized agglomerates (Fig. 4c and g), which appeared structurally featureless when viewed at high resolution. In addition to TEM-EDX analyses, the precipitates were probed using selected area electron diffraction (SEAD) (data not presented). However, the lack of ring structures noted indicated random diffraction commonly associated with amorphous structures.
FIG 5.

XPS Cr 2p region (a) and the wide-scan (b) spectra of the precipitates of the resting cell experiments at pH 10 (i) and 11 (ii).
The Cr 2p XPS region (Fig. 5a) was fitted with a component consisting of two peaks, at ∼576.5 and ∼586.0 eV BE, consistent with previously reported Cr(III) phases and a component of higher BE peaks, at ∼579.0 and ∼588.8 eV, consistent with Cr(VI) phases (67–69). The pH 10 precipitate was best fitted with the single Cr(III) component, while the pH 11 precipitates required fitting with a contribution from the Cr(VI) component, equating to 26% Cr(VI) of total Cr (Fig. 5 and Table 1). It is not clear whether the presence of Cr(VI) occurs as an adsorbed phase or within the bulk mineral, where due to the surface sensitivity of XPS, any adsorbed phase would be overrepresented in the spectra. The presence of Cr(VI) in the experiments exhibiting a lower rate of reduction is presumably due to incomplete reduction prior to sampling for solid-phase analysis.
The chemistry of the bulk sample was observed, by XPS, to be dominated by the C and O 1s regions, with smaller Si, Ca, Na, N, and Cr components. The relative contributions of these smaller components showed minimal variation between the two samples. It is important to note that the elemental composition noted by XPS reflects that of the bulk precipitates and not the Cr biominerals alone. Also, due to the surface sensitivity of XPS, typically sampling to a depth of <10 nm (70), the results are likely to overrepresent the finer-grained fraction as opposed to the bulk of larger Cr biominerals. The elemental chemistry of the samples was therefore analyzed using TEM-EDX to target the differing minerals present in the heterogeneous precipitates. The TEM-EDX spectra (Fig. 4b and f) do not show a significant Cr component and are composed primarily of Ca, P, and O, with minor contributions of S, Si, Fe, Na, and Mg. The larger micron-scale particles, from their corresponding EDX spectra (Fig. 4d and h), were principally composed of Cr and O, with minor contributions from Si and Ca. Relative intensities differed marginally between the pH replicates. The dominance of the Cr and O in the spectra would be consistent with the presence of a Cr(III) oxide or hydroxide, the latter being widely reported as the dominant form of Cr(III) in high-pH environments (1). However, the techniques employed in this study are not able to determine the exact phase of the Cr(III) and give little indication of possible differences indicated by the differing colors of the precipitates noted at pH 10 and 11.
Conclusions.
This study has demonstrated the ability of an haloalkaliphilic soda lake isolate, belonging to the Halomonas genus, to grow while reducing aqueous Cr(VI) from a COPR extract under alkaline conditions. Bacterial cells were also able to anaerobically reduce significant concentrations of Cr(VI) (2.5 mM) under nongrowing conditions, with an inverse relationship between pH and reaction rates over the pH range tested here. Cr(VI) reduction occurred up to pH 10.5 and ultimately resulted in the precipitation of predominantly Cr(III) minerals.
The application of this Mono Lake Halomonas isolate offers a potential in situ bioremediation treatment for the reduction of alkaline leachates with high Cr(VI) concentrations associated with COPR contamination. These findings therefore provide useful information for the development of in situ trials via bioaugmentation of environments affected by COPR. In addition, as this organism has been demonstrated previously to have the ability to reduce Tc(VII) (43) and is closely related to other nitrate-reducing (61) and aromatic-degrading (58) strains of the Halomonas genus, this isolate may be of use in the bioremediation of a variety of contaminants occurring in alkaline environments.
ACKNOWLEDGMENTS
M.P.W. acknowledges financial support from the BBSRC for a Ph.D. bursary and CASE partner Parsons Brinckerhoff.
We thank B. R. G. Johnson and Z. Aslam for their assistance with XPS and TEM analysis via the Leeds EPSRC Nanoscience and Nanotechnology Research Equipment Facility (LENNF). We especially thank N. Johnson of ERS Land Regeneration for help in obtaining COPR samples.
REFERENCES
- 1.Kimbrough DE, Cohen Y, Winer AM, Creelman L, Mabuni C. 1999. A critical assessment of chromium in the environment. Crit Rev Environ Sci Technol 29:1–46. doi: 10.1080/10643389991259164. [DOI] [Google Scholar]
- 2.Fruchter J. 2002. In situ treatment of chromium-contaminated groundwater. Environ Sci Technol 36:464A–472A. doi: 10.1021/es022466i. [DOI] [PubMed] [Google Scholar]
- 3.Kamaludeen S, Megharaj M, Juhasz A, Sethunathan N, Naidu R. 2003. Chromium-microorganism interactions in soils: remediation implications. Rev Environ Contam Toxicol 178:93–164. [DOI] [PubMed] [Google Scholar]
- 4.Rai D, Eary LE, Zachara JM. 1989. Environmental chemistry of chromium. Sci Total Environ 86:15–23. doi: 10.1016/0048-9697(89)90189-7. [DOI] [PubMed] [Google Scholar]
- 5.Rai D, Sass BM, Moore DA. 1987. Chromium(III) hydrolysis constants and solubility of chromium(III) hydroxide. Inorg Chem 26:345–349. doi: 10.1021/ic00250a002. [DOI] [Google Scholar]
- 6.Fendorf SE. 1995. Surface reactions of chromium in soils and waters. Geoderma 67:55–71. doi: 10.1016/0016-7061(94)00062-F. [DOI] [Google Scholar]
- 7.Wang Y-T. 2000. Microbial reduction of Cr(VI), p 225–235. In Lovley DR. (ed), Environmental microbe-metal interactions. ASM Press, Washington, DC. [Google Scholar]
- 8.Dayan AD, Paine AJ. 2001. Mechanisms of chromium toxicity, carcinogenicity and allergenicity: review of the literature from 1985 to 2000. Hum Exp Toxicol 20:439–451. doi: 10.1191/096032701682693062. [DOI] [PubMed] [Google Scholar]
- 9.Kotaś J, Stasicka Z. 2000. Chromium occurrence in the environment and methods of its speciation. Environ Pollut 107:263–283. doi: 10.1016/S0269-7491(99)00168-2. [DOI] [PubMed] [Google Scholar]
- 10.Dhal B, Thatoi HN, Das NN, Pandey BD. 2013. Chemical and microbial remediation of hexavalent chromium from contaminated soil and mining/metallurgical solid waste: a review. J Hazard Mater 250-251:272–291. doi: 10.1016/j.jhazmat.2013.01.048. [DOI] [PubMed] [Google Scholar]
- 11.Darrie G. 2001. Commercial extraction technology and process waste disposal in the manufacture of chromium chemicals from ore. Environ Geochem Health 23:187–193. doi: 10.1023/A:1012295927081. [DOI] [Google Scholar]
- 12.Burke T, Fagliano J, Goldoft M, Hazen RE, Iglewicz R, McKee T. 1991. Chromite ore processing residue in Hudson County, New Jersey. Environ Health Perspect 92:131–137. doi: 10.1289/ehp.9192131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antony MP, Tathavadkar VD, Calvert CC, Jha A. 2001. The soda-ash roasting of chromite ore processing residue for the reclamation of chromium. Metal Mater Trans B 32:987–995. doi: 10.1007/s11663-001-0087-6. [DOI] [Google Scholar]
- 14.Geelhoed JS, Meeussen JCL, Roe MJ, Hillier S, Thomas RP, Farmer JG, Paterson E. 2003. Chromium remediation or release? Effect of iron(II) sulfate addition on chromium(VI) leaching from columns of chromite ore processing residue. Environ Sci Technol 37:3206–3213. [DOI] [PubMed] [Google Scholar]
- 15.Geelhoed JS, Meeussen JCL, Hillier S, Lumsdon DG, Thomas RP, Farmer JG, Paterson E. 2002. Identification and geochemical modelling of processes controlling leaching of Cr(VI) and other major elements from chromite ore processing residue. Geochim Cosmochim Acta 66:3927–3942. doi: 10.1016/S0016-7037(02)00977-8. [DOI] [Google Scholar]
- 16.Farmer JG, Thomas RP, Graham MC, Geelhoed JS, Lumsdon DG, Paterson E. 2002. Chromium speciation and fractionation in ground and surface waters in the vicinity of chromite ore processing residue disposal sites. J Environ Monit 4:235–243. doi: 10.1039/b108681m. [DOI] [PubMed] [Google Scholar]
- 17.Hillier S, Roe MJ, Geelhoed JS, Fraser AR, Farmer JG, Paterson E. 2003. Role of quantitative mineralogical analysis in the investigation of sites contaminated by chromite ore processing residue. Sci Total Environ 308:195–210. doi: 10.1016/S0048-9697(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 18.Cheng Y, Holman H-Y, Lin Z. 2012. Remediation of chromium and uranium contamination by microbial activity. Elements 8:107–112. doi: 10.2113/gselements.8.2.107. [DOI] [Google Scholar]
- 19.Farmer JG, Paterson E, Bewley RJF, Geelhoed JS, Hillier S, Meeussen JCL, Lumsdon DG, Thomas RP, Graham MC. 2006. The implications of integrated assessment and modelling studies for the future remediation of chromite ore processing residue disposal sites. Sci Total Environ 360:90–97. doi: 10.1016/j.scitotenv.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 20.Whalley C, Hursthouse A, Rowlatt S, Iqbal-Zahid P, Vaughan H, Durant R. 1999. Chromium speciation in natural waters draining contaminated land, Glasgow, UK. Water Air Soil Pollut 112:389–405. doi: 10.1023/A:1005017506227. [DOI] [Google Scholar]
- 21.Bewley RJF, Jeffries R, Watson S, Granger D. 2001. An overview of chromium contamination issues in the south-east of Glasgow and the potential for remediation. Environ Geochem Health 23:267–271. doi: 10.1023/A:1012261432256. [DOI] [Google Scholar]
- 22.World Health Organization. 2008. Guidelines for drinking water quality. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 23.Lloyd JR. 2003. Microbial reduction of metals and radionuclides. FEMS Microbiol Rev 27:411–425. doi: 10.1016/S0168-6445(03)00044-5. [DOI] [PubMed] [Google Scholar]
- 24.Watts MP, Lloyd JR. 2012. Bioremediation via microbial metal reduction, p 161–201. In Gescher J, Kappler A (ed), Microbial metal respiration: from geochemistry to potential applications. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 25.Lovley DR, Phillips EJP. 1994. Reduction of chromate by Desulfovibrio vulgaris and its c3 cytochrome. Appl Environ Microbiol 60:726–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cervantes C, Campos-García J, Nies D, Silver S. 2007. Reduction and efflux of chromate by bacteria molecular microbiology of heavy metals, p 407–419. In Nies DH, Silver S (ed), Molecular microbiology of heavy metals, vol 6 Springer-Verlag, Berlin, Germany. [Google Scholar]
- 27.Wang Y-T, Shen H. 1995. Bacterial reduction of hexavalent chromium. J Ind Microbiol Biotechnol 14:159–163. [DOI] [PubMed] [Google Scholar]
- 28.Ishibashi Y, Cervantes C, Silver S. 1990. Chromium reduction in Pseudomonas putida. Appl Environ Microbiol 56:2268–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tebo BM, Obraztsova AY. 1998. Sulfate-reducing bacterium grows with Cr(VI), U(VI), Mn(IV), and Fe(III) as electron acceptors. FEMS Microbiol Lett 162:193–198. doi: 10.1111/j.1574-6968.1998.tb12998.x. [DOI] [Google Scholar]
- 30.Francis CA, Obraztsova AY, Tebo BM. 2000. Dissimilatory metal reduction by the facultative anaerobe Pantoea agglomerans SP1. Appl Environ Microbiol 66:543–548. doi: 10.1128/AEM.66.2.543-548.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeyasingh J, Philip L. 2005. Bioremediation of chromium contaminated soil: optimization of operating parameters under laboratory conditions. J Hazard Mater 118:113–120. doi: 10.1016/j.jhazmat.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Roh Y, Chon C-M, Moon J-W. 2007. Metal reduction and biomineralization by an alkaliphilic metal-reducing bacterium, Alkaliphilus metalliredigens (QYMF). Geosci J 11:415–423. doi: 10.1007/BF02857056. [DOI] [Google Scholar]
- 33.Wani R, Kodam K, Gawai K, Dhakephalkar P. 2007. Chromate reduction by Burkholderia cepacia MCMB-821, isolated from the pristine habitat of alkaline crater lake. Appl Microbiol Biotechnol 75:627–632. doi: 10.1007/s00253-007-0862-7. [DOI] [PubMed] [Google Scholar]
- 34.VanEngelen M, Peyton B, Mormile M, Pinkart H. 2008. Fe(III), Cr(VI), and Fe(III) mediated Cr(VI) reduction in alkaline media using a Halomonas isolate from Soap Lake, Washington. Biodegradation 19:841–850. doi: 10.1007/s10532-008-9187-1. [DOI] [PubMed] [Google Scholar]
- 35.Ge S, Zhou M, Dong X, Lu Y, Ge S. 2013. Distinct and effective biotransformation of hexavalent chromium by a novel isolate under aerobic growth followed by facultative anaerobic incubation. Appl Microbiol Biotechnol 97:2131–2137. doi: 10.1007/s00253-012-4361-0. [DOI] [PubMed] [Google Scholar]
- 36.Shapovalova AA, Khijniak TV, Tourova TP, Sorokin DY. 2009. Halomonas chromatireducens sp. nov., a new denitrifying facultatively haloalkaliphilic bacterium from solonchak soil capable of aerobic chromate reduction. Microbiology 78:102–111. doi: 10.1134/S0026261709010135. [DOI] [PubMed] [Google Scholar]
- 37.Long D, Tang X, Cai K, Chen G, Chen L, Duan D, Zhu J, Chen Y. 2013. Cr(VI) reduction by a potent novel alkaliphilic halotolerant strain Pseudochrobactrum saccharolyticum LY10. J Hazard Mater 256–257: 24–32. doi: 10.1016/j.jhazmat.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 38.Horikoshi K. 1999. Alkaliphiles: some applications of their products for biotechnology. Microbiol Mol Biol Rev 63:735–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowers KJ, Wiegel J. 2011. Temperature and pH optima of extremely halophilic archaea: a mini-review. Extremophiles 15:119–128. doi: 10.1007/s00792-010-0347-y. [DOI] [PubMed] [Google Scholar]
- 40.Duckworth AW, Grant WD, Jones BE, van Steenbergen R. 1996. Phylogenetic diversity of soda lake alkaliphiles. FEMS Microbiol Ecol 19:181–191. doi: 10.1111/j.1574-6941.1996.tb00211.x. [DOI] [Google Scholar]
- 41.Jones BE, Grant WD, Duckworth AW, Owenson GG. 1998. Microbial diversity of soda lakes. Extremophiles 2:191–200. doi: 10.1007/s007920050060. [DOI] [PubMed] [Google Scholar]
- 42.Shapovalova AA, Khijniak TV, Tourova TP, Muyzer G, Sorokin DY. 2008. Heterotrophic denitrification at extremely high salt and pH by haloalkaliphilic Gammaproteobacteria from hypersaline soda lakes. Extremophiles 12:619–625. doi: 10.1007/s00792-008-0166-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khijniak TV, Medvedeva-Lyalikova NN, Simonoff M. 2003. Reduction of pertechnetate by haloalkaliphilic strains of Halomonas. FEMS Microbiol Ecol 44:109–115. doi: 10.1016/S0168-6496(03)00018-7. [DOI] [PubMed] [Google Scholar]
- 44.Watts MP, Coker VS, Parry SA, Pattrick RAD, Thomas RAP, Kalin R, Lloyd JR. 2015. Biogenic nano-magnetite and nano-zero valent iron treatment of alkaline Cr(VI) leachate and chromite ore processing residue. Appl Geochem 54:27–42. doi: 10.1016/j.apgeochem.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watts MP, Coker VS, Parry SA, Thomas RAP, Kalin R, Lloyd JR. 2015. Effective treatment of alkaline Cr(VI) contaminated leachate using a novel Pd-bionanocatalyst: impact of electron donor and aqueous geochemistry. Appl Catal B 170-171:162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown E, Skougstad MW, Fishman MJ. 1979. Methods for determination of inorganic substances in water and fluvial sediments. Techniques of Water Resource Investigation 05-A1. US Geological Survey, Reston, VA. [Google Scholar]
- 47.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. 1985. Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 48.Shirley DA. 1972. High-resolution X-ray photoemission spectrum of the valence bands of gold. Phys Rev B 5:4709–4714. doi: 10.1103/PhysRevB.5.4709. [DOI] [Google Scholar]
- 49.Eden PA, Schmidt TM, Blakemore RP, Pace NR. 1991. Phylogenetic analysis of Aquaspirillum magnetotacticum using polymerase chain reaction-amplified 16S rRNA-specific DNA. Int J Syst Bacteriol 41:324–325. doi: 10.1099/00207713-41-2-324. [DOI] [PubMed] [Google Scholar]
- 50.Goodfellow M, Stackebrandt E (ed). 1991. Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Hoboken, NJ. [Google Scholar]
- 51.Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. 1985. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci U S A 82:6955–6959. doi: 10.1073/pnas.82.20.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. [DOI] [PubMed] [Google Scholar]
- 55.Tamura K, Nei M, Kumar S. 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A 101:11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oie CSI, Albaugh CE, Peyton BM. 2007. Benzoate and salicylate degradation by Halomonas campisalis, an alkaliphilic and moderately halophilic microorganism. Water Res 41:1235–1242. doi: 10.1016/j.watres.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 57.García MT, Mellado E, Ostos JC, Ventosa A. 2004. Halomonas organivorans sp. nov., a moderate halophile able to degrade aromatic compounds. Int J Syst Evol Microbiol 54:1723–1728. doi: 10.1099/ijs.0.63114-0. [DOI] [PubMed] [Google Scholar]
- 58.Alva VA, Peyton BM. 2003. Phenol and catechol biodegradation by the haloalkaliphile Halomonas campisalis: influence of pH and salinity. Environ Sci Technol 37:4397–4402. doi: 10.1021/es0341844. [DOI] [PubMed] [Google Scholar]
- 59.Coronado M-J, Vargas C, Hofemeister J, Ventosa A, Nieto JJ. 2000. Production and biochemical characterization of an α-amylase from the moderate halophile Halomonas meridiana. FEMS Microbiol Lett 183:67–71. doi: 10.1111/j.1574-6968.2000.tb08935.x. [DOI] [PubMed] [Google Scholar]
- 60.Gadda G, McAllister-Wilkins EE. 2003. Cloning, expression, and purification of choline dehydrogenase from the moderate halophile Halomonas elongata. Appl Environ Microbiol 69:2126–2132. doi: 10.1128/AEM.69.4.2126-2132.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boltyanskaya YV, Kevbrin VV, Lysenko AM, Kolganova TV, Tourova TP, Osipov GA, Zhilina TN. 2007. Halomonas mongoliensis sp. nov. and Halomonas kenyensis sp. nov., new haloalkaliphilic denitrifiers capable of N2O reduction, isolated from soda lakes. Microbiology 76:739–747. doi: 10.1134/S0026261707060148. [DOI] [PubMed] [Google Scholar]
- 62.Ye Q, Roh Y, Carroll SL, Blair B, Zhou J, Zhang CL, Fields MW. 2004. Alkaline anaerobic respiration: isolation and characterization of a novel alkaliphilic and metal-reducing bacterium. Appl Environ Microbiol 70:5595–5602. doi: 10.1128/AEM.70.9.5595-5602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He Z, Gao F, Sha T, Hu Y, He C. 2009. Isolation and characterization of a Cr(VI)-reduction Ochrobactrum sp. strain CSCr-3 from chromium landfill. J Hazard Mater 163:869–873. doi: 10.1016/j.jhazmat.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 64.Farmer JG, Graham MC, Thomas RP, Licona-Manzur C, Paterson E, Campbell CD, Geelhoed JS, Lumsdon DG, Meeussen JCL, Roe MJ, Conner A, Fallick AE, Bewley RJF. 1999. Assessment and modelling of the environmental chemistry and potential for remediative treatment of chromium-contaminated land. Environ Geochem Health 21:331–337. doi: 10.1023/A:1006788418483. [DOI] [Google Scholar]
- 65.Nassau K. 1978. The origins of color in minerals. Am Mineral 63:219–229. [Google Scholar]
- 66.Motzer WE. 2004. Chemistry, geochemistry, and geology of chromium and chromium compounds, p 23–88. In Guertin J, Jacobs JA, Avakian CP (ed), Chromium(VI) handbook. CRC Press, Boca Raton, FL. [Google Scholar]
- 67.Asami K, Hashimoto K. 1977. The X-ray photo-electron spectra of several oxides of iron and chromium. Corrosion Sci 17:559–570. doi: 10.1016/S0010-938X(77)80002-4. [DOI] [Google Scholar]
- 68.Aronniemi M, Sainio J, Lahtinen J. 2005. Chemical state quantification of iron and chromium oxides using XPS: the effect of the background subtraction method. Surf Sci 578:108–123. doi: 10.1016/j.susc.2005.01.019. [DOI] [Google Scholar]
- 69.Biesinger MC, Brown C, Mycroft JR, Davidson RD, McIntyre NS. 2004. X-ray photoelectron spectroscopy studies of chromium compounds. Surf Interface Anal 36:1550–1563. doi: 10.1002/sia.1983. [DOI] [Google Scholar]
- 70.Moulder JF, Stickle WF, Sobol PE, Bomben KD. 1992. Handbook of X-ray photoelectron spectroscopy, vol 40 Perkin Elmer, Eden Prairie, MN. [Google Scholar]