

Abstract
In terrestrial mammals, the oxygen storage capacity of the CNS is limited, and neuronal function is rapidly impaired if oxygen supply is interrupted even for a short period of time. However, oxygen tension monitored by the peripheral (arterial) chemoreceptors is not sensitive to regional CNS differences in partial pressure of oxygen (PO2) that reflect variable levels of neuronal activity or local tissue hypoxia, pointing to the necessity of a functional brain oxygen sensor. This experimental animal (rats and mice) study shows that astrocytes, the most numerous brain glial cells, are sensitive to physiological changes in PO2. Astrocytes respond to decreases in PO2 a few millimeters of mercury below normal brain oxygenation with elevations in intracellular calcium ([Ca2+]i). The hypoxia sensor of astrocytes resides in the mitochondria in which oxygen is consumed. Physiological decrease in PO2 inhibits astroglial mitochondrial respiration, leading to mitochondrial depolarization, production of free radicals, lipid peroxidation, activation of phospholipase C, IP3 receptors, and release of Ca2+ from the intracellular stores. Hypoxia-induced [Ca2+]i increases in astrocytes trigger fusion of vesicular compartments containing ATP. Blockade of astrocytic signaling by overexpression of ATP-degrading enzymes or targeted astrocyte-specific expression of tetanus toxin light chain (to interfere with vesicular release mechanisms) within the brainstem respiratory rhythm-generating circuits reveals the fundamental physiological role of astroglial oxygen sensitivity; in low-oxygen conditions (environmental hypoxia), this mechanism increases breathing activity even in the absence of peripheral chemoreceptor oxygen sensing. These results demonstrate that astrocytes are functionally specialized CNS oxygen sensors tuned for rapid detection of physiological changes in brain oxygenation.
SIGNIFICANCE STATEMENT Most, if not all, animal cells possess mechanisms that allow them to detect decreases in oxygen availability leading to slow-timescale, adaptive changes in gene expression and cell physiology. To date, only two types of mammalian cells have been demonstrated to be specialized for rapid functional oxygen sensing: glomus cells of the carotid body (peripheral respiratory chemoreceptors) that stimulate breathing when oxygenation of the arterial blood decreases; and pulmonary arterial smooth muscle cells responsible for hypoxic pulmonary vasoconstriction to limit perfusion of poorly ventilated regions of the lungs. Results of the present study suggest that there is another specialized oxygen-sensitive cell type in the body, the astrocyte, that is tuned for rapid detection of physiological changes in brain oxygenation.
Keywords: astrocyte, glia, hypoxia, oxygen, respiration
Introduction
Oxygen is essential for all complex forms of life (Lyons et al., 2014), and the brain is highly vulnerable to oxygen deprivation. Specialized peripheral oxygen-sensing elements have evolved to monitor and ensure adequate oxygenation of the arterial blood supplying the brain. Strategically located in the carotid bifurcation and the aortic arch, peripheral respiratory chemoreceptors detect decreases in the arterial partial pressure of oxygen (PO2) and trigger adaptive changes in breathing (Kumar and Prabhakar, 2012; Prabhakar, 2013). However, peripheral chemoreceptors are not sensitive to regional CNS differences in PO2 that may develop as a result of variable levels of neuronal activity or significant changes in perfusion, and mammals can survive complete loss of peripheral oxygen sensitivity.
It is generally believed that the CNS is devoid of a physiological oxygen sensor (i.e., a cell capable of sensing parenchymal PO2 changes within the physiological range) and that no functional brain oxygen sensor is capable of stimulating breathing. This view persists despite a number of previous studies reporting significant ventilatory responses to hypoxia in unanesthetized experimental animals with denervated (or silenced) peripheral oxygen chemoreceptors (Davenport et al., 1947; Miller and Tenney, 1975; Bisgard et al., 1980; Olson et al., 1988; Curran et al., 2000). The existence of a functional CNS oxygen sensor is also supported by the evidence showing that brainstem respiratory and cardiovascular control circuits are highly sensitive to oxygen deprivation (Sun and Reis, 1994; Ramirez et al., 1998; Solomon et al., 2000; Thoby-Brisson and Ramirez, 2000; Neubauer and Sunderram, 2004; Peña et al., 2004; Solomon, 2005; D'Agostino et al., 2009; Ruangkittisakul and Ballanyi, 2012; Marina et al., 2015). However, the cellular identity of such a sensor and molecular mechanisms underlying CNS oxygen sensitivity remain essentially unknown.
Ubiquitous CNS glial cells called astrocytes are able to detect various sensory modalities (Gourine et al., 2005a, 2010; Marty et al., 2005; Kasymov et al., 2013). Astrocytes enwrap all penetrating and intracerebral arterioles and capillaries, control the ionic and metabolic environment of the neuropil, and support synaptic activity by supplying neurons with a renewable source of transmitters (Haydon and Carmignoto, 2006; Magistretti, 2006). Astrocytes are also believed to provide a neurovascular coupling interface by controlling cerebrovascular tone and mediating cerebrovascular responses to heightened neuronal activity (Attwell et al., 2010).
There is evidence that tissue PO2 is an important factor that determines the polarity of cerebrovascular responses that develop in response to astroglial activation (Gordon et al., 2008), and, in this study, we tested the hypothesis that astrocytes are specialized to directly sense changes in brain oxygenation. Our experiments suggest that astrocytes are indeed sensitive to physiologically relevant decreases in brain parenchymal PO2 and identify a putative mechanism underlying this O2 sensitivity. Experimental blockade of astroglial signaling pathways demonstrated the significance of this oxygen sensitivity in one, clearly defined physiological paradigm; detection of brain hypoxia by astrocytes controls a vital homeostatic reflex, the hypoxic ventilatory response, by maintaining elevated respiratory activity at low levels of ambient O2.
Materials and Methods
The experiments were performed on Sprague Dawley rats and PINK1 (PTEN-induced putative kinase 1)-deficient mice (Yao et al., 2011; generated by Lexicon Genetics) in accordance with the European Commission Directive 2010/63/EU (European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes) and the UK Home Office (Scientific Procedures) Act (1986) with project approval from the respective Institutional Animal Care and Use Committees.
Experimental models for imaging
In vivo preparations.
Young male rats (100–120 g) were anesthetized with urethane (initial dose, 1.3 g/kg, i.p.; then 10–25 mg · kg−1 · h−1, i.v.). Adequate anesthesia was ensured by maintaining stable levels of arterial blood pressure and heart rate showing lack of responses to a paw pinch. The femoral artery and vein were cannulated for measurement of the arterial blood pressure and administration of anesthetic, respectively. The trachea was cannulated, and the animal was ventilated with room air using a positive pressure ventilator with a tidal volume of ∼1 ml/100 g and a ventilator frequency similar to the resting respiratory rate (∼60 strokes/min). The animal was then placed in a stereotaxic frame. The skin overlying the skull was removed, and a small craniotomy (∼2 mm2) was made in the parietal bone above the somatosensory cortex. Cortical astrocytes were labeled with sulforhodamine 101 (SR101) and the Ca2+ indicator Oregon Green BAPTA 1 AM (OGB-1). OGB-1 was dissolved initially in DMSO and 20% Pluronic F127. The solution containing OGB-1 (1 mm) and SR101 (8 μm) in artificial CSF (aCSF; 124 mm NaCl, 3 mm KCl, 2 mm CaCl2, 26 mm NaHCO3, 1.25 mm NaH2PO4, 1 mm MgSO4, 10 mm d-glucose saturated with 95% O2/5% CO2, pH 7.4) was delivered via glass micropipettes at four to six separate sites within the targeted area of the cortex. The exposed surface of the cortex was then protected with a glass coverslip secured to the skull using acrylic dental cement. In some of the experiments, test compounds [e.g., FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone)] were coinjected with the dye and applied to the exposed cortical surface to ensure adequate penetration into the tissue. During imaging, the animal was paralyzed with gallamine triethiodide (50 mg/kg, i.v.; then 10 mg · kg−1 · h−1, i.v.). PO2, PCO2, and pH of the arterial blood were measured regularly and kept within the physiological ranges by adjusting tidal volume and/or ventilator frequency. The body temperature was maintained at 37.0 ± 1°C.
Acute brain slices.
Animals were killed by halothane inhalation overdose, and the brain was removed quickly and placed in chilled (∼4°C) aCSF saturated with 95% O2/5% CO2, pH 7.4, with an additional 9 mm Mg2+. The brainstem was isolated, and a sequence of transverse (250 μm) slices was cut. Once cut, the slices were stored before use in aCSF saturated with 95% O2 and 5% CO2, pH 7.4, at room temperature. Optical recordings were made in a flow chamber at 35–37°C from the slices placed on an elevated grid to permit access of aCSF to both sides of the slice.
Organotypic brain slices.
Rat pups (P8–P10 of either sex) were killed by halothane overdose and transferred into a laminar flow hood. All procedures were performed aseptically. The brainstem was removed quickly and bathed in ice-cold HBSS without Ca2+ with added 20 mm glucose (total 25.6 mm), 10 mm MgCl2, 1 mm HEPES, 1 mm kynurenic acid, 0.005% phenol red, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. A sequence of transverse (250 μm) slices was cut and plated on Millicell-CM organotypic culture membrane inserts. Slices were cultured in medium containing 50% Optimem-1, 25% fetal bovine serum (FBS), 21.5% HBSS, 25 mm glucose, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. After 3 d, the plating medium was removed, and DMEM containing 10% FBS, 2 mm l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin was added and subsequently replaced twice a week. Astrocytes were targeted to express a single color genetically encoded Ca2+ indicator Case12 using an adenoviral vector (AVV) under the control of an enhanced glial fibrillary acidic protein (GFAP) promoter, as described previously (Guo et al., 2010). AVV–sGFAP–Case12 (sGFAP stands for “superGFAP” to delineate an enhanced version of human GFAP promoter, as described by Liu et al., 2008) was added to the medium at the time of slice culture preparation at 5 × 108 to 5 × 1010 transducing units/ml. Experiments were performed after 7–10 d of incubation.
Primary dissociated neuroglial cultures.
Cultures were prepared from the cortical, hippocampal, midbrain, and brainstem tissue of rat pups (P2–P3 of either sex) as described previously (Marriott et al., 1995; Domijan et al., 2014). After isolation, the cells were plated on poly-d-lysine-coated coverslips and maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air for a minimum of 12 d before the experiments. Viral vectors were added to the incubation medium at the time of culture preparation (AVV–sGFAP–Case12) or after 1 week of culturing (AVV–sGFAP–VNUT–GFP, AVV–sGFAP–EGFP–TeLC).
Carotid body cell cultures.
Rat pups (P5–P7 of either sex) were killed by isoflurane inhalation overdose before bilateral excision of the carotid bodies. Glomus cells were isolated enzymatically by exposing the carotid body tissue to a 0.1% trypsin–0.1% collagenase solution for 1 h at 37°C, followed by mechanical dissociation. The dispersed cell suspension was then collected and triturated in growth medium [F-12 nutrient medium supplemented with 10% (v/v) FBS, 80 U/L insulin, 6% (w/v) glucose, 2 mm glutamine, and 1% penicillin–streptomycin]. Dispersed cells were then placed onto poly-l-lysine-coated coverslips. Cultures were grown at 37°C in a humidified atmosphere of 95% air/5% CO2 for 72 h. Each culture contained multiple cell clusters containing ∼5–20 glomus cells that were identified using bright-field microscopy.
Imaging
Two-photon imaging in vivo.
Ca2+ responses in the cortex were visualized via a cranial window using an Olympus FV1000 microscope (Olympus), equipped with MaiTai HP DeepSee laser (Spectra-Physics). A 25× water-immersion objective [XLPlan N, 1.05 numerical aperture (NA); Olympus] was used. Fluorophores were excited in two-photon xyz-t mode at 800 nm. Red fluorescence (SR101) was separated from green fluorescence (OGB-1) via a dichroic mirror. Images were acquired up to 200 μm deep from the cortical surface. Z-stacks (∼15 μm) were acquired and maximum projected to enable tracking of cells that moved in the z-plane during the experimental challenge. For time-lapse imaging, stacks of 15 μm (five focal planes) were acquired at a rate of 0.75 Hz at 320 × 320 pixel resolution. The laser power was kept to a minimum to reduce photo toxicity (12 mW average power). Intensities of regions of interest corresponding to cellular somas were measured, and background fluorescence level was subtracted. Intensities of fluorescence were normalized to the average of the prestimulus baseline values. Time-lapse series were acquired for 20 min with a period of hypoxia (15 or 10% O2 in the inspired air) lasting 1 min. On average, responses of ∼10 labeled cells per animal were captured and analyzed. Changes in fluorescence are shown as the average across all the animals in each experimental group.
In vitro imaging.
Optical measurements of changes in intracellular calcium ([Ca2+]i), mitochondrial membrane potential (Δψm), reactive oxygen species (ROS) production, and the rate of lipid peroxidation were performed using a Zeiss 710 CLSM confocal microscope (Zeiss), an Olympus FV1000 confocal microscope, or an inverted epifluorescence Olympus microscope, equipped with a cooled CCD camera (Retiga; QImaging) as described previously (Abramov et al., 2007; Gourine et al., 2010; Vaarmann et al., 2010; Domijan et al., 2014). [Ca2+]i responses were visualized using conventional Ca2+ indicators (e.g., fura-2) or the genetically encoded Ca2+ indicator Case12 expressed in astrocytes using an AVV with enhanced GFAP promoter. Specificity of this vector as well as Ca2+ sensitivity and the dynamic range of Case12 were described previously (Gourine et al., 2010; Guo et al., 2010). Confocal imaging of hypoxia-induced [Ca2+]i responses in astrocytes transduced with AVV–sGFAP–Case12 was performed in acute brain slices of adult rats and organotypic brain slice cultures. For imaging, a section of the membrane with an organotypic slice or an acute slice was placed on an elevated grid in a flow chamber (2 ml volume). Recordings were performed at 35–37°C in aCSF. The rate of perfusion was 4 ml/min. Images were obtained using an upright confocal microscope with 40× water-immersion objective. The 488 nm argon laser line was used to excite Case12 fluorescence, which was measured using a bandpass filter of 505–550 nm. Illumination intensity was kept to a minimum (at 0.5–0.7% of laser output).
All the experiments in dissociated cell cultures were performed in a custom-made flow-through imaging chamber in a standard HBSS. For simultaneous measurements of changes in [Ca2+]i and Δψm, cell cultures were loaded with fura-2 (5 μm; 30 min incubation; Invitrogen) and rhodamine 123 (Rh123; 1 μg/ml; 15 min incubation; Invitrogen). After incubation with the dyes, cultures were washed five times before the experiment. Under these loading conditions, Rh123 is not toxic and reports changes in Δψm in living cells. FCCP (1 μm) was applied at the end of the experiment to produce complete mitochondrial depolarization required to calibrate the Rh123 signal. Changes in [Ca2+]i and Δψm were monitored in individual cells using excitation light provided by a xenon arc lamp with the beam passing sequentially through a monochromator at 340, 380, and 490 nm (Cairn Research). For both indicators, emitted fluorescence at 515 nm was registered.
Changes in mitochondrial ROS production in cultured cells were assessed using the mitochondrial superoxide indicator MitoSOX (5 μm; 10 min incubation; Invitrogen). MitoSOX fluorescence was excited at 563 nm, and emitted fluorescence was registered above 580 nm. The rate of lipid peroxidation in cultured cells was assessed using the ratiometric fluorescent sensor BODIPY 581/591 C11 (10 μm; 20 min incubation; Invitrogen). BODIPY 581/591 C11 fluorescence was excited at 488 and 563 nm, and emitted fluorescence at 505–550 nm and 570–630 nm was collected. Hypoxic conditions in vitro were induced by displacement of oxygen in the medium with nitrogen or argon. All test drugs were applied 15–30 min before the hypoxic challenge. Imaging data were collected and analyzed using build-in Zeiss, Olympus, or Andor software (Andor). All data presented were obtained from at least six separate experiments.
Total internal reflection fluorescence microscopy.
To identify ATP-containing vesicular compartments, we generated an AVV in which the vesicular nucleotide transporter (VNUT or SLC17A9; Sawada et al., 2008) is fused with green fluorescent protein (GFP) and then expressed it in astrocytes using an sGFAP promoter. Astrocytes plated on poly-d-lysine-coated coverslips were transduced with AVV–sGFAP–VNUT–GFP added to the medium at 5 × 108 to 5 × 1010 transducing units/ml. Putative ATP-containing compartments were also visualized using quinacrine staining (5 μm, 15 min incubation at 37°C). The acridine derivative quinacrine is a weak base that binds ATP with high affinity and can be used to visualize putative ATP-containing vesicles in living cells, including astrocytes (Coco et al., 2003). An Olympus total internal reflection fluorescence (TIRF) microscope was used to detect vesicular fusion events in astrocytes expressing VNUT–GFP or stained with quinacrine, as described in detail previously (Kasymov et al., 2013). Fluorescence was excited at 488 nm and collected at 500–530 nm. The imaging setup included a high-NA oil-immersion objective (60×, 1.65 NA), an inverted microscope (IX71; Olympus) and a cool-charge-coupled-device camera (Hamamatsu). Images were acquired and analyzed using Olympus Cellt̂ool software (Olympus). All the experiments were conducted at 37°C.
Measurements of PO2
In the in vivo experiments, PO2 in the brain (cortical) parenchyma was monitored using optical probes (250 μm tip diameter; OxyLite system; Oxford Optronix) placed 0.5–1 mm below the cortical surface. In the in vitro experiments, PO2 was monitored by the probes placed on the surface of the slice (in acute and organotypic slices) or in close proximity to the cellular layer (in culture). The operation of the sensor is based on optical fluorescence technology that allows real-time detection of changes in PO2 within the physiological range and precise PO2 measurements in conditions of hypoxia.
Viral gene transfer in vivo
Young adult male rats (200–270 g) were anesthetized with a mixture of ketamine (60 mg/kg, i.m.) and medetomidine (250 μg/kg, i.m.) and placed in a stereotaxic frame. The tooth bar was adjusted so that bregma was positioned 5 mm below lambda. Brainstem respiratory networks, including the rhythm-generating circuits of the pre-Bötzinger complex (preBötC), were targeted bilaterally with microinjections of the viral vectors (LVV–EF1α–TMPAP–EGFP, LVV–EF1α–EGFP, AVV–sGFAP–EGFP–TeLC, or AVV–sGFAP–CatCh–EGFP) using the following coordinates: mediolateral, ±2.0 mm and dorsoventral, −2.6 mm from calamus scriptorius. After microinjections, the wound was sutured and anesthesia was reversed with atipemazole (1 mg/kg). Immediate postoperative care was given, and animals were left to recover for 5–10 d before the experiments to ensure stable and high level of transgene expression.
Ablation of the carotid body chemoreceptors
Rats were anesthetized with a mixture of ketamine (60 mg/kg, i.m.) and medetomidine (250 μg/kg, i.m.). Using an aseptic technique, an anterior midline neck incision was performed, and the sternohyoid and sternocleidomastoid muscles were retracted. The carotid bifurcation was exposed, the occipital artery was retracted, the carotid sinus nerve and its branches were cut, and the carotid bodies were removed. Sham-operated rats underwent the same surgical procedures to expose the carotid bifurcation, but the carotid sinus nerves and the carotid bodies were left intact. After the surgery, the wound was sutured and anesthesia was reversed with atipemazole (1 mg/kg). No mortalities occurred, and animals gained weight normally. Carotid body ablations were performed 10 weeks before the main experiments.
Lentiviral vector for the expression of transmembrane prostatic acid phosphatase
To interfere with ATP-mediated signaling by promoting breakdown of the vesicular and/or released ATP, the brainstem respiratory rhythm-generating circuits were targeted with a lentiviral vector (LVV) to express transmembrane prostatic acid phosphatase (TMPAP; Zylka et al., 2008) under the control of an elongation factor 1α (EF1α) promoter. Generation of the vector (LVV–EF1α–TMPAP–EGFP) and validation of the transgene efficacy in preventing vesicular ATP accumulation in astrocytes and blocking ATP-mediated signaling between cultured astrocytes and in vivo have been described in detail previously (Marina et al., 2013; Wells et al., 2015). Control animals were transduced to express enhanced GFP (EGFP; vector LVV–EF1α–EGFP). Because the strategy was to maximize TMPAP expression and therefore ATP degradation, vectors were designed not to be cell selective. However, despite the use of a non-cell-specific promoter (EF1α), TMPAP and the control transgene were shown to be mainly expressed in astrocytes (Wells et al., 2015).
Assessment of the respiratory activity
Respiratory responses to hypoxia were determined in conscious and anesthetized rats and conscious mice (male PINK-1 knock-out and their wild-type counterparts) using whole-body plethysmography (Enhorning et al., 1998; Mortola and Frappell, 1998; Trapp et al., 2011). In brief, the animal was placed in a recording chamber (∼1 L for rats and ∼200 ml for mice) that was flushed continuously with humidified mixture of 79% N2 and 21% O2 (at 22–24°C). The animal was allowed at least 1 h to acclimatize to the chamber environment at normoxia/normocapnia (21% O2, 79% N2, and <0.3% CO2) before measurements of baseline ventilation were taken. Hypoxia was induced by lowering the O2 concentration in the chamber to 10% (with the balance being N2) for 10 min. The measurements of ventilatory responses to hypoxia were obtained during the last 5 min of the 10 min hypoxia period when breathing has stabilized. Before and after each experiment, the plethysmograph pressure signal was calibrated by repeated injections and withdrawal of air (0.05, 0.1, and 0.2 ml) from within the recording chamber. Interbreath intervals, absolute frequency, the relative amplitude of volume excursions associated with each breath, and relative changes in minute ventilation were determined.
Assessment of sleep efficiency
Sleep state was scored by an investigator who was unaware of the nature of the experimental groups. Established behavioral criteria (Gramsbergen et al., 1970) were used to calculate sleep efficiency (SE) score before, during, and after the hypoxic challenge with an epoch length of 5 s. Briefly, rats were considered to be awake (W: grooming, eyes open, gross body movement, such as crawling and changing position), in quiet sleep (QS: eyes closed, absence of movement except for periodic sighs and intermittent brief startles or a gross body movement lasting <2 s during a relatively long period of QS), or in active sleep [AS: eyes closed, frequent twitches of the whiskers, ears, and extremities or very brief (<2 s) and small movements of the head or limbs]. The state was recorded as indeterminate (IN) when it was difficult to establish the state. After rats had habituated to the plethysmography environment and were asleep for >30 min, room air supplying the chamber was exchanged for the 10% O2/90% N2 gas mixture for 10 min. SE score was then assessed for 10 min after the termination of the hypoxic stimulus. The SE score was calculated according to the following formula: SE = 100 × (QS + AS)/(QS + AS + W + IN) (Cohen et al., 2002).
Immunohistochemistry
At the end of the experiments, the rats were given an anesthetic overdose (pentobarbitone sodium, 200 mg/kg, i.p.) and perfused transcardially with 4% paraformaldehyde. The brains were removed and stored in the same fixative overnight at 4°C. After cryoprotection in 30% sucrose, the brainstem was isolated and a sequence of transverse slices (40 μm) was cut and processed for immunohistochemical detection of GFP, GFAP, neurokinin-1 receptor (NK-1R), and/or choline acetyltransferase (ChAT) expression. Free-floating brain slices were incubated in chicken anti-GFP (1:250; Aves Labs), Cy3-conjugated mouse anti-GFAP (1:1000; Sigma), rabbit anti-NK-1R (1:1000; Millipore), and/or goat anti-ChAT antibody (1:100; Millipore). For the detection of GFP, NK-1R, or ChAT immunoreactivities, the sections were subsequently incubated in specific secondary antibodies conjugated to the fluorescent probes [Alexa Fluor-488 donkey anti-chicken (Invitrogen), Cy3-conjugated donkey anti-rabbit (Jackson ImmunoResearch), or Alexa Fluor-564 donkey anti-goat (Invitrogen), respectively] for 1 h.
Data analysis
Built-in analysis software tools (Zeiss, Olympus, or Andor) were used to analyze the results of the imaging experiments. Records from the in vivo experiments were analyzed offline using Cambridge Electronic Design Spike 2 software. Astroglial responses ([Ca2+]i, Δψm, ROS production, lipid peroxidation, and vesicular fusion) in vivo and in vitro and the hypoxia-evoked changes in respiratory rate, tidal volume, and minute ventilation in vivo in the absence and presence of test drugs/treatments or expression of a particular transgene were compared by one-way or two-way ANOVA, followed by the post hoc Tukey–Kramer test or Student's paired or unpaired t test, as appropriate (NCSS 2007; NCSS). Data are reported as mean ± SEM. Differences with p < 0.05 were considered to be significant.
Results
Astrocytes are sensitive to physiological decreases in PO2
Using two-photon imaging in vivo (anesthetized, paralyzed, and artificially ventilated rats; n = 15), we detected robust [Ca2+]i responses in cortical astrocytes (SR101-labeled cells) when the concentration of oxygen in the inspired air was lowered for 60 s from 21% to 15% or 10% (Fig. 1A). Hypoxia-induced [Ca2+]i responses were only observed in SR101-labeled cells (p < 0.001, F(2,514) = 88.3); neighboring cortical cells that lacked SR101 labeling (Fig. 1A) did not respond to hypoxia. Measurements of tissue oxygenation in the cerebral cortex in these experimental conditions (anesthetized, paralyzed, and artificially ventilated rats, no respiratory compensation; n = 6) revealed that acute exposure to 15 and 10% O2 in the inspired air was associated with decreases in cortical parenchymal PO2 from 24 ± 3 to 11 ± 2 and 6 ± 1 mmHg, respectively (p < 0.001, F(2,15) = 18.4).
Figure 1.

Astrocytes are sensitive to physiological decreases in PO2. A, In vivo imaging of hypoxia-evoked astrocytic [Ca2+]i responses in somatosensory cortex of an anesthetized adult rat. Top, Pseudocolored images showing changes in OGB-1 fluorescence taken at the times indicated by arrows on the bottom panel. 5–7, Nonresponding cells that were not labeled with SR101. Bottom, Traces showing changes in astrocytic [Ca2+]i in response to hypoxia. Inset, Averaged changes in OGB-1 fluorescence induced by hypoxia in 10 SR101-labeled cells (SR101+) and five neighboring cortical cells that lacked SR101 labeling (SR101−) recorded in this experiment. B, Hypoxia-induced [Ca2+]i responses of carotid body glomus cells in culture, visualized using the Ca2+ indicator fura-2 (PO2 threshold of activation, 40 mmHg). Inset, Pseudocolored images of a cluster of glomus cells showing changes in fura-2 fluorescence in response to hypoxia. C, Hypoxia-induced [Ca2+]i responses of brainstem astrocytes (PO2 threshold of activation, 15 mmHg). In this example, astrocytes were identified and their responses to hypoxia were assessed in organotypic brainstem slice using the genetically encoded Ca2+ sensor Case12 expressed under the control of GFAP promoter (Gourine et al., 2010). D, Simultaneous imaging of hypoxia-induced changes in Δψm and [Ca2+]i in cultured brainstem astrocytes using Rh123 and fura-2 showing that mitochondrial depolarization precedes Ca2+ responses. Mitochondrial depolarization is induced by FCCP (1 μm) applied at the end of the experiment to calibrate the Rh123 signal (100%).
Decreases in PO2 reliably triggered activation of astrocytes, measured as increases in [Ca2+]i, in various reduced preparations, including acute brainstem slices of adult rats (n = 9 slices), organotypic brain slice cultures (n = 21 slices; Fig. 1C), and dissociated cell cultures (n = 54 cultures, 0.61 ± 0.15 fura-2 ratio, t test, p < 0.001; Fig. 1D). In culture conditions, cortical, hippocampal, midbrain, and brainstem astrocytes displayed similar sensitivity to PO2 decreases, responding with sustained [Ca2+]i elevations, [Ca2+]i oscillations, or a combination of these responses (Fig. 1C,D). Hypoxia-induced [Ca2+]i responses in astrocytes were not affected by the P2Y1 purinergic receptor (main metabotropic ATP receptor expressed by astrocytes) antagonist MRS2179 (2′-deoxy-N6-methyladenosine-3′,5′-bisphosphate; 30 μm; data not shown). [Ca2+]i elevations in cultured cortical astrocytes were also evoked reliably by application of the oxygen scavenger sodium dithionite (n = 16 cultures, 0.70 ± 0.19 fura-2 ratio, t test, p = 0.02; data not shown).
Simultaneous monitoring of oxygen tension in the incubation medium and fluorescent imaging of intracellular [Ca2+] in a prototypical somatic oxygen sensor, the glomus cell of the carotid body, revealed that these peripheral chemoreceptors are activated when PO2 falls below 37 ± 3 mmHg (n = 15 preparations; Fig. 1B). Under similar experimental conditions, astrocytes in organotypic brainstem slice cultures responded with increases in [Ca2+]i when PO2 was lowered below 17 ± 3 mmHg (n = 6 preparations; Fig. 1C). These results demonstrate that the PO2 threshold of astroglial activation is lower (p < 0.001, F(1,19) = 19.0) than that of the carotid body glomus cells, residing ∼10 mmHg below the normal level of brain oxygenation. In all the subsequent in vitro experiments, hypoxic conditions were mimicked by lowering PO2 in the perfusate or incubation medium to between 5 and 10 mmHg.
Next we tested the hypothesis that the hypoxia sensor in astrocytes resides in the mitochondria, in which oxygen is consumed. Mitochondrial function depends on Δψm, and inhibition of respiration decreases Δψm. In cultured astrocytes, simultaneous measurements of Δψm and [Ca2+]i using Rh123 and fura-2, respectively, revealed that a decrease in PO2 causes a 55 ± 15% decrease in Δψm and that this mitochondrial depolarization precedes increases in [Ca2+]i (Fig. 1D).
Mechanisms underlying astrocytic Ca2+ responses to decreases in PO2
Inhibition of mitochondrial respiration should increase mitochondrial production of ROS (Murphy, 2012). We measured ROS using the fluorescent probe MitoSOX and detected immediate increases in mitochondrial ROS production by cultured astrocytes in response to hypoxia (Fig. 2A). Hypoxia-induced ROS production in astrocytes was markedly reduced by pretreatment of cell cultures with the mitochondrial uncoupler FCCP at a low concentration (0.5 μm; n = 15, t test, p < 0.001; Fig. 2A,I) or by the mitochondrial antioxidant MitoQ (100 nm; n = 12, t test, p < 0.001; Fig. 2A). Hypoxia-induced [Ca2+]i responses in cultured astrocytes were also blocked effectively by FCCP (0.5 μm; 0.11 ± 0.05, n = 12, t test, p < 0.001; Fig. 2B), MitoQ (100 nm; 0.05 ± 0.04, n = 9, t test, p < 0.001; Fig. 2B), or the ROS scavenger α-tocopherol (vitamin E, 100 μm; 0.08 ± 0.07, n = 7, t test, p < 0.001; Fig. 2B). In vivo, astrocytic [Ca2+]i responses induced by decreases in the level of inspired O2 (from 21 to 10%) were abolished after local application of FCCP (10 μm; t = 20.6; df = 103, p < 0.001; Fig. 2C). FCCP treatment had no effect on frequency of the spontaneous somatic Ca2+ transients in cortical astrocytes observed in vivo at resting conditions before the hypoxic challenge (3.8 ± 1.7 events/10 min, n = 7 controls vs 4.4 ± 1.4 events/10 min, n = 6 FCCP treated, t test, p = 0.8). [Ca2+]i responses evoked by P2 receptor activation (application of ATP, 10 μm) in cultured astrocytes were preserved and even enhanced (possibly because of the inability of mitochondria to buffer cytosolic Ca2+ rises) in the presence of FCCP (Fig. 2D). These in vivo and in vitro data imply that blockade of hypoxia-induced [Ca2+]i responses in astrocytes is not attributable to potential toxic/adverse cellular effects of FCCP.
Figure 2.
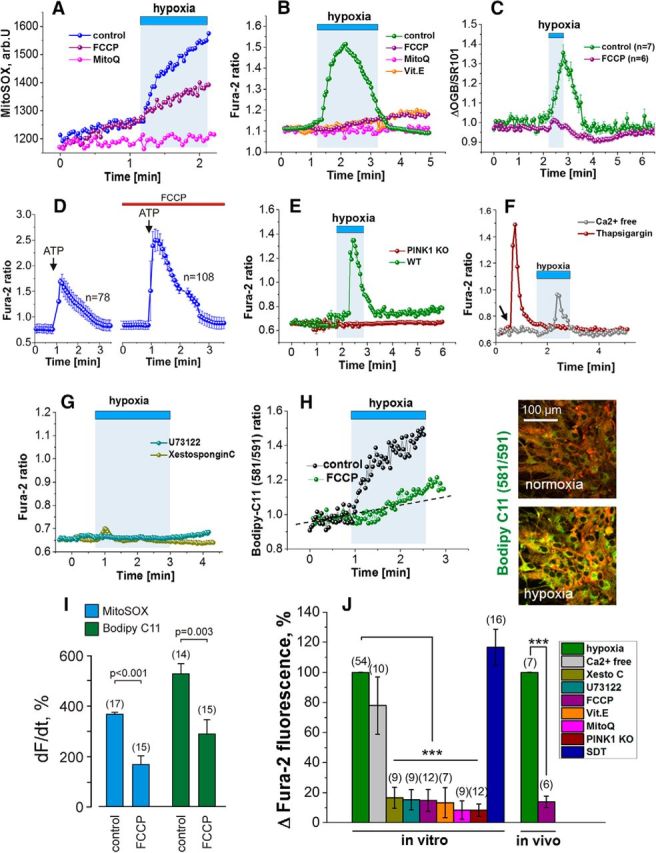
Mechanisms underlying astroglial Ca2+ responses to decreases in PO2. A, Representative traces of the hypoxia-induced changes in the rate of mitochondrial ROS production in individual astrocytes in culture. Responses are blocked by the mitochondrial ROS scavenger MitoQ or after mitochondrial depolarization induced by FCCP (0.5 μm). B, Hypoxia-induced [Ca2+]i responses in cultured astrocytes are blocked by FCCP (0.5 μm), MitoQ, or vitamin E (α-tocopherol). C, Group data obtained in anesthetized rats illustrating blockade of hypoxia-induced [Ca2+]i responses in cortical astrocytes after application of FCCP (10 μm). D, [Ca2+]i responses in cultured astrocytes induced by P2 receptor activation (ATP, 10 μm) in control conditions and after treatment with FCCP (0.5 μm). E, Hypoxia fails to evoke Ca2+ responses in cultured astrocytes of PINK1 knock-out (KO) mice. WT, Wild-type. F, Cultured astrocytes display Ca2+ responses to hypoxia after removal of external Ca2+, whereas depletion of the ER Ca2+ pools by thapsigargin abolishes the responses. Arrow indicates the time of thapsigargin application. G, Hypoxia fails to evoke Ca2+ responses in cultured astrocytes in the presence of PLC inhibitor (U73122) or IP3 antagonist [Xestospongin C (Xesto C)]. H, Hypoxia increases the rate of lipid peroxidation in cultured astrocytes. Right, Pseudocolored images showing changes in BODIPY 581/591 C11 fluorescence taken before and at the peak of the response induced by hypoxia. I, Summary data illustrating the effect of mitochondrial depolarization induced by FCCP (0.5 μm) on the rate of mitochondrial ROS production (reported by MitoSOX) and lipid peroxidation (reported by BODIPY 581/591 C11) in cultured astrocytes. J, Group data of the pharmacology of Ca2+ responses induced in astrocytes by decreases in PO2, suggesting that hypoxia leads to inhibition of mitochondrial respiration, ROS production, lipid peroxidation, activation of PLC, IP3 receptors, and recruitment of Ca2+ from the intracellular stores. SDT, Sodium dithionite. ***p < 0.001.
Hypoxia failed to elicit Ca2+ responses in astrocytes cultured from mice deficient in PINK1 (0.05 ± 0.02, n = 12, t test, p < 0.001; Fig. 2E), in which mitochondrial function is impaired and resting Δψm is lower compared with their wild-type counterparts (Gandhi et al., 2009), a condition that is mimicked by FCCP treatment of wild-type astrocytes. Lack of hypoxia-induced [Ca2+]i response in PINK1-deficient astrocytes supports the key role of mitochondria in the mechanisms underlying astroglial oxygen sensitivity. Combined, these results suggest that astrocytic [Ca2+]i responses triggered by decreases in PO2 are dependent on Δψm and mitochondrial ROS production.
In Ca2+-free media (with the addition of 0.5 mm EGTA), hypoxia still evoked [Ca2+]i responses in cultured astrocytes (0.47 ± 0.12, n = 10; Fig. 2F). In contrast, astrocytic Ca2+ responses to decreases in PO2 were reduced dramatically after pretreatment with the sarco/endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin (0.5 μm; 0.25 ± 0.05, n = 7, t test, p < 0.001; Fig. 2F), inhibition of phospholipase C (PLC), activity with U73122 (1-[6[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione; 5 μm; 0.1 ± 0.1, n = 9, t test, p < 0.001; Fig. 2G), or blockade of IP3 receptors with Xestospongin C (0.5 μm; 0.11 ± 0.06, n = 9, t test, p < 0.001; Fig. 2G) or 2-aminoethoxydiphenylborane (50 μm; 0.12 ± 0.08, n = 7, t test, p = 0.002; data not shown). We have demonstrated previously that PLC can be activated by lipid peroxidation (Vaarmann et al., 2010; Domijan et al., 2014). In cultured astrocytes, hypoxia increased the rate of lipid peroxidation as measured using the fluorescent dye BODIPY 581/591 C11 (526 ± 41%, n = 14, t test, p < 0.001; Fig. 2H), and these responses were reduced significantly by FCCP pretreatment (0.5 μm; 288 ± 60%, n = 15, t test, p = 0.003; Fig. 2H,I). Together (Fig. 2J), these data suggest that, in astrocytes, hypoxia leads to inhibition of mitochondrial respiration, facilitated mitochondrial ROS production, increased rate of lipid peroxidation, activation of PLC, IP3 receptors, and recruitment of Ca2+ from the intracellular stores.
Astrocytes respond to low PO2 with increased rate of exocytosis of ATP-containing vesicles
ATP-mediated purinergic signaling is responsible for communication between electrically non-excitable cells and is fundamental for astroglial signaling (Araque et al., 2014). We next determined whether [Ca2+]i responses induced in astrocytes by decreases in PO2 trigger release of ATP. We monitored the motility of VNUT-containing vesicles in cultured cortical astrocytes using TIRF microscopy and found that astrocytes respond to low PO2 with an increased rate of exocytosis of these vesicular compartments (p < 0.001, F(3,65) = 3.7; Fig. 3A,C,F). Increased fusion was also observed in astrocytes in which putative ATP-containing vesicles were identified using quinacrine staining (Fig. 3B,D,F). The majority of labeled puncta (78 ± 10%) underwent membrane fusion when PO2 was lowered by either displacing O2 in the incubation media with argon (Fig. 3A–D) or bath applying the O2 scavenger sodium dithionite (Fig. 3E). The peak frequency of hypoxia-induced fusion rate was reduced (n = 8 cultures; Tukey–Kramer post hoc test, p < 0.05) by the Ca2+ chelator BAPTA-AM (Fig. 3G), suggesting that facilitation of vesicular release in astrocytes at low PO2 requires intracellular Ca2+ signaling. The frequency of fusion events was not affected (n = 10 cultures; Tukey–Kramer post hoc test, p > 0.05) in the presence of the ATP degrading enzyme apyrase (25 U/ml) and the ATP receptor antagonist MRS2179 (30 μm; Fig. 3G), indicating that the potential autocrine/paracrine actions of ATP do not contribute significantly to either the hypoxia-evoked Ca2+ signals in astrocytes or the Ca2+-dependent fusion of astroglial ATP-containing vesicular compartments.
Figure 3.

Hypoxia facilitates exocytosis of ATP-containing vesicular compartments in astrocytes. A, TIRF images of VNUT–GFP-labeled vesicular compartments in cultured astrocytes at resting conditions and during hypoxic challenge. B, TIRF images of quinacrine-labeled vesicular compartments in cultured astrocytes at resting conditions and during hypoxic challenge. C, Plot of the TIRF intensity changes showing hypoxia-induced loss of VNUT–GFP fluorescence from a proportion of labeled organelles in an individual astrocyte. D, Plot of the TIRF intensity changes showing loss of quinacrine fluorescence from a proportion of labeled organelles in an individual astrocyte exposed to hypoxia. E, Plot of the TIRF intensity changes showing loss of quinacrine fluorescence from a proportion of labeled organelles in an individual astrocyte in response to application of the O2 scavenger sodium dithionite. F, Averaged temporal distribution of fusion event frequency detected before and during hypoxia in 10 VNUT–GFP-labeled and 10 quinacrine-loaded brainstem astrocytes. G, Summary data illustrating the peak frequency of hypoxia-induced fusion of ATP-containing compartments recorded in brainstem astrocytes in the absence and presence of MRS2179/apyrase or after 1 h incubation with BAPTA-AM. *p < 0.001 compared with the rate of fusion in normoxia; #p < 0.05 compared with the rate of fusion during hypoxia.
Central respiratory oxygen sensitivity is mediated by ATP actions in the brainstem
The functional significance of astroglial oxygen sensitivity leading to the release of ATP was next studied in vivo, using a clear experimental paradigm in which the output of the physiological system can be assessed easily. At the level of the brainstem, ATP stimulates breathing (Gourine et al., 2005a, 2010; Lorier et al., 2007; Huxtable et al., 2009), so we next determined whether hypoxia-induced release of ATP contributes to the central component of the hypoxic ventilatory response. In agreement with previous reports (Davenport et al., 1947; Miller and Tenney, 1975; Bisgard et al., 1980; Olson et al., 1988), conscious rats with denervated peripheral oxygen chemoreceptors (10 weeks after carotid body ablation) displayed robust respiratory responses to hypoxia (p < 0.001, F(2,48) = 56.4; Fig. 4A). Under urethane anesthesia (1.5 mg/kg), rats with denervated peripheral chemoreceptors showed a markedly lower respiratory response to hypoxia compared with that recorded in conscious rats (p < 0.001, F(2,48) = 19.5; Fig. 4A), suggesting that a key component of the signaling mechanism that underlies central respiratory oxygen sensitivity is impaired by general anesthesia.
Figure 4.
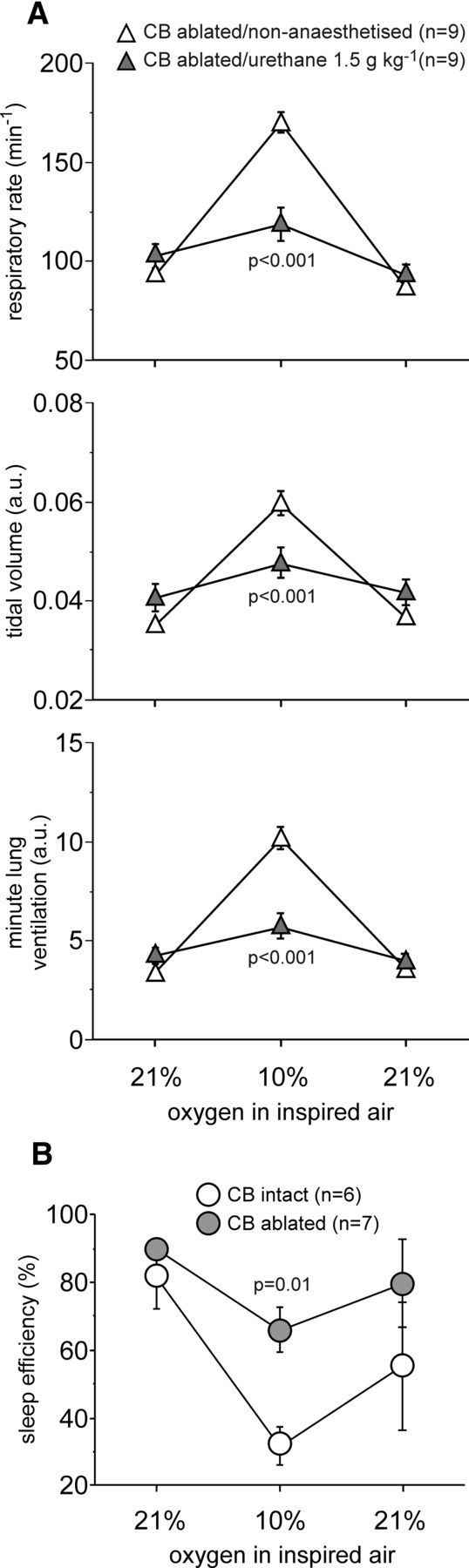
Hypoxia-induced respiratory and arousal responses in rats with denervated peripheral oxygen chemoreceptors. A, Summary data illustrating respiratory rate, tidal volume, and minute lung ventilation before, during, and after hypoxia in rats 10 weeks after carotid body (CB) ablation recorded in the non-anesthetized state during QS and under general anesthesia (urethane, 1.5 mg/kg). B, Summary data illustrating changes in SE induced by hypoxia in the carotid body ablated and sham-operated animals (10 weeks after the surgery).
To verify successful ablation of the carotid bodies, the animals were exposed to hypoxic conditions (10% oxygen in the inspired air) while asleep (during the light phase of the 24 h cycle), and changes in SE were assessed. Peripheral chemoreceptors contribute significantly to the hypoxia-induced arousal response (Miller and Tenney, 1975; Bowes et al., 1981). Thus, SE should be minimally affected by hypoxia in the absence of the peripheral oxygen sensitivity. SE was found to be the same in the carotid body-denervated and sham-operated animals under normoxic conditions (Fig. 4B). However, during hypoxia (10% inspired O2), SE was higher in rats with ablated carotid bodies compared with sham-operated animals (66 ± 7 vs 32 ± 6%; p = 0.01, F(2,33) = 6.3; Fig. 4B). These data confirm that, in rats, the sensory inputs from the peripheral oxygen chemoreceptors remain absent/impaired 10 weeks after carotid body ablation.
To interfere with ATP-mediated signaling, we used an LVV to overexpress a potent ectonucleotidase, TMPAP (Zylka et al., 2008), on the cellular membranes (Marina et al., 2013). TMPAP catalytic activity effectively prevents accumulation of ATP in astroglial vesicular compartments (Wells et al., 2015) and blocks ATP-mediated communication between astrocytes (Marina et al., 2013). Brainstem respiratory networks, including rhythm-generating circuits of the preBötC (Smith et al., 1991; Fig. 5A), were targeted bilaterally to express TMPAP or a control transgene, EGFP (Fig. 5B). Comparison of the respiratory responses in conditions of peripheral chemoreceptor ablation in rats of control (EGFP) and experimental (TMPAP) groups revealed that blockade of ATP-mediated signaling within the brainstem respiratory circuits reduces hypoxia-induced increases in the respiratory rate (p = 0.02, F(2,27) = 4.4) and, hence, minute ventilation (p = 0.02, F(2,27) = 4.3; Fig. 5C,D). There were no differences in tidal volume between the experimental groups (p = 0.13, F(2,27) = 2.2). The hypoxic ventilatory response was similarly reduced in rats expressing TMPAP in the brainstem that were also treated systemically with the A1 adenosine receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX; p = 0.02, F(1,48) = 5.7; Fig. 5D), indicating that the inability of the respiratory network to increase its activity reflected the loss of ATP actions rather than the accumulation and actions of adenosine produced after ATP breakdown.
Figure 5.
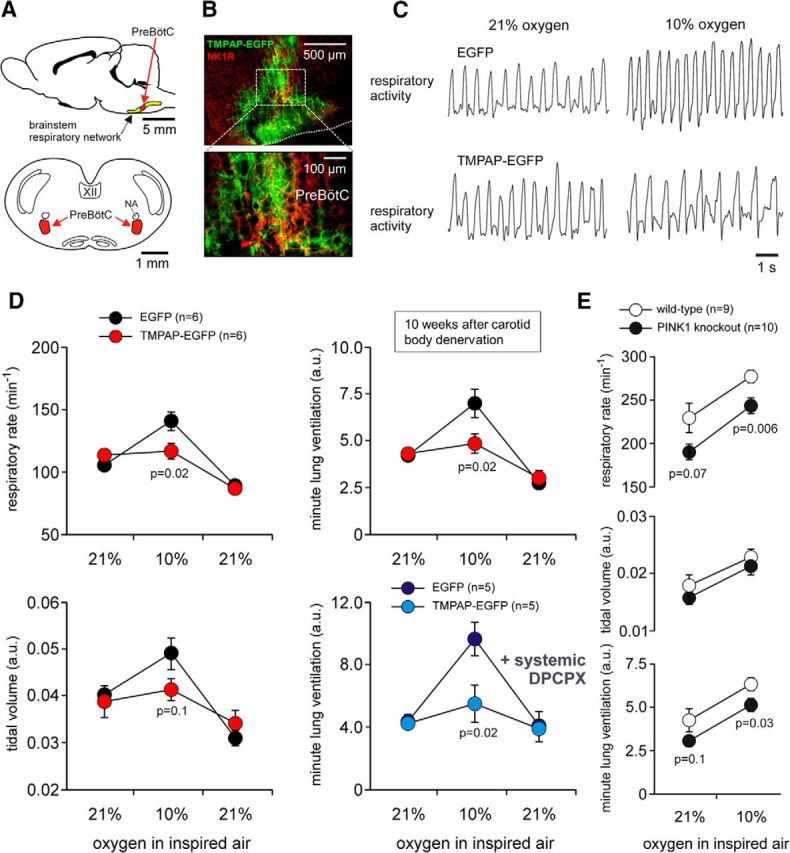
Central respiratory oxygen sensitivity is mediated by ATP actions within the brainstem respiratory circuits. A, Schematic drawings of the rat brain in sagittal and coronal projections illustrating the anatomical location of the brainstem respiratory network. NA, Nucleus ambiguus. B, Confocal images of TMPAP–EGFP expression in the respiratory areas of the ventrolateral medulla oblongata centered in (but not limited to) the preBötC, identified by NK-1R immunoreactivity. C, Plethysmography traces illustrating respiratory responses to hypoxia in conscious rats expressing EGFP or TMPAP–EGFP within the brainstem respiratory circuits 10 weeks after peripheral chemodenervation. D, Summary data illustrating changes in the respiratory rate, tidal volume, and minute lung ventilation during and after hypoxia in conscious rats expressing EGFP or TMPAP–EGFP within the brainstem respiratory circuits 10 weeks after peripheral chemodenervation. E, Resting ventilation and ventilatory responses to hypoxia in conscious PINK1-deficient mice and their wild-type counterparts.
Interestingly, during hypoxia, a lower rate of breathing (p = 0.006, F(1,34) = 11.7; but not tidal volume, p = 0.22, F(1,34) = 1.5) was recorded in PINK1-deficient mice (that lack oxygen-sensitive astrocytes; Fig. 2E) compared with their wild-type counterparts (Fig. 5E). This experiment was conducted in PINK1 knock-out and wild-type mice in which peripheral chemoreceptors were left intact. Compensation by the arterial oxygen sensors may explain a relatively modest effect of constitutive PINK1 deficiency. However, these data provide additional supportive evidence showing that failure of astrocytes to sense changes in PO2 is associated with lower respiratory activity.
Vesicular release of ATP by brainstem astrocytes mediates central respiratory oxygen sensitivity
Although astrocytes may release signaling molecules by several distinct mechanisms, observations in cultured astrocytes (Fig. 3) pointed to the involvement of vesicular exocytosis in hypoxia-induced release of ATP. To specifically block astroglial vesicular release, we generated an AVV for astrocyte-specific expression of the light chain of tetanus toxin (TeLC), which cleaves certain SNARE proteins required for vesicular docking and fusion. Tetanus toxin has been shown to inhibit both ATP and glutamate release by cultured astrocytes (Coco et al., 2003). The AVV to express TeLC in astrocytes was produced from a clone donated by Dr. J. Gogos (Columbia University, New York, NY) by homologous recombination as described previously (Duale et al., 2005; Liu et al., 2008). The layout of the vector is shown in Figure 6A. We generated a bicistronic construct in which the enhanced shortened GFAP promoter (GfaABC1D) is used to drive the expression of EGFP linked to TeLC via a “SKIP” sequence (Fig. 6A). This sequence makes use of the viral ribosomal skipping mechanism and was further enhanced by a number of modifications (Yang et al., 2008). In bicistronic constructs, two different peptides are produced by translation of the same mRNA. Commonly, this is achieved using an internal ribosome entry site to which the SKIP sequence provides a useful alternative. We aimed to attenuate expression of TeLC, which could be potentially toxic to cells, by placing it second in the cassette after the SKIP sequence. At the same time, expression of EGFP was expected to be high to aid visualization of the transduced cells.
Figure 6.

Blockade of vesicular release mechanisms in astrocytes by virally driven expression of tetanus toxin. A, Schematic of AVV–sGFAP–EGFP–TeLC vector. B, Plots of the TIRF intensity changes showing loss of quinacrine fluorescence from a proportion of labeled organelles in two individual cultured astrocytes transduced to express EGFP (control, left) or TeLC (right) in response to application of a Ca2+ ionophore ionomycin (1 μm). In astrocytes expressing TeLC, digitonin was applied at the end of the experiment to permeabilize the membranes. C, Averaged temporal distribution of ionomycin-induced (left) and sodium dithionite-induced (right) fusion events detected in quinacrine-loaded cultured astrocytes (n = 10 in each group) expressing EGFP or TeLC. D, Summary data illustrating peak frequency of ionomycin- and sodium dithionite-induced fusion events of putative ATP-containing compartments (labeled by quinacrine) detected in cultured astrocytes expressing EGFP (control) or TeLC. *p < 0.001 compared with the rate of fusion induced by ionomycin in control astrocytes. #p = 0.02 compared with the rate of fusion induced by sodium dithionite in control astrocytes. E, TeLC expression inhibits Ca2+ wave propagation induced by mechanical stimulation (MS, indicated by the arrow) in cultured astrocytes. 3D projections of the pixel intensity in the image series obtained before and at indicated time points after mechanical stimulation in astrocyte cultures transduced to express EGFP (top) or EGFP–TeLC (bottom) and loaded with Ca2+ indicator fura-2. F, Bar graph showing the extent of MS-evoked Ca2+ wave propagation (from the point of MS to the front of the wave at 20 s after MS) in control astrocytes, astrocytes expressing TeLC, and naive astrocytes in the presence of the ATP-degrading enzyme apyrase (25 U/ml). Numbers of individual tests are indicated. *p < 0.05.
To test the TeLC efficacy in blocking the mechanisms of vesicular exocytosis, TIRF microscopy was used to monitor fusion of quinacrine-labeled vesicular compartments in cultured astrocytes transduced to express TeLC. The facilitated rate of vesicular fusion induced by the Ca2+ ionophore ionomycin (1 μm; Fig. 6B–D) or the O2 scavenger sodium dithionite (Fig. 6C,D) was not observed in astrocytes transduced to express TeLC (p = 0.02, F(1,48) = 6.2). TeLC expression also effectively blocked signaling mechanisms underlying communication between cultured astrocytes. In response to mechanical stimulation, astrocytes in culture generate “waves” of Ca2+ excitation that propagate via the release of ATP (Bowser and Khakh, 2007; Fig. 6E). To trigger a Ca2+ wave, a single astrocyte was stimulated mechanically with a patch pipette, and [Ca2+]i responses evoked in the surrounding cells were recorded (Fig. 6E). In agreement with the previous report (Bowser and Khakh, 2007), the ATP degrading enzyme apyrase effectively blocked the spread of Ca2+ excitation among cultured astrocytes (t = −2.2; df = 12, p = 0.048; Fig. 6F). TeLC expression had the same effect, preventing the spread of Ca2+ excitation propagating among cultured astrocytes in response to mechanical stimulation (p = 0.03, F(1,30) = 5.3; Fig. 6E,F), an effect consistent with the inhibition of vesicular ATP release by astrocytes (Bowser and Khakh, 2007).
Bilateral expression of TeLC in astrocytes residing within the respiratory rhythm-generating circuits of the preBötC (Fig. 7A,B) reduced the hypoxia-induced increases in respiratory rate (p < 0.001, F(1,36) = 17.9) and minute ventilation (p < 0.001, F(1,36) = 17.5) in both the carotid body intact and peripherally chemodenervated animals (Fig. 7C). There were no differences in hypoxia-induced changes in tidal volume between the experimental groups (p = 0.76, F(1,36) = 0.1; Fig. 7C). Bilateral TeLC expression in astrocytes of the preBötC (in the carotid body intact animals) and bilateral carotid body ablation (in rats expressing control transgene in the preBötC) resulted in quantitatively similar reductions in the magnitude of the respiratory response to hypoxia (Fig. 7C).
Figure 7.
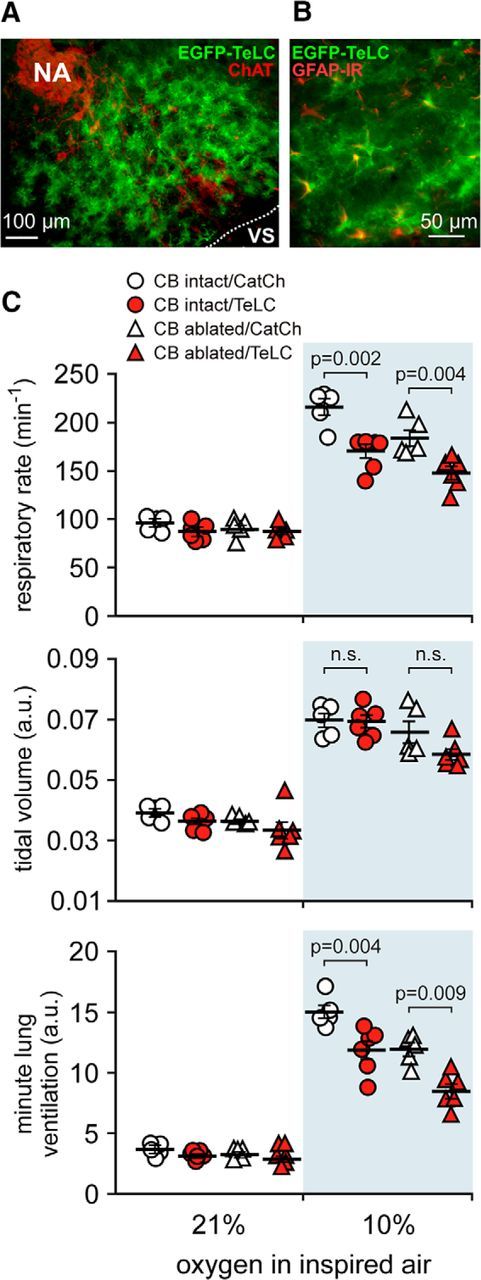
Blockade of vesicular release by brainstem astrocytes impairs central respiratory oxygen sensitivity. A, EGFP–TeLC expression in astrocytes in the brainstem region corresponding to the anatomical location of the preBötC [ventral to the compact formation of the nucleus ambiguus (NA) neurons expressing ChAT]. B, Higher-magnification image of EGFP–TeLC expression (green) in astrocytes of the preBötC region. IR, Immunoreactivity. C, Summary data illustrating hypoxia-induced changes in the respiratory rate, tidal volume, and minute lung ventilation in carotid body intact and peripherally chemodenervated (10 weeks) conscious rats expressing CatCh (calcium translocating channelrhodopsin variant that was fused with EGFP and used as a control here) or TeLC within the brainstem respiratory circuits.
Discussion
Most, if not all, animal cells possess some hypoxia-sensing mechanisms that trigger slow-timescale, adaptive transcriptional changes, for example, signaling via hypoxia-inducible factors (Ratcliffe, 2013). To date, only two types of mammalian cells have been demonstrated to be specialized for rapid functional oxygen sensing: (1) glomus cells of the carotid body that stimulate breathing when arterial oxygenation drops; and (2) pulmonary arterial smooth muscle cells responsible for hypoxic pulmonary vasoconstriction to limit perfusion of poorly ventilated regions of the lungs. Results of the present study suggest that there is yet another oxygen-sensitive cell type in the body, the astrocyte, that is tuned for rapid detection of physiological changes in brain oxygenation. Astrocytes respond to decreases in PO2 below ∼17 mmHg with elevations in intracellular calcium and release of ATP by exocytosis of VNUT-containing vesicular compartments. Normal arterial oxygen tension is ∼100 mmHg, whereas average PO2 of the brain parenchyma is between 20 and 30 mmHg (Sakadzić et al., 2010; Parpaleix et al., 2013; Marina et al., 2015). These values vary little between mammalian species (Erecińska and Silver, 2001). It appears that, although peripheral chemoreceptors of the carotid body have a threshold for activation (∼37 mmHg) that falls between the PO2 levels of the arterial blood and brain parenchyma, astrocytes become activated (display Ca2+ excitability) when PO2 decreases a few millimeters of mercury below the normal level of brain oxygenation. This study also demonstrates the physiological significance of astroglial O2 sensitivity in at least one functional paradigm: during systemic hypoxia, it contributes to and, in the absence of peripheral oxygen sensing, preserves a significant component of the homeostatic respiratory response.
In vivo imaging revealed that only SR101-labeled cortical cells (putative astrocytes) respond to systemic hypoxia with elevations in [Ca2+]i, suggesting that astroglial responses are unlikely to be secondary to neuronal activation. Indeed, [Ca2+]i responses to decreases in PO2 were observed reliably in astrocytes isolated in culture. Moreover, although the general neuronal response to hypoxia is a reduction in excitability, some central neurons, including presympathetic catecholaminergic cells of the brainstem, increase their activity in low PO2 conditions (Sun and Reis, 1994). Our recent study, which specifically investigated the mechanisms underlying the hypoxia-induced excitation of the brainstem catecholaminergic neurons, demonstrated that the sensitivity of these neurons to decreases in PO2 is indirect and mediated by previous release and actions of ATP and lactate (Marina et al., 2015). Although the source(s) of ATP and lactate were not determined in that study, the data reported here suggest that at least one of these signaling molecules (ATP) is released in hypoxic conditions by astroglial cells.
Despite an extensive literature (which is impossible to list here) reporting the effects of chronic or acute anoxia/ischemia on various aspects of astroglial biology, this unusually high sensitivity of astrocytes to physiologically relevant decreases in brain oxygenation has never been documented thoroughly. There is evidence that the oxidized/reduced nicotinamide adenine dinucleotide redox state modulates Ca2+ signaling in astrocytes (Requardt et al., 2012), and two previous studies conducted in primary astroglial cultures reported that hypoxia triggers relatively slow (tens of seconds) [Ca2+]i responses with Ca2+ release from the internal stores via activation of ryanodine receptors (Smith et al., 2005; Aley et al., 2006). The level of oxygen tension applied in these studies to mimic hypoxia was in the range of ∼20–25 mmHg (Smith et al., 2005; Aley et al., 2006), which is very close to normal (normoxic) brain parenchymal PO2 levels (Sakadzić et al., 2010; Parpaleix et al., 2013). A more recent study demonstrated that, in aging mice, lower resting PO2 in the parenchyma of the cerebellar cortex is associated with increased frequency of spontaneous Ca2+ waves in Bergmann glia (Mathiesen et al., 2013). Interestingly, hyperoxia (PO2 of 40–60 mmHg) appeared to reduce the frequency of spontaneous glial Ca2+ waves in the cerebellar cortex (Mathiesen et al., 2013). Here we report that astrocytes in both culture conditions and the organized tissue (acute brain slices and in vivo) respond with robust elevations in [Ca2+]i to hypoxic conditions when PO2 falls below ∼17 mmHg.
Pharmacological analysis of astroglial Ca2+ responses suggests that inhibition of mitochondrial respiration and increased rate of ROS production underlie astroglial oxygen sensitivity. Indeed, inhibition of mitochondrial respiration in conditions of low PO2 was found to facilitate ROS production, and this ROS production was sensitive to blockade by the mitochondrial antioxidant MitoQ. Hypoxia-induced Ca2+ responses in astrocytes were blocked by MitoQ and the ROS scavenger α-tocopherol. The facilitated rate of ROS production enhances lipid peroxidation, and there is evidence that the PLC-mediated signaling cascade in astrocytes can be activated by lipid peroxidation (Vaarmann et al., 2010; Domijan et al., 2014). Our data suggest that this mechanism links facilitated ROS production with Ca2+ recruitment from the internal stores via activation of IP3 receptors. There is also evidence that ryanodine receptors can be activated by ROS derived from mitochondria (Camello-Almaraz et al., 2006). Although the latter mechanism may be involved [and supported by the literature data discussed above (Smith et al., 2005; Aley et al., 2006)], our data strongly suggest that enhanced mitochondrial ROS production in conditions of low PO2 increases lipid peroxidation, leading to activation of PLC, IP3 receptors, and recruitment of Ca2+ from the internal stores.
Activation of astrocytes in conditions of decreased PO2 is associated with increased exocytosis of putative ATP-containing vesicles, an observation supported by the results of our previous study that used amperometric enzymatic biosensors and demonstrated facilitated release of ATP during brain hypoxia in vivo and in vitro (Gourine et al., 2005b). Hypoxia-induced fusion of putative ATP-containing vesicles in astrocytes appears to recruit SNARE proteins sensitive to cleavage by tetanus toxin. Therefore, in the subsequent experiments to determine the physiological significance of the identified mechanism of astroglial O2 sensitivity, we used two different strategies to interfere with astroglial signaling pathways. First, to promote rapid breakdown of the vesicular and released ATP in the brain area of interest, a LVV was used to drive the expression of a potent ectonucleotidase TMPAP (Zylka et al., 2008). Second, to block the mechanisms of vesicular exocytosis, astrocytes were targeted with AVV to express TeLC.
The central stimulatory effect of hypoxia on breathing appears to be mediated by astroglial ATP release, which acts within a restricted region of the brainstem that harbors the respiratory rhythm-generating circuits. The hypoxic ventilatory response in rats with denervated peripheral respiratory chemoreceptors was virtually abolished by TMPAP activity and reduced markedly by TeLC expression in astrocytes intermingled with the brainstem respiratory networks. The effect of TMPAP expression on centrally mediated hypoxic ventilatory response was more pronounced compared with that of TeLC, suggesting that virally driven expression of TeLC may not have affected all astrocytes in the targeted area, that mechanisms of ATP release other than vesicular are recruited, or that TeLC blockade of fusion of ATP-containing vesicles is partial, as suggested by the existing evidence (Coco et al., 2003). It is also possible that direct sensitivity of the respiratory neurons to the decreases in PO2 contributes to the hypoxic ventilatory response.
Indeed, a number of studies performed using in vitro, in situ, and anesthetized animal models reported that the brainstem respiratory and cardiovascular control circuits are sensitive and activated in response to oxygen deprivation (Sun and Reis, 1994; Ramirez et al., 1998; Solomon et al., 2000; Thoby-Brisson and Ramirez, 2000; Neubauer and Sunderram, 2004; Peña et al., 2004; Solomon, 2005; D'Agostino et al., 2009; Ruangkittisakul and Ballanyi, 2012; Marina et al., 2015). Despite this significant evidence, the existence of a functional CNS oxygen sensor capable of stimulating breathing is not accepted universally, perhaps reflecting questions about the physiological relevance of the hypoxia-evoked responses observed in the reduced preparations and the results obtained in anesthetized animal models. Indeed, we confirm here that the respiratory responses to hypoxia in peripherally chemodenervated rats are reduced dramatically when the animals are anesthetized, suggesting that the mechanism(s) and/or signaling pathway(s) of central respiratory oxygen sensitivity are very sensitive to general anesthesia. We hypothesize that anesthesia impairs the processes downstream of astroglial Ca2+ signaling because the data obtained show that astrocytes are able to sense changes in PO2 in rats kept under urethane anesthesia. Importantly, previous studies performed in unanesthetized animals, including dogs (Davenport et al., 1947; Curran et al., 2000), cats (Miller and Tenney, 1975), ponies (Bisgard et al., 1980), and rats (Olson et al., 1988), also documented significant hypoxic ventilatory responses after denervation (or silencing) of the peripheral oxygen chemoreceptors. Thus, there is a considerable body of evidence that supports the existence of a functional central oxygen sensor capable of stimulating breathing.
In conclusion, the results of the present study suggest that astrocytes are functionally specialized CNS oxygen sensors tuned to detect physiological decreases in brain oxygenation. At the level of the whole organism, detection of low PO2 by brainstem astrocytes stimulates the networks of the respiratory neurons and contributes to the overall respiratory response to hypoxia to ensure appropriate oxygenation of the arterial blood. Because Ca2+-dependent release of vasoactive substances by astrocytes is believed to have a significant effect on cerebral vasculature (Attwell et al., 2010), we propose that the mechanism of direct oxygen sensing by astrocytes described here may also be important for local control of cerebral microcirculation when PO2 in a particular microdomain of the brain decreases. Failure of this mechanism may have an adverse effect on neuronal function and contribute to the development of neurological (Lioy et al., 2011) and neurodegenerative (Gandhi et al., 2009) disease(s).
Footnotes
This study was supported by the Wellcome Trust, British Heart Foundation, and Medical Research Council. A.V.G is a Wellcome Trust Senior Research Fellow (reference 095064). We thank Dr. Michael P. Murphy (Medical Research Council Mitochondrial Biology Unit, Cambridge, UK) for providing MitoQ and helpful discussions and Prof. David Attwell (University College London, London, UK) for critical review of a previous version of this manuscript.
The authors declare no competing financial interests.
References
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley PK, Murray HJ, Boyle JP, Pearson HA, Peers C. Hypoxia stimulates Ca2+ release from intracellular stores in astrocytes via cyclic ADP ribose-mediated activation of ryanodine receptors. Cell Calcium. 2006;39:95–100. doi: 10.1016/j.ceca.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A. Gliotransmitters travel in time and space. Neuron. 2014;81:728–739. doi: 10.1016/j.neuron.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgard GE, Forster HV, Klein JP. Recovery of peripheral chemoreceptor function after denervation in ponies. J Appl Physiol Respir Environ Exerc Physiol. 1980;49:964–970. doi: 10.1152/jappl.1980.49.6.964. [DOI] [PubMed] [Google Scholar]
- Bowes G, Townsend ER, Kozar LF, Bromley SM, Phillipson EA. Effect of carotid body denervation on arousal response to hypoxia in sleeping dogs. J Appl Physiol Respir Environ Exerc Physiol. 1981;51:40–45. doi: 10.1152/jappl.1981.51.1.40. [DOI] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J Gen Physiol. 2007;129:485–491. doi: 10.1085/jgp.200709780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. Mitochondrial reactive oxygen species and Ca2+ signaling. Am J Physiol Cell Physiol. 2006;291:C1082–C1088. doi: 10.1152/ajpcell.00217.2006. [DOI] [PubMed] [Google Scholar]
- Coco S, Calegari F, Pravettoni E, Pozzi D, Taverna E, Rosa P, Matteoli M, Verderio C. Storage and release of ATP from astrocytes in culture. J Biol Chem. 2003;278:1354–1362. doi: 10.1074/jbc.M209454200. [DOI] [PubMed] [Google Scholar]
- Cohen G, Gressens P, Gallego J, Gaultier C. Depression of hypoxic arousal response in adolescent mice following antenatal vasoactive intestinal polypeptide blockade. J Physiol. 2002;540:691–699. doi: 10.1113/jphysiol.2001.014464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran AK, Rodman JR, Eastwood PR, Henderson KS, Dempsey JA, Smith CA. Ventilatory responses to specific CNS hypoxia in sleeping dogs. J Appl Physiol (1985) 2000;88:1840–1852. doi: 10.1152/jappl.2000.88.5.1840. [DOI] [PubMed] [Google Scholar]
- D'Agostino D, Mazza E, Jr, Neubauer JA. Heme oxygenase is necessary for the excitatory response of cultured neonatal rat rostral ventrolateral medulla neurons to hypoxia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R102–R118. doi: 10.1152/ajpregu.90325.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport HW, Brewer G, Chambers AH, Goldschmidt S. The respiratory responses to anoxemia of unanesthetized dogs with chronically denervated aortic and carotid chemoreceptors and their causes. Am J Physiol. 1947;148:406–416. doi: 10.1152/ajplegacy.1947.148.2.406. [DOI] [PubMed] [Google Scholar]
- Domijan AM, Kovac S, Abramov AY. Lipid peroxidation is essential for phospholipase C activity and the inositol-trisphosphate-related Ca2+ signal. J Cell Sci. 2014;127:21–26. doi: 10.1242/jcs.138370. [DOI] [PubMed] [Google Scholar]
- Duale H, Kasparov S, Paton JFR, Teschemacher AG. Differences in transductional tropism of adenoviral and lentiviral vectors in the rat brainstem. Exp Physiol. 2005;90:71–78. doi: 10.1113/expphysiol.2004.029173. [DOI] [PubMed] [Google Scholar]
- Enhorning G, van Schaik S, Lundgren C, Vargas I. Whole-body plethysmography, does it measure tidal volume of small animals? Can J Physiol Pharmacol. 1998;76:945–951. doi: 10.1139/y99-002. [DOI] [PubMed] [Google Scholar]
- Erecińska M, Silver IA. Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol. 2001;128:263–276. doi: 10.1016/S0034-5687(01)00306-1. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, Abramov AY. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Llaudet E, Dale N, Spyer KM. ATP is a mediator of chemosensory transduction in the central nervous system. Nature. 2005a;436:108–111. doi: 10.1038/nature03690. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Llaudet E, Dale N, Spyer KM. Release of ATP in the ventral medulla during hypoxia in rats: role in hypoxic ventilatory response. J Neurosci. 2005b;25:1211–1218. doi: 10.1523/JNEUROSCI.3763-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K, Kasparov S. Astrocytes control breathing through pH-dependent release of ATP. Science. 2010;329:571–575. doi: 10.1126/science.1190721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramsbergen A, Schwartze P, Prechtl HF. The postnatal development of behavioral states in the rat. Dev Psychobiol. 1970;3:267–280. doi: 10.1002/dev.420030407. [DOI] [PubMed] [Google Scholar]
- Guo F, Liu B, Tang F, Lane S, Souslova EA, Chudakov DM, Paton JF, Kasparov S. Astroglia are a possible cellular substrate of angiotensin(1–7) effects in the rostral ventrolateral medulla. Cardiovasc Res. 2010;87:578–584. doi: 10.1093/cvr/cvq059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Huxtable AG, Zwicker JD, Poon BY, Pagliardini S, Vrouwe SQ, Greer JJ, Funk GD. Tripartite purinergic modulation of central respiratory networks during perinatal development: the influence of ATP, ectonucleotidases, and ATP metabolites. J Neurosci. 2009;29:14713–14725. doi: 10.1523/JNEUROSCI.2660-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasymov V, Larina O, Castaldo C, Marina N, Patrushev M, Kasparov S, Gourine AV. Differential sensitivity of brainstem versus cortical astrocytes to changes in pH reveals functional regional specialization of astroglia. J Neurosci. 2013;33:435–441. doi: 10.1523/JNEUROSCI.2813-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Prabhakar NR. Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol. 2012;2:141–219. doi: 10.1002/cphy.c100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Paton JF, Kasparov S. Viral vectors based on bidirectional cell-specific mammalian promoters and transcriptional amplification strategy for use in vitro and in vivo. BMC Biotechnol. 2008;8:49. doi: 10.1186/1472-6750-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorier AR, Huxtable AG, Robinson DM, Lipski J, Housley GD, Funk GD. P2Y1 receptor modulation of the pre-Bötzinger complex inspiratory rhythm generating network in vitro. J Neurosci. 2007;27:993–1005. doi: 10.1523/JNEUROSCI.3948-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons TW, Reinhard CT, Planavsky NJ. The rise of oxygen in Earth's early ocean and atmosphere. Nature. 2014;506:307–315. doi: 10.1038/nature13068. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006;209:2304–2311. doi: 10.1242/jeb.02208. [DOI] [PubMed] [Google Scholar]
- Marina N, Tang F, Figueiredo M, Mastitskaya S, Kasimov V, Mohamed-Ali V, Roloff E, Teschemacher AG, Gourine AV, Kasparov S. Purinergic signalling in the rostral ventro-lateral medulla controls sympathetic drive and contributes to the progression of heart failure following myocardial infarction in rats. Basic Res Cardiol. 2013;108:317. doi: 10.1007/s00395-012-0317-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Ang R, Machhada A, Kasymov V, Karagiannis A, Hosford PS, Mosienko V, Teschemacher AG, Vihko P, Paton JF, Kasparov S, Gourine AV. Brainstem hypoxia contributes to the development of hypertension in the spontaneously hypertensive rat. Hypertension. 2015;65:775–783. doi: 10.1161/HYPERTENSIONAHA.114.04683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott DR, Hirst WD, Ljungberg MC. Astrocytes. In: Cohen J, Wilkin GP, editors. Neural cell culture: a practical approach. Oxford: Oxford UP; 1995. pp. 85–96. [Google Scholar]
- Marty N, Dallaporta M, Foretz M, Emery M, Tarussio D, Bady I, Binnert C, Beermann F, Thorens B. Regulation of glucagon secretion by glucose transporter type 2 (Glut2) and astrocyte-dependent glucose sensors. J Clin Invest. 2005;115:3545–3553. doi: 10.1172/JCI26309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiesen C, Brazhe A, Thomsen K, Lauritzen M. Spontaneous calcium waves in Bergman glia increase with age and hypoxia and may reduce tissue oxygen. J Cereb Blood Flow Metab. 2013;33:161–169. doi: 10.1038/jcbfm.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Tenney SM. Hypoxia-induced tachypnea in carotid-deafferented cats. Respir Physiol. 1975;23:31–39. doi: 10.1016/0034-5687(75)90069-9. [DOI] [PubMed] [Google Scholar]
- Mortola JP, Frappell PB. On the barometric method for measurements of ventilation, and its use in small animals. Can J Physiol Pharmacol. 1998;76:937–944. doi: 10.1139/y99-001. [DOI] [PubMed] [Google Scholar]
- Murphy MP. Modulating mitochondrial intracellular location as a redox signal. Sci Signal. 2012;5:pe39. doi: 10.1126/scisignal.2003386. [DOI] [PubMed] [Google Scholar]
- Neubauer JA, Sunderram J. Oxygen-sensing neurons in the central nervous system. J Appl Physiol (1985) 2004;96:367–374. doi: 10.1152/japplphysiol.00831.2003. [DOI] [PubMed] [Google Scholar]
- Olson EB, Jr, Vidruk EH, Dempsey JA. Carotid-body excision significantly changes ventilatory control in awake rats. J Appl Physiol. 1988;64:666–671. doi: 10.1063/1.341959. [DOI] [PubMed] [Google Scholar]
- Parpaleix A, Goulam Houssen Y, Charpak S. Imaging local neuronal activity by monitoring PO2 transients in capillaries. Nat Med. 2013;19:241–246. doi: 10.1038/nm.3059. [DOI] [PubMed] [Google Scholar]
- Peña F, Parkis MA, Tryba AK, Ramirez JM. Differential contribution of pacemaker properties to the generation of respiratory rhythms during normoxia and hypoxia. Neuron. 2004;43:105–117. doi: 10.1016/j.neuron.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Sensing hypoxia: physiology, genetics and epigenetics. J Physiol. 2013;591:2245–2257. doi: 10.1113/jphysiol.2012.247759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez JM, Quellmalz UJ, Wilken B, Richter DW. The hypoxic response of neurones within the in vitro mammalian respiratory network. J Physiol. 1998;507:571–582. doi: 10.1111/j.1469-7793.1998.571bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe PJ. Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol. 2013;591:2027–2042. doi: 10.1113/jphysiol.2013.251470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requardt RP, Hirrlinger PG, Wilhelm F, Winkler U, Besser S, Hirrlinger J. Ca2+ signals of astrocytes are modulated by the NAD+/NADH redox state. J Neurochem. 2012;120:1014–1025. doi: 10.1111/j.1471-4159.2012.07645.x. [DOI] [PubMed] [Google Scholar]
- Ruangkittisakul A, Ballanyi K. Anoxia response in physiological potassium of the isolated inspiratory center in calibrated newborn rat brainstem slices. Adv Exp Med Biol. 2012;758:91–98. doi: 10.1007/978-94-007-4584-1_12. [DOI] [PubMed] [Google Scholar]
- Sakadzić S, Roussakis E, Yaseen MA, Mandeville ET, Srinivasan VJ, Arai K, Ruvinskaya S, Devor A, Lo EH, Vinogradov SA, Boas DA. Two-photon high-resolution measurement of partial pressure of oxygen in cerebral vasculature and tissue. Nat Methods. 2010;7:755–759. doi: 10.1038/nmeth.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, Omote H, Yamamoto A, Moriyama Y. Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci U S A. 2008;105:5683–5686. doi: 10.1073/pnas.0800141105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith IF, Boyle JP, Kang P, Rome S, Pearson HA, Peers C. Hypoxic regulation of Ca2+ signaling in cultured rat astrocytes. Glia. 2005;49:153–157. doi: 10.1002/glia.20083. [DOI] [PubMed] [Google Scholar]
- Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon IC. Glutamate neurotransmission is not required for, but may modulate, hypoxic sensitivity of pre-Bötzinger complex in vivo. J Neurophysiol. 2005;93:1278–1284. doi: 10.1152/jn.00932.2004. [DOI] [PubMed] [Google Scholar]
- Solomon IC, Edelman NH, Neubauer JA. Pre-Bötzinger complex functions as a central hypoxia chemosensor for respiration in vivo. J Neurophysiol. 2000;83:2854–2868. doi: 10.1152/jn.2000.83.5.2854. [DOI] [PubMed] [Google Scholar]
- Sun MK, Reis DJ. Hypoxia selectively excites vasomotor neurons of rostral ventrolateral medulla in rats. Am J Physiol. 1994;266:R245–R256. doi: 10.1152/ajpregu.1994.266.1.R245. [DOI] [PubMed] [Google Scholar]
- Thoby-Brisson M, Ramirez JM. Role of inspiratory pacemaker neurons in mediating the hypoxic response of the respiratory network in vitro. J Neurosci. 2000;20:5858–5866. doi: 10.1523/JNEUROSCI.20-15-05858.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Tucker SJ, Gourine AV. Respiratory responses to hypercapnia and hypoxia in mice with genetic ablation of Kir5.1 (Kcnj16) Exp Physiol. 2011;96:451–459. doi: 10.1113/expphysiol.2010.055848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaarmann A, Gandhi S, Abramov AY. Dopamine induces Ca2+ signaling in astrocytes through reactive oxygen species generated by monoamine oxidase. J Biol Chem. 2010;285:25018–25023. doi: 10.1074/jbc.M110.111450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA, Christie IN, Hosford PS, Huckstepp RT, Angelova PR, Vihko P, Cork SC, Abramov AY, Teschemacher AG, Kasparov S, Lythgoe MF, Gourine AV. A critical role for purinergic signalling in the mechanisms underlying generation of BOLD fMRI responses. J Neurosci. 2015;35:5284–5292. doi: 10.1523/JNEUROSCI.3787-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Cohen CJ, Peng PD, Zhao Y, Cassard L, Yu Z, Zheng Z, Jones S, Restifo NP, Rosenberg SA, Morgan RA. Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther. 2008;15:1411–1423. doi: 10.1038/gt.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Gandhi S, Burchell VS, Plun-Favreau H, Wood NW, Abramov AY. Cell metabolism affects selective vulnerability in PINK1-associated Parkinson's disease. J Cell Sci. 2011;124:4194–4202. doi: 10.1242/jcs.088260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylka MJ, Sowa NA, Taylor-Blake B, Twomey MA, Herrala A, Voikar V, Vihko P. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron. 2008;60:111–122. doi: 10.1016/j.neuron.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]