

Abstract
Agmatine, a significant polyamine in bacteria and plants, mostly arises from the decarboxylation of arginine. The functional importance of agmatine in fungi is poorly understood. The metabolism of agmatine and related guanidinium group-containing compounds in Aspergillus niger was explored through growth, metabolite, and enzyme studies. The fungus was able to metabolize and grow on l-arginine, agmatine, or 4-guanidinobutyrate as the sole nitrogen source. Whereas arginase defined the only route for arginine catabolism, biochemical and bioinformatics approaches suggested the absence of arginine decarboxylase in A. niger. Efficient utilization by the parent strain and also by its arginase knockout implied an arginase-independent catabolic route for agmatine. Urea and 4-guanidinobutyrate were detected in the spent medium during growth on agmatine. The agmatine-grown A. niger mycelia contained significant levels of amine oxidase, 4-guanidinobutyraldehyde dehydrogenase, 4-guanidinobutyrase (GBase), and succinic semialdehyde dehydrogenase, but no agmatinase activity was detected. Taken together, the results support a novel route for agmatine utilization in A. niger. The catabolism of agmatine by way of 4-guanidinobutyrate to 4-aminobutyrate into the Krebs cycle is the first report of such a pathway in any organism. A. niger GBase peptide fragments were identified by tandem mass spectrometry analysis. The corresponding open reading frame from the A. niger NCIM 565 genome was located and cloned. Subsequent expression of GBase in both Escherichia coli and A. niger along with its disruption in A. niger functionally defined the GBase locus (gbu) in the A. niger genome.
INTRODUCTION
The amino acid l-arginine contains a guanidinium group [—HN—C(=NH)—NH2] and occurs universally across all kingdoms of life. Arginase constitutes the major catabolic path for fungal l-arginine (1, 2). Agmatine, a significant polyamine of lower organisms and plants, mostly arises from the decarboxylation of arginine (3, 4). Arginine decarboxylase has been extensively studied in lower organisms (4) and plants (5, 6). On the contrary, with the sporadic exceptions of Ceratocystis minor and Verticillium dahliae (7), Gigaspora rosea (8), and Panus tigrinus (9), fungi do not have arginine decarboxylase. Other minor pathways of agmatine metabolism in fruit bodies of P. tigrinus have been proposed (9). An alternate route for putrescine biosynthesis via agmatine was engineered in Saccharomyces cerevisiae by introducing Escherichia coli arginine decarboxylase and agmatinase genes (10). More recently, an arginase-independent catabolism of arginine (also not involving agmatine) was reported in Kluyveromyces lactis; an aminotransferase was proposed to convert l-arginine to ketoarginine in the first step (11).
Very little is known about agmatine catabolism in fungi and aspergilli in particular (Fig. 1). Functional agmatinases that hydrolyze agmatine to putrescine are mostly reported from bacteria (12, 13). Published Aspergillus genomes do contain a putative agmatinase sequence(s) (Table 1), but none has been functionally annotated. Putrescine in fungi either can form spermidine/spermine or is converted to 4-aminobutyrate (GABA) via 4-aminobutyraldehyde (GABald) (14, 15). Another route to form putrescine from agmatine occurs in plants and archaebacteria; this involves two enzymes, namely, agmatine deiminase and N-carbamyl-putrescine hydrolase (16, 17). An amine oxidase isolated from Aspergillus niger can oxidize agmatine to 4-guanidinobutaraldehyde (GBald) (18). The fate of GBald in fungi is unclear, but it is oxidized to 4-guanidinobutyrate (GB) by an aldehyde dehydrogenase in Arthrobacter sp. (19), Pseudomonas aeruginosa (20), and Tinca tinca (21). 4-Guanidinobutyrase (GBase), which hydrolyzes GB to GABA, has recently been reported in A. niger (GenBank accession no. KF991130 for the nucleotide sequence and GenBank accession no. AHL44994.1 for protein sequence) and K. lactis (11). GB could be used as the sole nitrogen source by Penicillium roqueforti (22, 23) and K. lactis but not by S. cerevisiae (11). Finally, fungal GABA metabolism feeds into the Krebs cycle at succinate (15).
FIG 1.

Catabolic steps in agmatine metabolism across various organisms. The different enzymatic steps are numbered as follows: 1, amine oxidase (3, 25, 26); 2, agmatine deiminase (27–31); 3, agmatinase (13, 32); 4, GBald dehydrogenase (33–35); 5, GBase (11, 19, 36, 37); 6, GABA transaminase (15, 38, 39); 7, SSA dehydrogenase (15, 40); 8, N-carbamyl putrescine hydrolase (27–30); 9, di-/polyamine oxidase (3, 18, 26); 10, GABald dehydrogenase (19, 20); 11, arginine decarboxylase (7, 9, 41, 42); 12, arginase (2, 43, 45); 13, ornithine decarboxylase (2); 14, urease (2, 46). TCA, tricarboxylic acid. The metabolites and enzymes reported in A. niger prior to this work are shown in black.
TABLE 1.
Sequences annotated to be putative agmatinases in Aspergillus genomes
The NCBI protein database was queried for putative agmatinase sequences from each Aspergillus sp. None of these were annotated as GBase. Only sequences that showed ≥85% identity (by analysis with the blastp program) with the A. niger NCIM 565 GBase sequence (GenBank accession no. AHL44994.1) are in bold.
All four A. niger ATCC 1015 sequences are annotated as hypothetical proteins.
Annotated as a hypothetical protein.
Unlike in bacteria, the functional importance of agmatine and its metabolism in fungi is not well understood. Toward this objective, A. niger (as well as its arginase-knockout strain) was grown on agmatine and its catabolic route was established by analyzing the metabolites and enzymes. 4-Guanidinobutyrase is an essential ureohydrolase of this pathway. A novel route for agmatine utilization in A. niger, by way of GB to GABA, leading to the Krebs cycle, is reported here.
MATERIALS AND METHODS
Chemicals and reagents.
The guanidinium compounds (l-arginine, agmatine, GB, 3-guanidinopropionate, guanidinoacetate, l-homoarginine, and d-arginine) and pyrroloquinoline quinone (PQQ) were obtained from Sigma-Aldrich Co., St. Louis, MO, USA. All other chemicals were of analytical grade and were obtained either from Sigma-Aldrich or from local suppliers. Water purified from Millipore Milli-Q Ultrapure water purification systems from Millipore, MA, USA, was used throughout this study. Silica thin-layer chromatography (TLC) plates were from Merck (product code 1055540007), and silica gel (60 to 120 mesh) was from Sisco Research Laboratories, India.
Strains and plasmids.
Aspergillus niger NCIM 565 was obtained from the National Collection of Industrial Microorganisms at the National Chemical Laboratory, Pune, India. A. niger strains D-42 (arginase knockout) and ΔXCA-29 (constitutively overexpressing arginase) were derived from the wild-type (WT) parent strain A. niger NCIM 565, as described previously (1). The kusA disruption strain (kusA::amdS; pyrG−) of A. niger (strain MA169.4) (24) was from the Fungal Genetics Stock Center, Manhattan, KS, USA. The plasmids pKA5 (containing E. coli arginine decarboxylase and agmatinase [13]) and pETNat (containing A. niger arginase cDNA [25]) were used to transform E. coli XL1-Blue and BL21(DE3), respectively. Two plasmids, namely, pCBΔXCGB (with the A. niger GBase open reading frame [ORF]) and pETAnGB (with GBase cDNA), were constructed (see below) for GBase expression in A. niger and E. coli BL21(DE3), respectively.
Media and culture conditions.
The A. niger strains (except for A. niger MA169.4) were maintained on potato dextrose agar (PDA). The inoculum for liquid cultures was generated using spores from such PDA plates. Shake flask cultivation was carried out in either minimal medium (26) or nitrogen-free minimal medium supplemented with the respective guanidinium compounds (with N in amounts equivalent to that in 28 mM NH4NO3); they were individually added after filter sterilization. The final pH of the medium was between 5.5 and 6.0. The mycelia were harvested, blot dried on filter paper, immediately frozen in liquid nitrogen, and stored at −20°C until further use. Stored mycelia were used within 15 days. Enzyme was extracted from the mycelia as described earlier (27). A. niger MA169.4 was maintained on minimal agar medium and was cultivated in shake flasks in either complete medium (24) or nitrogen-free minimal medium supplemented with the respective guanidinium compounds (as mentioned above).
Detection of metabolites.
A. niger samples (5.0 ml) were collected from the growth medium and centrifuged at 2,000 × g to remove the mycelia, and the clear spent medium was used to analyze the metabolites. Various guanidinium compounds and metabolites either were resolved on TLC plates or were quantified directly. The TLC plates were developed in two solvent systems (system I, acetone–n-butanol–acetic acid–water, 35:35:7:23 [vol/vol]; system II acetone–n-butanol–diethylamine–water, 10:10:2:5 [vol/vol]). The amino group-containing compounds were visualized with ninhydrin (1.0% in isopropanol) (28). The Ehrlich reagent (1.0 g p-dimethylaminobenzaldehyde dissolved in 25 ml concentrated HCl and 75 ml methanol) was used to detect urea and citrulline. A solution of 2,4-dinitrophenylhydrazine (0.4%) in 2.0 M hydrochloric acid was used to detect aldehydes (29). All guanidinium compounds were detected by spraying the Sakaguchi reagent (30).
The guanidinium compounds were also derivatized with benzoin (31), and the fluorescence either was detected (at 312 nm) on TLC plates or was quantified in a spectrofluorometer (RF-5301PC series from Shimadzu; excitation at 325 nm and emission at 443 nm).
4-Guanidinobutyraldehyde (GBald) is not commercially available and, hence, was synthesized (32, 33). GBald was resolved from unreacted l-arginine by silica gel chromatography with methanol as the solvent. The isolated GBald invariably contained traces of GB (a possible oxidation product). GBald was estimated by monitoring its azine derivative at 306 nm (34, 35).
Enzyme and protein estimations.
To assay different enzyme activities, the crude A. niger mycelial extracts were prepared in appropriate buffers. Ureohydrolases were extracted in 200 mM potassium phosphate buffer (with 2.0 mM 2-mercaptoethanol and 1.0 mM phenylmethylsulfonyl fluoride at a final pH of 7.2), whereas for aldehyde dehydrogenases, 100 mM sodium pyrophosphate (with 14 mM 2-mercaptoethanol, 1.0 mM EDTA, and 0.1% Triton X-100 at a final pH of 9.0) was used. These enzyme extracts were routinely desalted (on Sephadex G-25) to eliminate assay interference by small molecules. The protein concentration was estimated (36) using bovine serum albumin as a reference. For each enzyme monitored, specific activity is defined as the number of units per milligram of protein.
The arginase standard assay mixture at pH 10.6 consisted of l-arginine (150 mM), glycine (150 mM), MnSO4 (1.0 mM), and an appropriate amount of enzyme. For other ureohydrolases, standard assay mixtures at pH 7.5 consisted of HEPES (100 mM), MnSO4 (1.0 mM), the corresponding guanidinium substrate (25 mM), and an appropriate amount of enzyme. The urea formed was estimated by a modified Archibald method (37, 38). One unit of ureohydrolase activity was defined as amount of enzyme required to produce 1 μmol of urea per min in the standard assay. Amine oxidase (with benzylamine as the substrate) was routinely assayed as reported earlier (18). One unit of amine oxidase was defined as the amount of enzyme that produced 1.0 nmol of benzaldehyde per min in the standard assay. Amine oxidase (with agmatine as the substrate) was also assayed by monitoring the azine derivative of GBald at 306 nm (34, 35); however, an acetaldehyde standard curve was employed, as pure GBald was not available. One unit of enzyme activity is defined as the amount of enzyme required to produce 1.0 nmol of GBald (acetaldehyde equivalents) per min in the standard assay. Arginine decarboxylase was assayed with l-arginine as described before (39). Both arginine and agmatine (the product) were monitored on TLC plates as their fluorescent derivatives (31, 40). The E. coli extract containing arginine decarboxylase and agmatinase served as the respective enzyme-positive controls; E. coli XL1-Blue transformed with pKA5 was used for this purpose (13). The A. niger succinic semialdehyde (SSA) dehydrogenase (SSADH) activity was determined by monitoring the formation of NADH at 340 nm (41). GBald dehydrogenase was assayed by a method essentially similar to that used for SSA dehydrogenase, except that GBald was used as the substrate in place of SSA.
The enzyme activity data presented are typical reproductions of at least three independent measurements. Experimentally determined values are presented as points, while the lines join two data points.
MS/MS analysis of GBase protein.
GBase protein was purified to homogeneity from GB-grown A. niger mycelia. The purified GBase was desalted, concentrated, and taken in water with an Amicon Ultra-15 centrifugal filter unit with an Ultracel-10 membrane (molecular mass cutoff, ∼10 kDa; Millipore) before loading on an SDS-polyacrylamide gel. In-gel digestion with trypsin was done as reported previously (42), with some modification. The trypsin-digested GBase was mixed with matrix solution (α-cyano-4-hydroxycinnamic acid [CHCA]; λ maxima at 337 nm and 355 nm). The sample was air-\dried at room temperature before it was subjected to matrix-assisted laser desorption ionization–tandem time of flight mass spectrometry (MS) analysis (AB Sciex, Framingham, MA). A frequency-tripled neodymium-doped yttrium aluminum garnet laser (355 nm, 3.49 eV) was used as the laser source. The acquired spectra were processed using attached 4000 series Explorer software (version 3.5.3). The peptide fragment ions were subjected to similarity searches (MS/MS ion search) using the Mascot server (version 2.1; Matrix Science) against the sequences in the NCBInr database with fungi as the queried organism. The number of missed cleavages was set at 2. MS/MS spectra with a Mascot score higher than the significant score, using a confidence interval greater than 95%, were considered.
Cloning and expression of the GBase ORF in A. niger.
The peptide fragment data from MS/MS analysis of the purified GBase protein were used to identify the GBase locus (gbu) in the A. niger genome (http://genome.jgi-psf.org/Aspni5/Aspni5.home.html). The corresponding GBase ORF from A. niger NCIM 565 genomic DNA (1,384 bp) was PCR amplified using primers GBNdF1 and GBNoR1 (Table 2). The PCR product (obtained by use of a 25-μl reaction mixture containing 3.5 mM MgCl2, 200 μM deoxynucleoside triphosphates, 0.5 μM primers, and 5 units of Pfu DNA polymerase from MBI Fermentas) was purified with a GeneJET gel extraction kit (MBI Fermentas, Lithuania). The NdeI-NotI fragment from this amplicon was cloned in front of the constitutive A. niger citA promoter to obtain pCBΔXCGB (see Fig. S1 in the supplemental material). This plasmid was linearized with ScaI and used to transform A. niger NCIM 565 protoplasts (43). The transformants were single spored and characterized for GBase expression.
TABLE 2.
Primers used in this work
| Primer | Sequence (5′-3′)a | Restriction site |
|---|---|---|
| GBNdF1 | 5′-CAAATTCCATATGAGATCCCTCACTTGGG-3′ | NdeI |
| GBNoR1 | 5′-CATAACTTGCGGCCGCATACTTCATTCACAGC-3′ | NotI |
| GBEco2 | 5′-CGGAATTCAGAAGCTGAACATTG-3′ | EcoRI |
| GBXhR2 | 5′-GCAGTACAAGCTCAAGGACG-3′ | |
| GBXbF1 | 5′-TACAAATCTAGAAAGCACTATGG-3′ | XbaI |
| GBEco1 | 5′-GTCGAATTCTGTGATCGGGATG-3′ | EcoRI |
| GBM1F1 | 5′-GATTGCTGCTCCGAATTTCC-3′ | |
| GBM1R1 | 5′-GCTATCCGTCTCACTCTTAC-3′ | |
| GBRTR1 | 5′-TGGTGTGATCGGGATGTCG-3′ |
Restriction sites are underlined.
Construction of A. niger GBase knockout.
A total of 1,442 bp (the terminal 253 bp of the GBase ORF along with 1,189 bp of the 3′ flanking region) was PCR amplified from A. niger NCIM 565 genomic DNA with primers GBEco2 and GBXhR2 (Table 2). The amplicon was cloned into pBScGDH (44) between the EcoRI and XhoI sites to obtain pBSGB3′F. At the same time, 1,548 bp (the initial 514 bp of the GBase ORF along with 1,034 bp of the 5′ flanking region) was also PCR amplified (with primers GBXbF1 and GBEco1; Table 2) and cloned into pBSGB3′F between the EcoRI and XbaI sites to obtain pBSGB5′3′F. A. niger pyrA was inserted at the EcoRI site of pBSGB5′3′F to form the gbu disruption cassette (pBSGBPyrADel). A. niger MA169.4 was transformed with either a circular or an XmnI-linearized disruption cassette. The transformants (gbu::pyrA) were selected on YDA (yeast nitrogen base without amino acids and ammonium sulfate but containing dextrose, ammonium nitrate, and agar) plates without uracil, thus scoring for uracil prototrophy.
Cloning and expression of GBase cDNA in E. coli.
Total RNA was extracted from A. niger mycelia grown on GB by use of an RNeasy plant minikit (Qiagen, Leusden, The Netherlands). Reverse transcriptase PCR (RT-PCR) was performed with 6.5 μg of total RNA using a Transcriptor first-strand cDNA synthesis kit (Roche, Mannheim, Germany). Two short introns were predicted in the early part of the GBase ORF (GenBank accession no. KF991130). Only this region was reverse transcribed with a combination of primers GBNdF1 and GBRTR1 (Table 2). The NdeI-BmtI fragment of this amplicon was moved along with the BmtI-NotI fragment from pCBΔXCGB to construct complete GBase cDNA (1,269 bp) in pETAnGB (see Fig. S1 in the supplemental material). This plasmid was transformed into competent E. coli BL21(DE3) cells, and GBase expression was followed (45).
RESULTS AND DISCUSSION
Arginase provides the sole catabolic route for arginine in A. niger (1). An in silico analysis retrieved four more putative ureohydrolase sequences from the genome of this fungus (Table 1). Such sequences from A. niger and other aspergilli are mostly annotated as putative agmatinases. The possible occurrence of additional ureohydrolases and the catabolism of other guanidinium compounds were thus anticipated. The metabolism of agmatine and related guanidinium group-containing compounds by A. niger was therefore explored through growth, metabolite, and enzyme studies.
Growth on various guanidinium compounds.
The nitrogen-free minimal medium supplemented with l-arginine, agmatine, or GB supported good growth of A. niger. Growth on NH4NO3 as the nitrogen source served as a positive control. Scant or negligible growth was observed on media containing 3-guanidinopropionate, guanidinoacetate, and two arginine analogs, namely, d-arginine and l-homoarginine (Fig. 2). The A. niger strains D-42 (arginase knockout) and ΔXCA-29 (constitutively overexpressing arginase) (1) were chosen to assess the arginase contribution to guanidinium compound metabolism in this fungus. The growth phenotypes of these two strains were similar to the growth phenotype of the parent strain (WT), except that strain D-42 was unable to grow on arginine. The results are consistent with the fact that the fungus is able to utilize agmatine and GB in an arginase-independent manner.
FIG 2.

Growth of A. niger on various guanidinium compounds. (Top) Spores (equal numbers) of A. niger (the WT, D-42, and ΔXCA-29 strains) were spot inoculated on media containing different nitrogen sources (20 mM with respect to total N), namely, l-arginine (Arg), agmatine (Agm), 4-guanidinobutyrate (GB), 3-guanidinopropionate (GP), d-arginine (D-Arg), l-homoarginine (H-Arg), and guanidinoacetate (GAA). (Bottom) The corresponding growth patterns on urea and the respective amino products (namely, l-ornithine [Orn], putrescine [Put], and 4-aminobutyric acid [GABA]) of ureohydrolase action. The results for controls with ammonium nitrate (NH4NO3) and no nitrogen source (−N) are also shown. Growth was recorded at two time points.
Ureohydrolases are known to act on l-arginine, agmatine, and GB to form ornithine, putrescine, and GABA, respectively, with the other product always being urea (Fig. 1, steps 3, 5, and 12). The suitability of these metabolites as potential nitrogen sources was examined. The three strains of A. niger were able to grow equally well on ornithine, putrescine, GABA, and urea (Fig. 2). The possible occurrence of agmatinase and GBase in this fungus could therefore be anticipated and was tested (see below). For this, the fungus was conveniently grown in liquid culture to observe possible metabolites appearing in the culture medium and detect enzyme activities in the mycelia.
Metabolites observed in culture media.
The culture media were sampled periodically during the growth of A. niger (WT and D-42 strains) on different nitrogen sources. These samples were analyzed for the presence of various metabolites.
(i) GB as an intermediate in agmatine catabolism.
l-Arginine, agmatine, and GB disappeared from the A. niger culture medium with time (Fig. 3); since strain D-42 was unable to grow on arginine, only agmatine and GB disappearance was observed in this case (Fig. 4). The results are consistent with the growth of A. niger on these nitrogen sources (Fig. 2). A transient appearance of GB was observed (at between 30 and 50 h) only in the agmatine-containing culture medium. No GB could be found in the culture media when the fungus was grown on arginine or ammonium nitrate (Fig. 3). As agmatine and l-arginine could not be directly resolved, their fluorescent benzoin derivatives were analyzed (see Fig. S2 in the supplemental material). Agmatine was not detected in the spent media of arginine-grown samples. A faint fluorescent spot (Rf, 0.94) was, however, observed in agmatine-grown samples at 30 h; the Rf of this spot corresponds to that of standard GBald, thereby suggesting that it may also be a metabolic intermediate in the agmatine catabolic pathway.
FIG 3.

Detection of guanidinium compounds by TLC. A. niger strains (the WT and D-42 strains) were grown in liquid culture with agmatine (A), GB (B), l-arginine (C), and NH4NO3 (D) as the sole nitrogen source. Aliquots of these culture media (2.0 μl each) were spotted on TLC plates, developed in solvent system I, and visualized by use of the Sakaguchi reagent. Results for samples obtained after every 10 h of growth (lanes 1 to 10) are shown. Culture media with no nitrogen (lane M), the standards (l-arginine, ornithine, agmatine, putrescine, GB, GABA, urea, citrulline, SSA, and GBald [lanes 1 to 10, respectively]), and a mixture of l-arginine, agmatine, and GB (lane 11) were tested.
FIG 4.

Fate of guanidinium compounds and detection of urea in A. niger culture media. A. niger strains (the WT and D-42 strains) were grown in liquid culture with l-arginine (circles), agmatine (inverted triangles), GB (squares), or NH4NO3 (diamonds) as the sole nitrogen source. Aliquots of the culture media were sampled at various time points and analyzed for the respective guanidinium compounds (filled symbols) and urea (the corresponding open symbols). GB appeared (↓) and disappeared (↑) from the culture medium (Fig. 4) only when the fungus was grown on agmatine.
Potential metabolic intermediates, like citrulline, SSA, putrescine, and GABA, were not detected in the culture media at any time point. Amino compounds (except for agmatine or arginine, whenever one of these was added to the media) were not detected. Interestingly, urea was found in the culture media when the organism was grown on l-arginine, agmatine, or GB; urea transiently appeared when these compounds disappeared from the media (Fig. 4). Besides urea as the common product, ureohydrolase action does produce an amine, such as ornithine (by arginase), putrescine (by agmatinase), or GABA (by GBase). As none of these products appeared in the respective culture media, the transient accumulation of urea (and not ornithine or GABA) suggests that A. niger prefers the prior catabolism of amino products over urea. However, aspergilli do possess a functional urease, and the A. niger enzyme has a reasonable Km for its substrate (3.0 mM) (46). Deferred urea utilization could be due to the paucity of urease activity per se under those growth conditions.
Table 3 summarizes the metabolites found in the culture media. The data suggest that GB and also, possibly, GBald are intermediates in the catabolism of agmatine. Urea was the common metabolite detected on l-arginine, agmatine, and GB. Urea formation from agmatine and GB (even with A. niger strain D-42) clearly implied the possible presence of a ureohydrolase(s) other than arginase.
TABLE 3.
Summary of metabolites observed in the culture medium of A. niger WT and D-42 strains
| Metabolite | Detection of metabolite with the following nitrogen source and straina: |
|||||||
|---|---|---|---|---|---|---|---|---|
|
l-Arginine |
Agmatine |
GB |
NH4NO3 |
|||||
| WT | D-42 | WT | D-42 | WT | D-42 | WT | D-42 | |
| l-Arginine | + | + | − | − | − | − | (+) | (+) |
| Agmatine | − | − | + | + | − | − | − | − |
| GABA | − | − | − | − | − | − | − | − |
| GB | − | − | + | + | + | + | − | − |
| GBald | − | − | + | + | − | − | − | − |
| SSA | − | − | − | − | − | − | − | − |
| l-Ornithine | − | − | − | − | − | − | (+) | (+) |
| Putrescine | − | − | − | − | − | − | − | − |
| Urea | + | − | + | + | + | + | − | − |
+ and −, presence and absence of the particular metabolite, respectively; (+), detection of traces. The D-42 strain does not grow on l-arginine.
(ii) Detection of PQQ in the culture medium.
Only when A. niger (and also the D-42 strain) was grown on agmatine as the sole nitrogen source, the culture medium turned pink at later time points. The possible identity of this pink product as pyrroloquinoline quinone (PQQ) was considered. The pink product was purified on a DEAE-Sephacel column and spectrally characterized (Fig. 5). The UV-visible (UV-Vis) and fluorescence spectra of this compound matched those reported for PQQ (47, 48). Upon electrospray ionization MS analysis, the m/z value (313.89) of the compound was the same as that of authentic PQQ. PQQ is a cofactor for some amine oxidases (47–49). Detection of PQQ, only when grown on agmatine, implicates a role for amine oxidase in A. niger agmatine catabolism.
FIG 5.

Identification of PQQ in A. niger spent medium. (Left) The UV-Vis spectra (solid line, pH 1.0; dotted line, pH 7.0; dashed line, pH 12.0) and the fluorescence spectra at pH 7.0 (black line, emission λ at 448 nm; gray line, excitation λ at 323 nm) of the isolated pink compound are shown. (Right) The mass spectrum of the pink compound is compared with that of standard PQQ. AU, relative abundance units.
Enzymology of agmatine metabolism.
The detection of relevant enzymes constitutes important evidence in support of an operational metabolic pathway. Therefore, various enzyme activities were monitored in the desalted crude mycelial extracts (see Materials and Methods) of A. niger grown (for 24 h) on l-arginine, agmatine, GB, or NH4NO3.
(i) Ureohydrolases.
The ability of A. niger to utilize arginine, agmatine, and GB (Fig. 2) in association with the detection of urea in the culture media (Fig. 4 and Table 3) points to the possible occurrence of more than one ureohydrolase. Of these, arginase is well studied (1, 25), and it was detected on all the nitrogen sources tested (Table 4). Scoring for the presence of agmatinase (on agmatine) and GBase (on GB) activities was therefore of interest. Since agmatinase activity was not found in agmatine-grown mycelia, an alternate route(s) for agmatine catabolism was anticipated. However, GBase activity was consistently observed when the fungus was grown on either GB or agmatine (Table 4). Under similar conditions, the A. niger D-42 strain (arginase knockout) was devoid of agmatinase and arginase activities, but GBase activity was readily detected. Arginase therefore may not have a direct role to play in agmatine/GB catabolism. Arginase-independent catabolism of arginine was recently reported in K. lactis; this pathway involved ketoarginine and GBase, but no agmatine was involved (11). The A. niger ureohydrolase data presented here clearly implicate a role for GBase in agmatine catabolism.
TABLE 4.
Enzyme activities in A. niger mycelia
| Enzyme | Activity (U mg−1 protein)a |
|||
|---|---|---|---|---|
| l-Arginine | Agmatine | GB | NH4NO3 | |
| Ureohydrolase | ||||
| 4-Guanidinobutyrase | ND | 0.7 | 0.2 | ND |
| Arginase | 10.0 | 2.3 | 4.0 | 5.0 |
| Agmatinase | ND | ND | ND | ND |
| Amine oxidase | ||||
| Benzylamine | ND | 81.0 | ND | ND |
| Agmatine | ND | 5.0 | ND | ND |
| Aldehyde dehydrogenaseb | ||||
| GBald | 0.3 | 11.8 | 9.6 | ND |
| Propionaldehyde | 1.9 | 64.9 | 21.5 | ND |
| Acetaldehyde | 1.1 | 59.3 | 18.8 | ND |
| SSA | ND | 5.0 | 28.4 | ND |
The data on enzyme activity in crude extracts are representative of those from three independent experiments (performed in duplicate), and the variation was less than 10%. ND, not detected.
A high background dehydrogenase activity (NADH appearance) was observed with agmatine.
(ii) Amine oxidase.
The utilization of agmatine without an agmatinase contribution (see above) and the associated detection of PQQ in the spent medium suggested that an amine oxidase may participate in the first step of its catabolism (Fig. 1). A. niger expresses amine oxidases to utilize amines as the nitrogen source; many of them are well characterized. A broadly specific amine oxidase acting on agmatine is reported from A. niger (18, 50). A. niger crude extracts, obtained from agmatine-grown mycelia, contained an amine oxidase that could oxidize agmatine and benzylamine (Table 4). The absence of this activity in mycelia grown on other nitrogen sources, like GB, arginine, or NH4NO3, is strongly indicative of its role in initiating agmatine catabolism in A. niger. Detection of GBald (Table 3), the product of agmatine oxidation, lends further support to this view.
(iii) Aldehyde dehydrogenases.
An amine oxidase acting on agmatine forms GBald. Consecutive steps catalyzed by amine oxidase and GBald dehydrogenase define a metabolic path from agmatine to GB (Fig. 1). A dehydrogenase converting GBald to GB could be anticipated from the unambiguous detection of GB in the culture medium (Fig. 2). Its biochemical characterization was limited by the fact that GBald is not commercially available. Claiming a specific A. niger GBald dehydrogenase activity is fraught with two difficulties: (i) some GB was always found in synthesized and purified GBald samples; GBald may be unstable and susceptible to air oxidation. (ii) A. niger displays significant dehydrogenase activities with promiscuous substrate specificity (51). While NAD-dependent GBald oxidation could be demonstrated in extracts of mycelia grown on agmatine, substantial dehydrogenase activity was also observed with acetaldehyde and propionaldehyde as the substrates (Table 4). Resolving/purifying these aldehyde dehydrogenases is required to establish the presence of a GBald-specific enzyme in A. niger. Such a GBald-specific aldehyde dehydrogenase is known in Pseudomonas spp. (52, 53).
GABA may arise from glutamate, agmatine, or 4-guanidinobutyrate, but its catabolism is well established in A. niger and involves GABA transaminase and SSADH (15). GB (provided externally or arising internally from agmatine; Table 3) is converted by A. niger GBase to GABA (Table 4). SSADH is thus the second aldehyde dehydrogenase expected to participate in agmatine catabolism. Indeed, SSADH activity was found in A. niger mycelia grown on agmatine or GB (Table 4). A specific SSADH from GABA-grown A. niger has already been characterized (41, 51).
Deconstructing A. niger agmatine metabolism.
Spermidine is the major polyamine in ascomycete fungi, while putrescine and spermine contribute as minor polyamines (14). The presence of agmatine is not documented in these fungi. With the rare exceptions of Ceratocystis minor and Verticillium dahliae (7) and of Panus tigrinus (9), there are no reports on arginine decarboxylase (and, by implication, agmatine biosynthesis) in the fungal kingdom. In several attempts, we did not detect any arginine decarboxylase activity in arginine-grown A. niger mycelia. Also, when queried with known arginine decarboxylase sequences, no promising hits were retrieved from the A. niger genome (http://genome.jgi-psf.org/Aspni7/Aspni7.home.html). Both biochemical data and the results of in silico analysis suggest that A. niger may not have a functional arginine decarboxylase, the major route for agmatine biosynthesis (Fig. 1). The inability of strain D-42 to grow on arginine is also consistent with this idea.
Constitutive overexpression of arginase did not impart any growth advantage to A. niger on other guanidinium compounds (Fig. 2, strain ΔXCA-29). Agmatine is not a substrate for purified A. niger arginase. A highly specific arginase and the absence of agmatinase rule out the formation of putrescine as the first product of agmatine in A. niger. The presence of significant amine oxidase activity in agmatine-grown A. niger mycelia along with the detection of PQQ (a cofactor associated with some amine oxidases) in the corresponding spent media is consistent with agmatine oxidation by an amine oxidase. Agmatine is converted to GBald by this step. There are two possible routes from GBald to GABA (Fig. 6), whereas the subsequent metabolism of GABA in A. niger is well established. The detection of GB (only in the culture medium supplemented with agmatine; Table 3) and the induction of GBald dehydrogenase, GBase, and SSADH activities on agmatine strongly favor the GB route (Fig. 6). Without firm evidence to exclude the possibility of the presence of a GBald-specific ureohydrolase, the existence of the alternate path for GBald cannot be discounted. Both GBald and GABald dehydrogenase activities were ascribed to the same enzyme in P. aeruginosa (52), whereas GBald and GABald dehydrogenase represent two distinct enzymes in other systems (53, 54). A well-designed isotope-labeling study could substantiate this aspect further. Regardless of how GBald is processed, GABA is the common downstream metabolite. The sequential action of GABA transaminase and SSADH converts GABA to succinate, which in turn enters the Krebs cycle (15).
FIG 6.
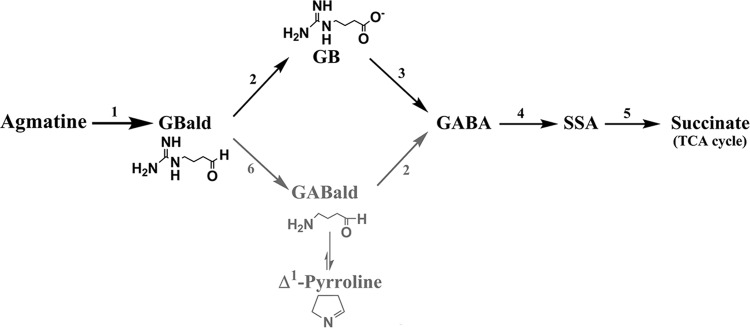
Proposed pathway for agmatine catabolism in A. niger. The different enzymatic steps are numbered as follows: 1, agmatine oxidase; 2, GBald/GABald dehydrogenase; 3, GBase; 4, GABA transaminase; 5, SSA dehydrogenase; 6, GBald ureohydrolase. Likely steps and metabolites in the alternate path for GBald are in gray.
While A. niger may not biosynthesize agmatine, this fungus is capable of growth by assimilating externally provided agmatine (Fig. 2). This metabolic versatility is consistent with its saprophytic lifestyle. Three alternate routes of agmatine catabolism are possible across organisms. GB and GBase participate in only one of these pathways; incidentally, this route begins with agmatine oxidation. The growth, metabolite, and enzyme activity data, taken together, define a novel pathway for agmatine catabolism in A. niger. Although individual enzymatic steps are reported in diverse organisms (Fig. 1), A. niger provides the first functional example of this agmatine catabolic pathway.
Functional annotation of A. niger GBase.
The detection of GBase activity in A. niger mycelia grown on either GB or agmatine (Table 4) further led to its purification and characterization. The peptide fragments of the GBase protein (see “MS/MS analysis of GBase protein” in Materials and Methods) matched those of hypothetical protein AN7488.2 (from A. nidulans FGSC A4; NCBInr database for fungi). A blastp analysis of this sequence retrieved a highly similar sequence (Table 5) from the A. niger genome (http://genome.jgi-psf.org/Aspni5/Aspni5.home.html; annotated as a putative arginase/agmatinase/formiminoglutamase). The corresponding ORF from A. niger NCIM 565 genomic DNA (1,384 bp) was cloned in front of the citA promoter for its constitutive expression (see Fig. S1 in the supplemental material). When transformed with linearized pCBΔXCGB (see Materials and Methods), 28 stable A. niger transformants were obtained. Four of them were analyzed for GBase expression in detail. These transformants were constitutive for GBase and did not require induction with GB or agmatine for expression (Table 6).
TABLE 5.
A. niger GBase sequence retrieved by MS/MS analysis
| Sequence type | Sequence |
|---|---|
| Peptide fragments retrieved by MS/MS analysisa | DLGGWYGRK |
| QMAGTSYNTR | |
| SPVTPASSEYGSK | |
| LVTLGGDHSVALPALR | |
| SKLVTLGGDHSVALPALR | |
| DCGDIPITPFDNGLAER | |
| LNIVGADIVEVSPSYDNK | |
| ALYQIYQKPITVLHFDAHLDTWNPVR | |
| Retrieved GBase sequence from A. niger genomeb | MRSLTWAACLALAGTAFAHAHHDHEEVPEHVREELLRKWDQEFTFSGIASFAHLKPVKCLIEPDERYDIAVIGAPFDTAVSYRPGARFGPRSIRAASARQMAGTSYNTRAGINPYASWATVKDCGDIPITPFDNGVAERQMYEAFLELGSRPALTSADSKYGTKGISAGKSKLVTLGGDHSVALPALRALYQIYQKPITVLHFDAHLDTWNPVRYSAYWTSEQAHFNHGSFFHKASREGLICNSTSAHAGLRTRLTGVDDSDYTNPGPEQGFMRIHADDIDDLGPMGIVNRIIERIGLDSDQPVYLSVDIDVLDPATAPGTGTPEPGGWTTREFIRILRGIEKLNIVGADIVEVSPSYDNKGETTALAAAQVAFEIITSMVKAGAGEELGGWYGRKVTPVEETVVVEEGEDPVEKKEAKDEL |
The eight peptide fragments are presented in order of frequency of their occurrence (highest to lowest).
The A. niger GBase sequence was retrieved (protein identifier, 55649; location, chr_4_2:3573655-3575104) by querying the A. niger genome database (http://genome.jgi-psf.org/Aspni5/Aspni5.home.html) with the AN7488.2 hypothetical protein of A. nidulans (see Materials and Methods). Peptide fragments identified through MS/MS analysis are highlighted in gray. The single underlining and double underlining indicate that the overlapping SKLVTLGGDHSVALPALR and LVTLGGDHSVALPALR sequences were found as peptide fragments; both are distinct from the subsequent fragment, ALYQIYQKPITVLHFDAHLDTWNPVR.
TABLE 6.
GBase activity in A. niger transformants grown in minimal medium
| Transformant | Sp act (U mg−1 protein) |
|---|---|
| XCGB-13 | 0.36 |
| XCGB-03 | 0.30 |
| XCGB-19 | 0.25 |
| XCGB-17 | 0.31 |
| A. niger NCIM 565 | 0.00 |
| A. niger NCIM 565a | 0.16 |
| A. niger MA169.4a | 0.23 |
| A. niger Δgbu-90b | 0.00 |
With GB as the sole nitrogen source.
No growth on GB.
Disruption of the A. niger gbu locus was also attempted in a ΔkusA background (24). First, it was ensured that A. niger MA169.4 (kusA::amdS; pyrG−) grows on GB as the sole nitrogen source and displayed GBase activity (Table 6). Upon transformation with pBSGBPyrADel (see Materials and Methods), more than 150 transformants were obtained. Twelve of them were unable to grow on GB as the sole nitrogen source; one of them (the Δgbu-90 strain) was chosen for detailed characterization. Integration of the disruption cassette in the A. niger Δgbu-90 genome was confirmed by genomic PCR using the primer pair GBM1F1 and GBM1R1 (which produced an amplicon of 2.8 kb from A. niger Δgbu-90 versus a 1.6-kb product from the A. niger MA169.4 genome); this amplicon was also confirmed through restriction digestion with EcoRV (see Fig. S3 in the supplemental material). The growth of the Δgbu-90 strain on various metabolites of the proposed agmatine catabolic pathway was tested. As expected, the Δgbu-90 strain was unable to grow on GB but did grow on urea and GABA (the two products of the GBase reaction) (Fig. 7). In contrast, A. niger MA169.4 was able to utilize GB as the sole nitrogen source. This confirms that GBase is the second functional ureohydrolase in the A. niger genome (Table 1) and it is solely responsible for GB utilization. The growth (albeit poor; Fig. 7) of the Δgbu-90 strain on agmatine indicates that the agmatine amino group alone can serve as a source of nitrogen for this fungus. Agmatine metabolism by the Δgbu-90 strain in liquid culture was accompanied by the concomitant accumulation of GB but the absence of urea. However, about 5 mM urea was found in the medium after 60 h of A. niger MA169.4 growth. Taken together, these observations strongly indicate that efficient growth on agmatine requires a functional GBase as well.
FIG 7.
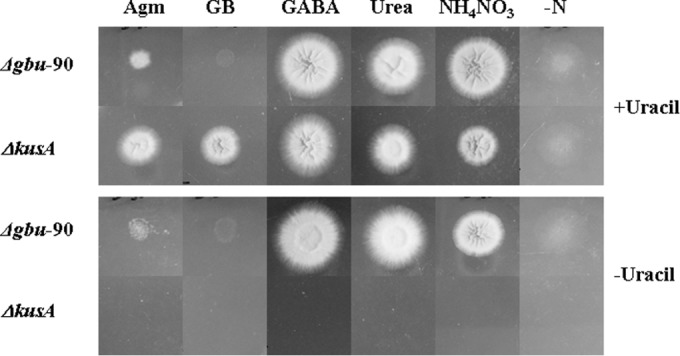
Phenotypic analysis of A. niger Δgbu-90 (GBase disruptant) and its parent, A. niger MA169.4 (ΔkusA). Spores (equal numbers) were spotted on minimal media containing different nitrogen sources (15 mM with respect to total N), namely, agmatine (Agm), 4-guanidinobutyrate (GB), 4-aminobutyric acid (GABA), and urea. Controls for growth on ammonium nitrate (NH4NO3) and no nitrogen source (−N) are also shown. Growth was recorded at 60 h. Medium without uracil (10 mM) served to show that A. niger Δgbu-90 is a uracil prototroph and also for utilization of uracil as a nitrogen source.
Two small introns (127 to 183 bp and 311 to 368 bp) were predicted in the A. niger GBase ORF (GenBank accession no. KF991130). These were confirmed by GBase cDNA synthesis by RT-PCR and subsequent expression of this cDNA (pETAnGB) in E. coli. Upon isopropyl-β-d-1-thiogalactopyranoside induction, E. coli cells transformed with pETAnGB displayed significant GBase activity (∼3.0 U mg−1 protein); no activity was detected in uninduced transformants or cells without the plasmid. It may be noted that the only ureohydrolase activity reported in E. coli is that of agmatinase (13). The heterologous expression of GBase activity in E. coli thus functionally annotated the gbu ORF of A. niger NCIM 565.
Conclusions.
The results presented here support a novel route for agmatine utilization in A. niger. The catabolism of agmatine proceeds by way of 4-guanidinobutyrate to 4-aminobutyrate into the Krebs cycle. This study also led to the discovery of GBase as the second ureohydrolase activity from A. niger. The corresponding GBase ORF was cloned and functionally expressed. An A. niger Δgbu strain was constructed, and it was unable to utilize GB. A. niger GBase orthologs (with amino acid sequence identities of ≥85%) were found to be present in all the available Aspergillus genomes (Table 1, accession numbers in bold). Finally, the similar patterns of expression of ureohydrolase activities in A. nidulans (see Table S1 in the supplemental material) lend support to the possibility that the A. niger agmatine catabolic pathway presented here may be common across aspergilli.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. M. Boyle, Virginia-Maryland Regional College of Veterinary Medicine, Blacksburg, VA, for providing plasmid pKA5.
Sunil Kumar received a UGC fellowship and Tejaswani Saragadam received institute teaching assistantship during the course of this work.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03987-14.
REFERENCES
- 1.Dave K, Ahuja M, Jayashri TN, Sirola RB, Punekar NS. 2012. A novel selectable marker based on Aspergillus niger arginase expression. Enzyme Microb Technol 51:53–58. doi: 10.1016/j.enzmictec.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Davis RH. 1986. Compartmental and regulatory mechanisms in the arginine pathways of Neurospora crassa and Saccharomyces cerevisiae. Microbiol Rev 50:280–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouchereau A, Guenot P, Larher F. 2000. Analysis of amines in plant materials. J Chromatogr B Biomed Sci Appl 747:49–67. doi: 10.1016/S0378-4347(00)00286-3. [DOI] [PubMed] [Google Scholar]
- 4.Tabor CW, Tabor H. 1985. Polyamines in microorganisms. Microbiol Rev 49:81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alcazar R, Altabella T, Marco F, Bortolotti C, Reymond M, Koncz C, Carrasco P, Tiburcio AF. 2010. Polyamines: molecules with regulatory functions in plant abiotic stress tolerance. Planta 231:1237–1249. doi: 10.1007/s00425-010-1130-0. [DOI] [PubMed] [Google Scholar]
- 6.Gupta K, Dey A, Gupta B. 2013. Plant polyamines in abiotic stress responses. Acta Physiol Plant 35:2015–2036. doi: 10.1007/s11738-013-1239-4. [DOI] [Google Scholar]
- 7.Khan AJ, Minocha SC. 1989. Biosynthetic arginine decarboxylase in phytopathogenic fungi. Life Sci 44:1215–1222. doi: 10.1016/0024-3205(89)90317-2. [DOI] [PubMed] [Google Scholar]
- 8.Sannazzaro AI, Alvarez CL, Menendez AB, Pieckenstain FL, Alberto EO, Ruiz OA. 2004. Ornithine and arginine decarboxylase activities and effect of some polyamine biosynthesis inhibitors on Gigaspora rosea germinating spores. FEMS Microbiol Lett 230:115–121. doi: 10.1016/S0378-1097(03)00880-2. [DOI] [PubMed] [Google Scholar]
- 9.Boldt A, Miersch J, Reinbothe H. 1971. Metabolism of agmatine in fruit-bodies of the fungus Panus tigrinus (Bull ex Fr.) Sing. (Tricholomataceae). Phytochemistry 10:731–738. doi: 10.1016/S0031-9422(00)97140-1. [DOI] [Google Scholar]
- 10.Klein RD, Geary TG, Gibson AS, Favreau MA, Winterrowd CA, Upton SJ, Keithly JS, Zhu G, Malmberg RL, Martinez MP, Yarlett N. 1999. Reconstitution of a bacterial/plant polyamine biosynthesis pathway in Saccharomyces cerevisiae. Microbiology 145(Pt 2):301–307. [DOI] [PubMed] [Google Scholar]
- 11.Romagnoli G, Verhoeven MD, Mans R, Fleury RY, Bel-Rhlid R, van den Broek M, Maleki Seifar R, Ten Pierick A, Thompson M, Muller V, Wahl SA, Pronk JT, Daran JM. 2014. An alternative, arginase-independent pathway for arginine metabolism in Kluyveromyces lactis involves guanidinobutyrase as a key enzyme. Mol Microbiol 93:369–389. doi: 10.1111/mmi.12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goda S, Sakuraba H, Kawarabayasi Y, Ohshima T. 2005. The first archaeal agmatinase from anaerobic hyperthermophilic archaeon Pyrococcus horikoshii: cloning, expression, and characterization. Biochim Biophys Acta 1748:110–115. doi: 10.1016/j.bbapap.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 13.Satishchandran C, Boyle SM. 1986. Purification and properties of agmatine ureohydrolyase, a putrescine biosynthetic enzyme in Escherichia coli. J Bacteriol 165:843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis RH, Morris DR, Coffino P. 1992. Sequestered end products and enzyme regulation: the case of ornithine decarboxylase. Microbiol Rev 56:280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar S, Punekar NS. 1997. The metabolism of 4-aminobutyrate (GABA) in fungi. Mycol Res 101:403–409. doi: 10.1017/S0953756296002742. [DOI] [Google Scholar]
- 16.Janowitz T, Kneifel H, Piotrowski M. 2003. Identification and characterization of plant agmatine iminohydrolase, the last missing link in polyamine biosynthesis of plants. FEBS Lett 544:258–261. doi: 10.1016/S0014-5793(03)00515-5. [DOI] [PubMed] [Google Scholar]
- 17.Nakada Y, Itoh Y. 2003. Identification of the putrescine biosynthetic genes in Pseudomonas aeruginosa and characterization of agmatine deiminase and N-carbamoylputrescine amidohydrolase of the arginine decarboxylase pathway. Microbiology 149:707–714. doi: 10.1099/mic.0.26009-0. [DOI] [PubMed] [Google Scholar]
- 18.Yamada H, Adachi O. 1971. Amine oxidase (Aspergillus niger). Methods Enzymol 17:705–709. [Google Scholar]
- 19.Arakawa N, Igarashi M, Kazuoka T, Oikawa T, Soda K. 2003. d-Arginase of Arthrobacter sp. KUJ 8602: characterization and its identity with Zn2+-guanidinobutyrase. J Biochem 133:33–42. [DOI] [PubMed] [Google Scholar]
- 20.Nakada Y, Itoh Y. 2005. Pseudomonas aeruginosa PAO1 genes for 3-guanidinopropionate and 4-guanidinobutyrate utilization may be derived from a common ancestor. Microbiology 151:4055–4062. doi: 10.1099/mic.0.28258-0. [DOI] [PubMed] [Google Scholar]
- 21.Campo ML, Fuentes JM, Soler G. 1992. An arginine regulated γ-guanidobutyrate ureahydrolase from tench liver (Tinca tinca L.). Arch Int Physiol Biochim Biophys 100:55–60. doi: 10.3109/13813459209035259. [DOI] [PubMed] [Google Scholar]
- 22.Brunel-Capelle G, Brunel A, Bailly-Fenech G. 1969. Sur la γ-guanidinobutyrique-ureohydrolase de Penicillium roqueforti. C R Hebd Seances Acad Sci 269:2095–2098. [PubMed] [Google Scholar]
- 23.Brunel A, Brunel-Capelle G, Miquel A. 1967. Biosynthese induite de la l-arginine-ureohydrolase (arginase), dela γ-guanidinobutyrique-ureohydrolase et de la guanidinoacetique-ureohydrolase (glycocyaminase) chez Penicillium roqueforti. C R Hebd Seances Acad Sci 264:2777–2780. [Google Scholar]
- 24.Carvalho ND, Arentshorst M, Jin Kwon M, Meyer V, Ram AF. 2010. Expanding the ku70 toolbox for filamentous fungi: establishment of complementation vectors and recipient strains for advanced gene analyses. Appl Microbiol Biotechnol 87:1463–1473. doi: 10.1007/s00253-010-2588-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jayashri TN, Anuradha R, Punekar NS. 2009. Single-stranded megaprimer splicing through OE-PCR: construction of full-length Aspergillus niger arginase cDNA. Indian J Biochem Biophys 46:266–268. [Google Scholar]
- 26.Punekar NS, Vaidyanathan CS, Rao NA. 1984. Mechanisms of citric acid fermentation by Aspergillus niger. J Sci Ind Res 43:398–404. [Google Scholar]
- 27.Choudhury R, Noor S, Varadarajalu LP, Punekar NS. 2008. Delineation of an in vivo inhibitor for Aspergillus glutamate dehydrogenase. Enzyme Microb Technol 42:151–159. doi: 10.1016/j.enzmictec.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Zolg W, Ottow JC. 1973. Improved thin-layer technique for detection of arginine dihydrolase among the Pseudomonas species. Appl Microbiol 26:1001–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brady OL, Elsmie GV. 1926. The use of 2,4-dinitrophenylhydrazine as a reagent for aldehydes and ketones. Analyst 51:77–78. doi: 10.1039/an9265100077. [DOI] [Google Scholar]
- 30.Irreverre F. 1965. A modified Sakaguchi spray. Biochim Biophys Acta 111:551–552. doi: 10.1016/0304-4165(65)90069-3. [DOI] [PubMed] [Google Scholar]
- 31.Kai M, Miura T, Kohashi K, Ohkura Y. 1981. New method for the fluorimetric determination of guanidino compounds with benzoin. Chem Pharm Bull (Tokyo) 29:1115–1120. doi: 10.1248/cpb.29.1115. [DOI] [Google Scholar]
- 32.Tanaka Dagger K, Nakai R, Sen K, Shimizu E, Karasawa D, Yorifuji T. 2002. Purification and characterization of aminobutyraldehyde dehydrogenase from Arthrobacter sp. TMP-1. J Biochem Mol Biol Biophys 6:171–175. doi: 10.1080/1025814021000000916. [DOI] [PubMed] [Google Scholar]
- 33.Vanderbilt AS, Gaby NS, Rodwell VW. 1975. Intermediates and enzymes between α-ketoarginine and γ-guanidinobutyrate in the l-arginine catabolic pathway of Pseudomonas putida. J Biol Chem 250:5322–5329. [PubMed] [Google Scholar]
- 34.Paz MA, Blumenfeld OO, Rojkind M, Henson E, Furfine C, Gallop PM. 1965. Determination of carbonyl compounds with N-methyl benzothiazolone hydrazone. Arch Biochem Biophys 109:548–559. doi: 10.1016/0003-9861(65)90400-5. [DOI] [PubMed] [Google Scholar]
- 35.Toraya T, Fujimura M, Ikeda SI, Fukui S, Yamada H. 1976. Affinity chromatography of amine oxidase from Aspergillus niger. Biochim Biophys Acta 420:316–322. doi: 10.1016/0005-2795(76)90323-8. [DOI] [PubMed] [Google Scholar]
- 36.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 37.Archibald RM. 1945. Colorimetric determination of urea. J Biol Chem 157:507–518. [Google Scholar]
- 38.Boyde TR, Rahmatullah M. 1980. Optimization of conditions for the colorimetric determination of citrulline, using diacetyl monoxime. Anal Biochem 107:424–431. doi: 10.1016/0003-2697(80)90404-2. [DOI] [PubMed] [Google Scholar]
- 39.Wu WH, Morris DR. 1973. Biosynthetic arginine decarboxylase from Escherichia coli. Purification and properties. J Biol Chem 248:1687–1695. [PubMed] [Google Scholar]
- 40.Kai M, Miyazaki T, Sakamoto Y, Ohkura Y. 1985. Use of benzoin as pre-column fluorescence derivatization reagent for the high-performance liquid chromatography of angiotensins. J Chromatogr 322:473–477. doi: 10.1016/S0021-9673(01)97713-1. [DOI] [PubMed] [Google Scholar]
- 41.Kumar S, Punekar NS. 1998. Inhibition of succinic semialdehyde dehydrogenase by N-formylglycine. J Enzyme Inhib 13:369–376. doi: 10.3109/14756369809021482. [DOI] [PubMed] [Google Scholar]
- 42.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1:2856–2860. [DOI] [PubMed] [Google Scholar]
- 43.Ahuja M, Punekar NS. 2008. Phosphinothricin resistance in Aspergillus niger and its utility as a selectable transformation marker. Fungal Genet Biol 45(7):1103–1110. doi: 10.1016/j.fgb.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 44.Walvekar AS, Choudhury R, Punekar NS. 2014. Mixed disulfide formation at Cys141 leads to apparent unidirectional attenuation of Aspergillus niger NADP-glutamate dehydrogenase activity. PLoS One 9:e101662. doi: 10.1371/journal.pone.0101662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 46.Smith PT, King AD Jr, Goodman N. 1993. Isolation and characterization of urease from Aspergillus niger. J Gen Microbiol 139:957–962. doi: 10.1099/00221287-139-5-957. [DOI] [PubMed] [Google Scholar]
- 47.Ameyama M, Hayashi M, Matsushita K, Shinagawa E, Adachi O. 1984. Microbial production of pyrroloquinoline quinone. Agric Biol Chem 48:561–565. doi: 10.1271/bbb1961.48.561. [DOI] [Google Scholar]
- 48.Duine JA. 1991. Quinoproteins: enzymes containing the quinonoid cofactor pyrroloquinoline quinone, topaquinone or tryptophan-tryptophan quinone. Eur J Biochem 200:271–284. doi: 10.1111/j.1432-1033.1991.tb16183.x. [DOI] [PubMed] [Google Scholar]
- 49.Frebort I, Toyama H, Matsushita K, Kumagai H, Pec P, Luhova L, Adachi O. 1994. Two distinct quinoprotein amine oxidases are functioning in Aspergillus niger, p 241–245. In Marino G, Sannia G, Bossa F (ed), Biochemistry of vitamin B6 and PQQ. Birkhäuser Verlag, Basel, Switzerland. [Google Scholar]
- 50.Yamada H, Adachi O, Odata K. 1965. Amine oxidases of microorganisms. Agric Biol Chem 29:117–123. doi: 10.1271/bbb1961.29.117. [DOI] [Google Scholar]
- 51.Kumar S, Kumar S, Punekar NS. 2015. Characterization of succinic semialdehyde dehydrogenase from Aspergillus niger. Indian J Exp Biol 53:67–74. [PubMed] [Google Scholar]
- 52.Jann A, Matsumoto H, Haas D. 1988. The fourth arginine catabolic pathway of Pseudomonas aeruginosa. J Gen Microbiol 134:1043–1053. [DOI] [PubMed] [Google Scholar]
- 53.Yorifuji T, Koike K, Sakurai T, Yokoyama K. 1986. 4-Aminobutyraldehyde and 4-guanidinobutyraldehyde dehydrogenases for arginine degradation in Pseudomonas putida. Agric Biol Chem 50:2009–2016. doi: 10.1271/bbb1961.50.2009. [DOI] [Google Scholar]
- 54.Matsuda H, Suzuki Y. 1984. γ-Guanidinobutyraldehyde dehydrogenase of Vicia faba leaves. Plant Physiol 76:654–657. doi: 10.1104/pp.76.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.