

Abstract
Synaptodendritic pruning and alterations in neurotransmission are the main underlying causes of HIV-associated neurocognitive disorders (HAND). Our studies in humans and nonhuman primates indicated that the protein ferritin heavy chain (FHC) is a critical player in neuronal changes and ensuing cognitive deficit observed in these patients. Here we focus on the effect of HIV proteins and inflammatory cytokines implicated in HAND on neuronal FHC levels, dendritic changes, and neurocognitive behavior. In two well characterized models of HAND (HIV transgenic and gp120-treated rats), we report reductions in spine density and dendritic branches in prefrontal cortex pyramidal neurons compared with age-matched controls. FHC brain levels are elevated in these animals, which also show deficits in reversal learning. Moreover, IL-1β, TNF-α, and HIV gp120 upregulate FHC in rat cortical neurons. However, although the inflammatory cytokines directly altered neuronal FHC, gp120 only caused significant FHC upregulation in neuronal/glial cocultures, suggesting that glia are necessary for sustained elevation of neuronal FHC by the viral protein. Although the envelope protein induced secretion of IL-1β and TNF-α in cocultures, TNF-α blockade did not affect gp120-mediated induction of FHC. Conversely, studies with an IL-1β neutralizing antibody or specific IL-1 receptor antagonist revealed the primary involvement of IL-1β in gp120-induced FHC changes. Furthermore, silencing of neuronal FHC abrogates the effect of gp120 on spines, and spine density correlates negatively with FHC levels or cognitive deficit. These results demonstrate that viral and host components of HIV infection increase brain expression of FHC, leading to cellular and functional changes, and point to IL-1β-targeted strategies for prevention of these alterations.
SIGNIFICANCE STATEMENT This work demonstrates the key role of the cytokine IL-1β in the regulation of a novel intracellular mediator [i.e., the protein ferritin heavy chain (FHC)] of HIV-induced dendritic damage and the resulting neurocognitive impairment. This is also the first study that systematically investigates dendritic damage in layer II/III prefrontal cortex neurons of two different non-infectious models of HIV-associated neurocognitive disorders (HAND) and reveals a precise correlation of these structural changes with specific biochemical and functional alterations also reported in HIV patients. Overall, these data suggest that targeting the IL-1β-dependent FHC increase may represent a valid strategy for neuroprotective adjuvant therapies in HAND.
Keywords: chemokine, cytokine, dendritic spine, gp120, HAND, HIV cognitive impairment
Introduction
Despite the benefit of combination antiretroviral therapy (cART), the neurological complications of HIV remain an important and unmet medical need (McArthur et al., 2010; Nightingale et al., 2014). Although neuronal loss occurs in CNS during advanced or uncontrolled HIV infection, synaptic/dendritic injury more closely correlates with neurocognitive impairment (Masliah et al., 1997; Everall et al., 1999; Ellis et al., 2007) and is potentially reversible. Changes in synaptic architecture in specific brain areas, such as the prefrontal cortex (PFC) and hippocampus, are normally associated with cognitive function and memory formation (Herndon et al., 1997; Nimchinsky et al., 2004; Dumitriu et al., 2010; Bloss et al., 2011; Morrison and Baxter, 2012). Dendritic pruning and excitatory synapse loss have been observed in brain tissue of HIV-positive (HIV+) patients, despite cART, as well as in culture/animal models of HIV (Ellis et al., 2007; Hargus and Thayer, 2013; Pitcher et al., 2014).
The chemokine/receptor pair CXCL12/CXCR4 is implicated in essential CNS processes (Li and Ransohoff, 2008; Nicolai et al., 2010), and disruption of this critical signaling axis leads to neuronal injury, particularly during HIV infection (Pitcher et al., 2014). A frequent comorbidity of HIV infection is abuse of opiates, associated typically with rapid disease progression and greater neurological dysfunction (Nath et al., 2002; Hauser et al., 2006; Bokhari et al., 2011). Our previous work demonstrated that morphine and selective μ-opioid receptor agonists dysregulate the CXCL12/CXCR4 axis by modulating neuronal levels of the iron storage protein, ferritin heavy chain (FHC; Li et al., 2006; Patel et al., 2006; Sengupta et al., 2009). Importantly, FHC is required for the ability of morphine to reduce dendritic spine density in cortical neurons (Pitcher et al., 2014). However, elevated levels of neuronal FHC can be found in brain cortical tissue of HIV+ patients in the absence of drug abuse, suggesting that HIV infection alone can also alter FHC in neurons (Pitcher et al., 2014).
During HIV infection, infected and activated microglia/macrophages contribute to a neurotoxic environment by releasing a variety of substances, including viral proteins and inflammatory cytokines, such as TNF-α and IL-1β (Kaul et al., 2001; Lindl et al., 2010). These cytokines can also directly damage neurons and their dendritic arbor (Pulliam et al., 1998; Viviani et al., 2006), but the mechanisms involved are not characterized fully. Interestingly, both cytokines are known regulators of FHC protein expression in non-neuronal cells (Torti and Torti, 2002). It is presently unclear whether these cytokines stimulate FHC expression in neurons.
Here we identify TNF-α and IL-1β as host components involved in the upregulation of neuronal FHC by HIV viral proteins and demonstrate the pivotal role of IL-1β in the regulation of FHC by the HIV-1 gp120. Furthermore, by using both neuronal/glial cultures and two non-infectious rodent models, we show that FHC is a key intracellular mediator of gp120-induced dendritic damage and the resulting neurocognitive impairment. In addition to providing a novel mechanistic link between dendritic injury and the HIV-1 envelope protein, this study is also the first to investigate systematically dendritic damage in PFC pyramidal neurons and to show a direct correlation of these structural changes with specific biochemical and functional alterations also reported in HIV patients.
Materials and Methods
Animals.
Animals were purchased from Harlan Laboratories (Holtzman and Fisher 344) or Taconic Farms [Sprague Dawley (SD)] and kept in Association for Assessment and Accreditation of Laboratory Animal Care-accredited university facilities in accordance with the National Institutes of Health guidelines and institutional approval by the Institutional Animal Care and Use Committee. As described previously (Pitcher et al., 2014), E17 Holtzman pregnant rats (or their P4 litters, either sex) were used for neuronal and glial cultures, respectively. Male HIV-transgenic (Tg) rats, Hsd:HIV-1(F344), and F344/NHsd age- and sex-matched controls were used to determine the in vivo effect of viral protein expression on dendritic branching and spines. The HIV Tg rats express seven of the nine HIV-1 proteins, including the envelope protein gp120 (Reid et al., 2001). Specifically, a 3 kbp SphI–MscI fragment encompassing the 3′ region of gag and the 5′ region of pol was removed from pNL4-3, an infectious proviral plasmid, to make the non-infectious HIV-1gag-pol clone pEVd1443 (Reid et al., 2001). Because of immunodeficiency, these animals were housed in isolation in our barrier facility. As reported in the literature, HIV Tg rats develop inflammatory and neuropathological features similar to those observed in HIV-associated neurocognitive disorders (HAND), including elevated levels of proinflammatory cytokines and behavioral problems indicative of cognitive deficits (Vigorito et al., 2007; Lashomb et al., 2009; Repunte-Canonigo et al., 2014). Moreover, this non-infectious model recapitulates other important features of cART-treated HIV patients, such as immune dysregulation and controlled viral replication (Peng et al., 2010). Finally, SD rats were used for the studies focused on gp120, as described next.
Cannula implant and gp120 infusions.
Male SD rats (200–400 g) were implanted stereotaxically, under isoflurane anesthesia, with stainless-steel cannulas placed 0.96 mm posterior and 3.18 mm lateral to bregma, 3.47 mm below the surface of the cranium at an angle of 22°. Four stainless-steel screws (#0-80) were placed around the cannula, and acrylic dental cement was used to anchor the cannula. Animals were allowed 7 d to recover before receiving a bilateral infusion of 1 μl of gp120IIIB (50 ng/μl in 0.1% BSA) once daily for 7 d via a 10 μl Hamilton syringe (needle diameter, 0.48 mm; infusion rate, 0.374 μl/min). Fluorescent tracers were used to assess diffusion after bilateral intracerebroventricular infusion via implanted stainless-steel cannulas and spread of the fluorescent tracers throughout the ventricular system (for representative images, see Fig. 2A). Animals were food restricted and began behavioral testing 21 d after the final infusion.
Figure 2.

Rats treated with gp120IIIB for 7 d exhibited decreases in dendritic spine density and branching. A, Coronal sections through the forebrain, midbrain, and brainstem show the results of bilateral intracerebroventricular infusion of fluorescent tracers into the rat brain using stainless-steel cannulas, as described in Materials and Methods. Evidence of tracer diffusion was observed throughout lateral, third, and fourth ventricles. B, Overall dendritic spine density of layer II/III pyramidal neurons in the medial PFC of gp120-treated SD rats is significantly reduced compared with vehicle-treated controls. Data shown in B–E come from four neurons per brain (i.e., control, n = 24; gp120, n = 20). C, In gp120-treated rats, apical and basal dendritic spine density is significantly reduced by a similar magnitude. D, The number of basal branch points, but not apical branch points, is significantly less in gp120-treated rats compared with control animals. E, Both apical and basal dendrite length are not different between control and gp120-treated rats. **p < 0.01; red symbols denote p < 0.001.
Neuronal cultures.
Rat cortical neurons were cultured in serum-free media in the presence of a glial feeder layer, as we described previously (Shimizu et al., 2011). This culture system primarily preserves the in vivo environment because of the presence of glia; however, it still enables direct investigation of neurons that can be separated from the glia before experiments. Selected studies were also performed in neurons grown in the absence of glia for the entire culture period, as indicated and defined as Neurobasal culture, and using previously established protocols (Sengupta et al., 2009). Neurons were typically used in weeks 2–3 of culture. For spine analyses, neurons were transfected with scrambled or silencing vector at the time of plating by Nucleofection (Amaxa), as described previously (Sengupta et al., 2009); experiments were conducted at 21 d in vitro (DIV). FHC shRNA and scrambled shRNA were from GenScript (custom order; shRNA vector also expressed GFP).
Western blots.
Total protein extracts were obtained using standard protocols, as we described previously (Sengupta et al., 2009; Cook et al., 2010). Briefly, cells or tissue were lysed in lysis buffer containing 150 mm NaCl, 50 mm Tris, 0.5% Na-deoxycholate, 0.1% SDS, 10 mm Na4P2O7, 5 mm EDTA, 1% Triton X-100, 1 mm DTT, and protease/phosphatase inhibitors. Equal amounts of protein (40–50 μg) were loaded into each lane for SDS-PAGE and transferred to PVDF membranes for immunoblotting. The following antibodies were used: anti-FHC (catalog #3998; 1:1000; Cell Signaling Technology) and anti-β-actin (catalog #A2066; 1:5000; Sigma-Aldrich).
ELISA.
TNF-α and IL-1β secreted protein levels were determined from coculture supernatants using commercially available rat TNF-α and IL-1β kits (Quantikine; R&D Systems). The assays were performed following the instructions of the manufacturer. Absorbance was read at 450 nm using Wallac Victor2 1420 Multilabel Counter (PerkinElmer Life and Analytical Sciences). The concentration of secreted TNF-α and IL-1β are expressed as picograms per milliliter; values were normalized to total protein content for each well.
Behavioral testing.
Animals were tested between 10:00 A.M. and 2:00 P.M. in an attention set-shifting task based on methodology described by Birrell and Brown (2000). On day 1 of training, food-restricted rats are habituated to the testing chamber for 20 min. The testing chamber (17 × 22 × 10 inches) is divided lengthwise by a Plexiglas wall with a guillotine-style door creating a waiting area, one-third of the chamber, and a testing area, the remaining two-thirds of the chamber. The far wall of the testing area (farthest from the waiting area) is bisected by a Plexiglas divider that extended 9.25 inches, thus creating two distinct yet easily accessible sides of the testing area. The following day, rats are trained to dig in terra cotta pots to obtain a food reward (one-third of a Frosted Cheerio to limit satiation). The food reward is buried progressively deeper in the pots, filled with corncob bedding, until the rat reliably digs to retrieve the food reward. On day 3, rats undergo exemplar training: discriminating between sensory cues to determine which pot contains the food reward. Three pairs of pots that differ in only one dimension (sensory modality) are used during exemplar training: two pots with differing scents (almond vs snow), two pots with differing digging media (black string vs white string), and two pots with different textures on the exterior of the pots (suede vs cotton). The scent-paired pots are placed in the testing area (one on each side of the divider), and one of those pots is baited with food reward. At trial onset, the guillotine door is raised, allowing the rat to leave the waiting area, explore the testing area, and dig in one of the pots. If the rat begins digging in the incorrect pot, the pots are removed from the testing area, the rat is placed back into the waiting area, and the trial is recorded as incorrect. A dig is defined as vigorous displacement of the digging medium. If the rat digs in the correct pot, it is permitted to eat the food reward before being placed back in the waiting area. The pots are removed, the appropriate pot is rebaited with a food reward, and the pots are placed back in the testing area. The side of the baited pot is selected on a pseudorandom basis. An animal reaches criterion after digging for reward in the correct pot on six consecutive trials. The same procedure is repeated with the other two sets of pots. When a rat is exposed to a pair of pots for the first time, there are four discovery trials that allow the animal to explore both pots, dig without consequence, and do not count as choices.
Day 4 is the testing day, which consists of eight stages. Stage 1 begins with a simple discrimination in which the pair of pots differs in only one dimension (e.g., scent). Stimulus modalities for each rat were assigned randomly and counterbalanced across subjects. After reaching criterion, stage 2 is a complex discrimination in which the pots differ in two dimensions (e.g., scent and digging media), but only one is relevant for reward discovery. Stage 3 is a reversal trial in which the previously unrewarded cue in the same dimension that was used in stage 2 now becomes the rewarded cue. Stage 4 is an intradimensional shift in which a new set of pots with entirely new scents, media, and textures are used, but the rewarded pot will remain in the same dimension as the previous stages. Stage 5 is another reversal trial conducted in the same manner as the reversal trial of stage 3. After this is an extra-dimensional shift in stage 6. During this stage, the previously rewarded dimension now becomes irrelevant, while one of the cues in the previously irrelevant dimension becomes rewarded (e.g., if scent was the rewarded dimension for the first five stages, the digging media becomes the rewarded dimension). Trials were separated by 3 s with a maximum of 230 trials for a single animal. Stage 7 is another reversal trial. Overall, the attention set-shifting task requires recognition of rule changes and cognitive flexibility. It is analogous to the Wisconsin Card Sort used in human testing (Dias et al., 1996, 1997; Birrell and Brown, 2000; Goetghebeur et al., 2010).
Immunocytochemistry.
Immunocytochemistry was performed as described previously (Khan et al., 2003; Pitcher et al., 2014). Cells were washed with PBS, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100 for 5 min. Blocking was performed with 5% normal goat serum for 30 min. Cells were stained with anti-MAP2 (1:1000; Millipore) and anti-FHC (1:250; Abcam) and signal detected with goat anti-mouse Alexa Fluor 488 (1:250; Invitrogen) and goat anti-rabbit (1:250; Invitrogen), respectively. Cells were counterstained with phalloidin (25 μg/ml in 1% BSA for 15 min; Invitrogen) to visualize dendritic spines. After staining, coverslips were rinsed in H2O and mounted using VECTASHIELD mounting media (Vector Laboratories). Relative FHC staining intensity was assessed using FV10-ASW version 3.01 (Olympus).
Dendritic spine analysis.
For in vitro experiments, neurons were cultured in the presence of glia for 21 d and were identified using either MAP2 staining or incorporated GFP expression (FHC shRNA experiments). Images were acquired using an Olympus FV1000 MPE laser-scanning confocal microscope using 100× objective (1.40 numerical aperture). Dendritic spines were identified as phalloidin- or GFP-positive inclusions connected clearly to the dendrite by a thin shaft. For each neuron, three to four dendrite segments totaling at least 200 μm were added to measure spine density, and each neuron was represented as a single data point. FHC shRNA experiments included three to four coverslips from three separate preparations, whereas FHC staining levels with dendritic spine number experiments included five coverslips from each treatment group from one preparation. For in vivo studies, dendrites were visualized using a Golgi stain kit, according to the instructions of the manufacturer (FD NeuroTechnologies) as reported previously (Pitcher et al., 2014). Briefly, brains were removed rapidly, rinsed in H2O, and incubated in Golgi stain for 2 weeks. Tissue was then cryoprotected in sucrose at 4°C for 7 d and frozen in isopentane. Brains were sectioned by cryostat at a thickness of 180 μm and mounted on gelatin-coated microscope slides. Sections were allowed to dry overnight, and a signal intensification step was performed before dehydrating, clearing in xylene, and coverslipping with Permount (Thermo Fisher Scientific). Pyramidal neurons in layer II/III PFC were identified and reconstructed in their entirety using a Zeiss Axio Imager M.2 microscope mounting a 100× oil-immersion objective and equipped with Neurolucida software. Total spine density for each neuron was recorded and reported as a single data point. Number, length, and branching points of basal/apical dendrites were also scored. As indicated in the figure legends, animal numbers per group ranged from four to six in the different experiments; therefore, 16–24 neurons per group were analyzed. Of note, Figure 2B–E shows data from the same control and gp120-treated animals that underwent behavioral testing (see Fig. 3A); importantly, identical results were also obtained from additional vehicle-treated (n = 4) or gp120-treated (n = 4) rats that were only used for Golgi staining (data not shown).
Figure 3.
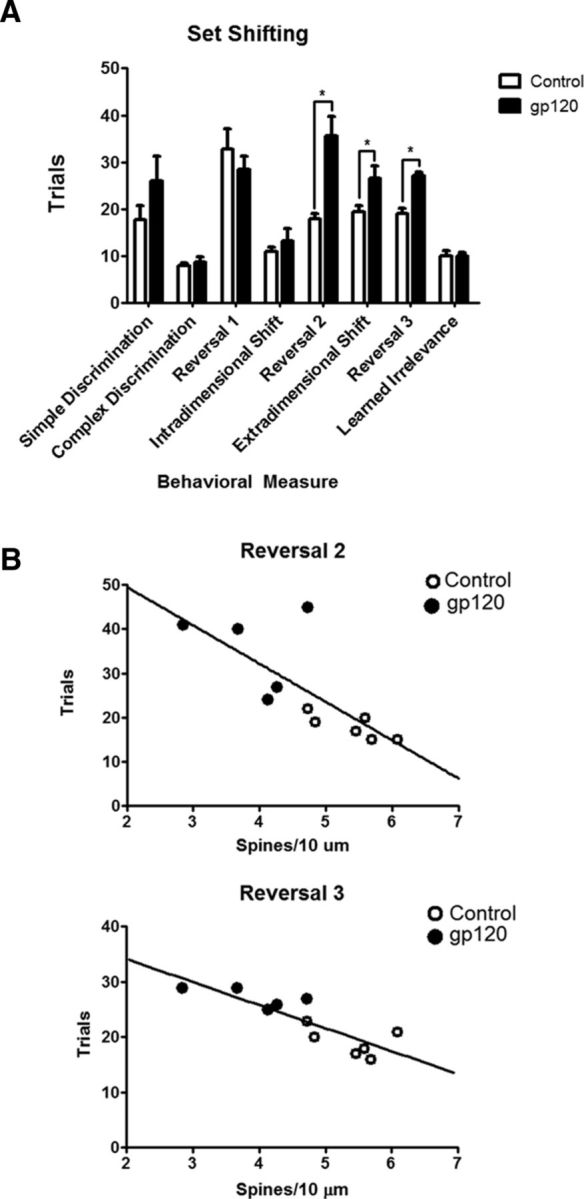
gp120-treated rats display deficits in flexible attention, which are correlated with reductions in dendritic spine density. A, Vehicle- and gp120-treated SD rats were tested in a flexible attention shifting task. Animals treated with gp120 (n = 5) are deficient in reversal learning and extra-dimensional shift compared with control rats (n = 6). B, Plotting each animal's overall dendritic spine density to their trials to criterion revealed a significant inverse correlation for Reversal 2 (n = 11, Pearson's r = −0.7578, p = 0.0069) and Reversal 3 (n = 11, Pearson's r = −0.8493, p = 0.0009). *p < 0.05.
Materials.
Cell culture media were purchased from Invitrogen. Morphine sulfate was obtained from Sigma-Aldrich. HIV-1IIIB gp120 was acquired from Immunodiagnostics. Recombinant TNF-α, IL-1β, monoclonal anti-rat IL-1β/IL-1F2 antibody, monoclonal anti-rat TNF-α antibody, and recombinant rat IL-1Ra/IL-1F3 were purchased from R&D Systems. The following reagent was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, National Institutes of Health/National Institute of Allergy and Infectious Diseases: HIV-1BaL gp120.
Statistics.
Data are reported as mean ± SEM. Statistical significance was determined by Student's t test or one-way ANOVA, followed by Newman–Keuls post hoc test. The number of trials to reach criterion for each discrimination during the behavioral task were analyzed using a Student's t test. The correlation between FHC and spine density and between spine density and trials to criterion were calculated as Pearson's r using GraphPad Prism (GraphPad Software). A p < 0.05 was considered statistically significant. p values and n are reported in the figure legends; for the correlation analyses, r and p values are also reported in the text.
Results
Dendritic alterations and FHC changes in HIV-1 Tg rats
The development of different animal models of HAND has allowed the study of individual viral proteins, as well as the whole virus, on CNS complications of HIV, leading to major advancements in the field. The HIV-1 Tg rat (Reid et al., 2001) expresses seven of the nine HIV-1 genes, both peripherally and centrally (including the PFC), and it is a good model to investigate the combined effect of multiple viral proteins in a non-infectious setting. Importantly, whole-brain lysates from these animals show elevated levels of TNF-α and IL-1β protein and mRNA (Royal et al., 2012). Also, gp160, the precursor for gp120 and gp41, has been detected in the frontal cortex of these Tg rats (Royal et al., 2012). Studies by others have shown that these animals manifest behavioral, neurochemical, and genetic profile abnormalities consistent with the neurocognitive deficit reported in HAND patients (Vigorito et al., 2007; Lashomb et al., 2009; Repunte-Canonigo et al., 2014). Synaptic injury, including reduction in dendritic arbor complexity and spine density, is a hallmark of HAND, and it is distributed widely throughout the HIV brain, although it appears predominant in some regions, such as the striatum, hippocampus, and PFC (Masliah et al., 1997; Everall et al., 1999). Therefore, we asked whether the changes in FHC we reported previously in the infected human brain, as well as the associated dendritic alterations (Pitcher et al., 2014), are also present in the HIV-1 Tg rat.
As detailed in Materials and Methods, dendrites of both HIV-1 Tg rats and age/sex-matched controls were analyzed among layer II/III pyramidal neurons of the rat medial PFC by reconstructing neurons in their entirety. Compared with controls, dendritic spine density was significantly lower in HIV-1 Tg rats (Fig. 1A). Spine density was reduced to a similar extent in apical and basal dendrites (Fig. 1B) of HIV-1 Tg rat PFC neurons. No differences in branch lengths were noted between wild-type (WT) and Tg rats (Fig. 1C). The overall number of branches in the Tg neurons was lower than in control neurons (Fig. 1D), particularly in the basal dendritic arbor. As expected, levels of FHC were increased in the PFC of HIV-1 Tg rats compared with controls (Fig. 1E); of note, here we report data from the frontal cortex of the hemisphere contralateral to the one processed for dendritic spine analysis. The graph shows that spine density correlates negatively with FHC level (Fig. 1F, Pearson's r = −0.7815, p = 0.0220). These findings suggest that FHC is involved in the regulation of dendritic spine density in the brain of HIV-1 Tg rats. The data also suggest that viral proteins may regulate FHC levels. This in agreement with our previous FHC studies in humans and nonhuman primates (Pitcher et al., 2014) and other in vitro/in vivo reports on the effect of gp120 on dendritic and synaptic injury (Viviani et al., 2006; Ellis et al., 2007; Hargus and Thayer, 2013).
Figure 1.

Dendritic spine density and branching are reduced in the PFC of the HIV Tg rat and correlates with FHC changes. A, Overall dendritic spine density of layer II/III pyramidal neurons in the medial PFC of HIV Tg rats is significantly reduced compared with F344 WT controls. Data shown in A–D come from four animals per group; four neurons were analyzed for each brain (16 neurons per group). B, Both apical and basal dendritic spine density is decreased by a similar magnitude in HIV Tg rats. C, Apical and basal dendrite length is unaltered in HIV Tg rats compared with WT controls. D, Analysis of branching in apical and basal dendrites revealed a significant decrease in the number of branch points in basal dendrites of HIV Tg animals but not apical dendrites. E, Frontal cortex lysates of HIV Tg rats used for Neurolucida analysis are shown to have increased FHC levels compared with control animals (n = 4 per group). F, Plotting each animal's FHC protein levels to their overall spine density revealed a significant inverse correlation, indicating a negative relationship between PFC FHC protein levels and spine density (n = 8; Pearson's r = −0.7815, p = 0.0220). *p < .0.05; red symbols denote p < 0.001.
Changes in dendritic spine density and branching in PFC neurons of gp120-treated rats correlate with deficits in flexible attention
The envelope protein gp120 is one of the most studied HIV-1 proteins that contributes to neuronal damage through both direct and indirect mechanisms (Bezzi et al., 2001; Kraft-Terry et al., 2009). Although cellular pathways implicated in the effects of gp120 on neuronal and glial cells have been investigated widely and mostly characterized, questions remain about the specific mediators involved in gp120-induced dendritic alterations and synaptic pruning (Ellis et al., 2007). Furthermore, the functional effect of temporally restricted (and/or limited) exposure of brain cells to gp120, which better reflects the clinical situation of most cART-treated patients, is still unknown. To address these issues, we studied changes in dendritic branching and spine numbers in the PFC of rats treated with HIV-gp120IIIB daily for 1 week and processed for spine analysis 4 weeks after the end of treatment. To directly correlate changes in the spines with cognitive behavior in each subject, these animals were also examined for performance in a flexible attention task. Dendrites of both gp120- and vehicle (BSA)-treated age/sex-matched controls were analyzed among layer II/III pyramidal neurons of the rat medial PFC, like in the previous set of studies.
As illustrated in Figure 2, dendritic spine density was significantly lower in gp120-treated rats compared with controls. Spine density was reduced to a similar extent in apical and basal dendrites of gp120-treated PFC neurons (Fig. 2C), and no differences in branch lengths were noted between the treated and control groups (Fig. 2D). However, in the basal dendritic arbor, the overall number of branches in the gp120-treated neurons was lower than in control neurons (Fig. 2E).
The attention set shifting (Birrell and Brown, 2000) was used to examine flexible attention of these animals. Five animals were tested in the task after exposure to gp120IIIB, and six animals were tested after similar treatment with the vehicle. The gp120-treated rats exhibited impaired attention set-shifting performance compared with controls (Fig. 3A). More specifically, these animals were impaired in the reversal learning component of the task (Reversal 2, t(4.620) = 4.035, p = 0.012, α = 0.05; Reversal 3, t(9) = 5.767, p < 0.001, α = 0.05). Importantly, the performance of the gp120-treated rats in Reversal 2 (Pearson's r = −0.7578, p = 0.0069, α = 0.05) and Reversal 3 (Pearson's r = −0.8493, p = 0.0009, α = 0.05) correlated with the changes in the spines (Fig. 3B). Previous research has shown that deficits in reversal learning occur after damage to the PFC (McAlonan and Brown, 2003). Additionally, rats treated with gp120 were impaired in extra-dimensional shifts (t(9) = 4.392, p = 0.026, α = 0.05), a component of the task also dependent on intact function of the medial PFC (Birrell and Brown, 2000). Additional tests indicated no significant difference in the performance of simple discriminations (t(9) = 1.439, p = 0.184, α = 0.05), a finding that suggests that gp120-induced deficits manifest in more complex aspects of the task. Overall, these data suggest that the structural changes induced by the envelope protein in the PFC compromise performance in PFC-dependent tasks.
Glia cells are required for sustained modulation of neuronal FHC by HIV-gp120
The HIV envelope protein has been shown to cause neuronal injury through a variety of mechanisms (Kraft-Terry et al., 2009), including alteration of chemokine receptor signaling and dendritic pruning (Meucci and Miller, 1996; Bardi et al., 2006; Viviani et al., 2006). To test whether changes in neuronal FHC may be responsible for gp120-mediated neuronal dysfunction, rat cortical neurons were exposed to either the X4-using gp120IIIB or the R5-using gp120BaL (200 pm for 3, 6, or 24 h); neuronal levels of FHC protein were assessed by Western blot analysis. For these studies, morphine was used as positive control because of its ability to increase FHC in pure neuronal cultures and neuronal/glial cocultures (Sengupta et al., 2009; Pitcher et al., 2014). Unlike morphine, both gp120IIIB and gp120BaL failed to affect neuronal FHC protein levels (Fig. 4A,B) under the same experimental conditions in pure neuronal cultures. However, either type of gp120 (200 pm, up to 24 h) was able to increase neuronal expression of FHC when neurons were exposed to these proteins in the presence of their glial feeder layer (Fig. 4C; only data for IIIB shown here), suggesting that a secreted mediator is driving this effect.
Figure 4.
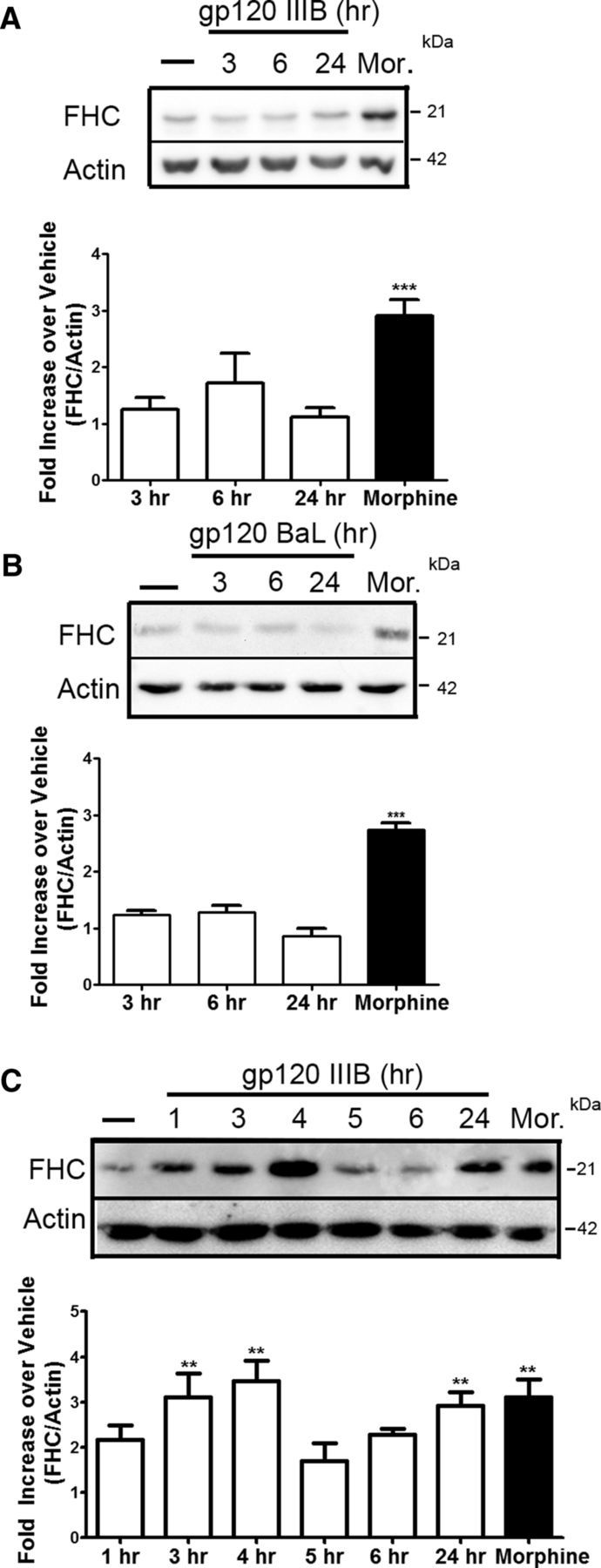
Glia cells modulate the effect of the HIV envelope protein gp120 on neuronal FHC protein. A, Cultured Neurobasal rat neurons were treated with gp120IIIB (200 pm) for the time points indicated or with morphine (Mor.; 1 μm, 24 h). gp120IIIB failed to induce any significant changes in FHC protein levels (n = 6; compared with vehicle). B, Similar experiments were conducted with the CCR5-using gp120BaL (200 pm), and no significant changes are observed at any of the time points (n = 3; compared with vehicle). C, Cortical neurons cultured in the presence of glia were treated with gp120IIIB (200 pm) for the indicated time points or with morphine (1 μm, 24 h). Significant increases in FHC neuronal protein occur at 3, 4, and 24 h (n = 3; compared with vehicle). **p < 0.01; ***p < 0.001.
Role of FHC in gp120-induced spine loss
To assess the potential relationship between gp120, FHC, and dendritic spine loss, cortical neurons cultured in the presence of a glial feeder layer were treated with either gp120IIIB or gp120BaL for 24 h and then stained with antibodies for MAP2, FHC, and phalloidin. There was a significant inverse correlation between the relative intensity of FHC staining found in neurons and their dendritic spine density (Fig. 5A; Pearson's r = −0.7441, p = 0.0015). Similar to the in vivo studies, treatment of neuronal/glial cocultures with either gp120 reduced the number of dendritic spines on the cortical neurons (Fig. 5B shows gp120IIIB data only). Downregulation of neuronal FHC expression by shRNA abolished this effect (Fig. 5B). As reported in Materials and Methods, only neurons (not glia) were transfected with the FHC silencing vector in these experiments. These findings demonstrate the critical role of FHC in the spine changes triggered by gp120 in the cocultures, which is in line with our previous studies in HIV+ drug users and nonhuman primates (Pitcher et al., 2014).
Figure 5.

FHC silencing abrogates the effect of gp120 on spines. A, Analysis of control and gp120 (either IIIB or BaL, 200 pm; 24 h)-treated cocultures shows a significant negative correlation between the FHC relative expression of each neuron [i.e., relative fluorescence units (RFU)] and their dendritic spine density (n = 15, Pearson's r = −0.7441, p = 0.0015). B, The ability of gp120IIIB to regulate dendritic spine density was blocked in FHC-deficient neurons, as shown using FHC-specific or scrambled control shRNA (n = 12). Veh, Vehicle. Blue symbols denote significance; ***p < 0.001.
TNF-α and IL-1β increase protein levels of neuronal FHC
Extracellular levels of proinflammatory cytokines, namely TNF-α and IL-1β, are typically increased in neuroinflammatory conditions, including HAND (Tyor et al., 1992). These cytokines are known regulators of FHC protein expression in non-neuronal cells (Torti et al., 1988; Wei et al., 1990) and involved previously in neuronal or synaptic injury (Viviani et al., 2006; Yang et al., 2013). Therefore, we asked whether they could directly regulate FHC expression in neurons. Rat cortical neurons were exposed to either TNF-α or IL-1β in the absence of glia. Based on previous literature (Bałkowiec-Iskra et al., 2011; Sticozzi et al., 2013), we tested the effect of these cytokines at a concentration of 0.5, 10, or 30 ng/ml (data not shown) and found that both cytokines upregulate neuronal FHC protein levels under these conditions. As shown in Figure 6A, reporting data for 10 ng/ml, maximal responses were not significantly different from those evoked by morphine, used here as a positive control (Sengupta et al., 2009). For these studies, neurons were treated with the cytokines for 24 h, a time point that consistently showed FHC upregulation in the gp120-treated cocultures. In agreement with reports in other cell types (Miller et al., 1991; Rogers, 1996; Thomson et al., 2005), some differences were noted between the two cytokines: FHC transcript levels increased after TNF-α, but not IL-1β, treatment, at either the 6 h (Fig. 6B) or 24 h (data not shown) time point. These experiments suggest that these two cytokines may play an important role in the induction of FHC observed during HIV infection, albeit via different mechanisms.
Figure 6.

The inflammatory cytokines IL-1β and TNF-α upregulate FHC protein levels in neurons, but only TNF-α alters FHC mRNA. A, Rat cortical Neurobasal neurons were treated with IL-1β (10 ng/ml), TNF-α (10 ng/ml), or morphine (Mor.; 1 μm) for 24 h. Both inflammatory cytokines cause a significant increase in neuronal FHC protein (n = 3; compared with vehicle). B, Neurobasal neurons were treated with either IL-1β or TNF-α (10 ng/ml) for 6 h, and total RNA was collected. FHC transcript levels were assessed via qPCR, and significant alterations are observed only for TNF-α (n = 3). FHC mRNA was quantified using the ΔΔCT method, and data are represented as relative to GAPDH. Veh, Vehicle. *p < 0.05; **p < 0.01; ***p < 0.001.
X4- and R5-tropic gp120 induced secretion of IL-1β and TNF-α in neuronal/glial cocultures
Chronic inflammation persists in HIV patients despite cART. Glia cells are a main source of cytokines in the CNS. Because our data show that these cells are required for the effect of gp120 on neuronal FHC, IL-1β and TNF-α may be involved in gp120-induced upregulation of FHC. Therefore, we measured levels of the two cytokines in the cocultures after exposure to either gp120IIIB or gp120BaL (200 pm for 24 h). According to these ELISA studies, levels of IL-1β and TNF-α were significantly higher in the culture media of gp120-treated cultures compared with controls; the magnitude of the response induced by the X4 and R5 envelope protein is similar (Fig. 7).
Figure 7.

X4- and R5-using gp120 induces the secretion of both TNF-α and IL-1β in bilaminar neuronal/glial cocultures. Cocultures were treated with vehicle, gp120IIIB (200 pm), or gp120BaL (200 pm) for 24 h. Levels of secreted TNF-α and IL-1β protein were analyzed from supernatants using ELISA. Both X4- and R5-using gp120 significantly increases soluble TNF-α and IL-1β protein levels. The magnitude of response is similar between the two types of gp120 (n = 3). N.D., Not detectable; Veh., vehicle. ***p < 0.001.
IL-1β is responsible for gp120-induced upregulation of neuronal FHC
Both IL-1β and TNF-α are ideal candidates for the secreted mediator responsible for the ability of gp120 to increase neuronal FHC. However, despite TNF-α being secreted in our cocultures, a neutralizing TNF-α antibody had no effect on upregulation of neuronal FHC by gp120IIIB (Fig. 8A). On the contrary, the presence of an IL-1β neutralizing antibody in gp120IIIB (or gp120BaL)-exposed cocultures reduced levels of neuronal FHC to nearly control values (Fig. 8B,D). To verify the involvement of IL-1β receptor, we also used the IL-1 receptor antagonist IL-1Ra, an endogenous inhibitor of IL-1 signaling. Similar to the neutralizing antibody, IL-1Ra completely blocked the ability of gp120 to upregulate protein levels of neuronal FHC (Fig. 8C,D). Together, these findings highlight the importance of IL-1β in mediating the upregulation of neuronal FHC protein after exposure to gp120, independently of coreceptor (CXCR4/CCR5) preference.
Figure 8.

IL-1β is responsible for gp120-mediated upregulation of neuronal FHC. A, Neuronal/glial cocultures were treated with gp120IIIB (200 pm, 24 h) in the presence or absence of a TNF-α neutralizing antibody (40 μg/ml, 24 h). The neutralizing TNF-α antibody is unable to prevent gp120-induced increases in neuronal FHC (n = 3; compared with vehicle). B, C, Cocultures were treated with gp120IIIB (200 pm, 24 h) in the presence or absence of either a neutralizing IL-1β antibody (50 μg/ml, 24 h) or the IL-1 receptor antagonist (1 μg/ml, 24 h). Neutralization of IL-1β or antagonism of IL-1R1β reduces significantly the ability of gp120IIIB to upregulate protein levels of neuronal FHC (n = 3; compared with vehicle). D, Neurons cultured in the presence of a glial feeder layer (8 DIV) were treated with gp120BaL (200 pm, 24 h) in the presence or absence of the IL-1β neutralizing antibody (50 μg/ml, 24 h) or IL-1Ra (1 μg/ml, 24 h). Similar to the effects observed with gp120IIIB, antagonism or neutralization of IL-1β prevents neuronal FHC upregulation induced by gp120BaL (n = 3; compared with vehicle). ***p < 0.001.
Discussion
HIV continues to have serious consequences in the CNS with mild forms of neurocognitive impairment on the rise as individuals live longer with the disease. Although a number of host and viral factors have been implicated previously in HAND, effective adjuvant treatments addressing neurocognitive deficit are still lacking and much needed. This study provides new insight into the mechanism of action of established players of HIV-induced brain dysfunction and suggests that IL-1β-targeting therapies may be worth pursuing to treat these CNS disorders. Briefly, in a small animal model that mimics relevant clinical and pathological conditions of HIV+ patients on cART (e.g., low-level chronic inflammation, limited expression of different viral proteins in the brain, synaptodendritic injury, and neurocognitive impairment), alterations of the dendritic arbor in PFC pyramidal neurons correlate with increased levels of FHC protein. Additionally, gp120-treated rats show decreased performance in an established flexible attention task that relies on intact function of the orbitofrontal cortex and medial PFC (Birrell and Brown, 2000; McAlonan and Brown, 2003). In light of previous studies demonstrating the impact of FHC on dendritic spines (Pitcher et al., 2014), these findings highlight the role of the envelope protein in dendritic damage that correlates with specific behavioral abnormalities. Moreover, our data show that IL-1β is necessary for gp120-mediated upregulation of FHC and that FHC mediates the effect of gp120 on dendritic spines.
Previous work implicated FHC in morphine-induced CXCR4 dysregulation and spine loss (Sengupta et al., 2009; Pitcher et al., 2014); knockdown of neuronal FHC prevents the inhibitory action of morphine on the CXCL12/CXCR4 axis and the resulting effects on dendritic spines. We also observed increases in FHC and subsequent downregulation of CXCR4 activation in brains of HIV+ patients in the absence of drug abuse, which suggested that viral or host factors induced by HIV may also target FHC. Here, we identify two proinflammatory cytokines (TNF-α and IL-1β) involved in this effect. TNF-α and IL-1β are known to regulate FHC in non-neuronal cells. Interestingly, the molecular mechanisms involved in FHC regulation in neurons are similar to the ones observed in other cell types. Indeed, TNF-α increased FHC by acting at the transcriptional level, whereas IL-1β, which did not affect levels of FHC mRNA in neurons, likely acts via posttranscriptional modifications (Miller et al., 1991; Rogers et al., 1994). This suggests that inflammation of the CNS, seen in HIV or other neuropathologies, may play a crucial role in CXCR4 dysregulation via increases in neuronal FHC.
In addition to investigating viral-induced host factors, we assessed the ability of the HIV-1 envelope protein to modulate neuronal FHC. Although gp120 directly affects neuronal signaling (Meucci et al., 1998; Bardi et al., 2006; Khan et al., 2008), it did not significantly alter the levels of FHC in pure neuronal cultures. The inability of either X4- or R5-using proteins to directly change levels of FHC in neurons suggests that known intracellular mediators of gp120, such as p38 and caspase-3 (Singh et al., 2005; Bardi et al., 2006), may not be directly involved in the regulation of FHC. Interestingly, the presence of glia distinctly modified the ability of gp120 to upregulate neuronal FHC. An important finding from these studies is that IL-1β, and not TNF-α, is responsible for gp120-mediated upregulation of FHC, despite both cytokines being secreted by the viral protein.
In line with our findings on FHC, recent work has shown that the ability of gp120 to downregulate dendritic spines in neuronal cultures depends on release of IL-1β from glial cells (Viviani et al., 2006); the secreted IL-1β induces tyrosine phosphorylation of the NMDA receptor (NMDAR), specifically the NR2A/2B subunit (Viviani et al., 2006). Additionally, the IL-1 receptor forms a complex that activates the AP-1 transcription factor, which can activate several proinflammatory genes, including COX-2 (Mishra et al., 2012). It is believed that the COX-2 pathway increases glutamate release; coupled with increased sensitivity of NMDAR to glutamate, this leads to synapse loss (Mishra et al., 2012). The relationship of these pathways with FHC are still unclear but likely involve CXCR4. Upregulation of FHC in neurons is known to inhibit homeostatic CXCR4 signaling (Pitcher et al., 2014). Recent work from our group has shown that the CXCL12/CXCR4 axis positively regulates spine number in cultured neurons and in vivo, whereas overexpression of FHC decreased dendritic spine density (Pitcher et al., 2014). Additional studies focused on the molecular players responsible for CXCL12/CXCR4 modulation of dendritic spine density are ongoing. Potential candidates include the Rac1/p21-activated kinase pathway (targeted by both CXCL12 and morphine; Volinsky et al., 2006; Impey et al., 2010; Majumdar et al., 2011) and other translation factors (e.g., CYFIP) identified recently as essential regulators of spine formation and cytoskeleton remodeling (De Rubeis et al., 2013). Interestingly, very recent work has shown that CXCL12 stabilizes filamentous actin (F-actin) at the leading process of migrating interneurons via regulation of calpain protease (Lysko et al., 2014). Because the cytoskeleton of dendritic spines is formed primarily by F-actin, similar mechanisms may be involved in the effect of CXCL12 on spines in mature neurons. Finally, CXCR4 also regulates NMDAR expression in neurons (Nicolai et al., 2010), which provides another link to the action of IL-1β warranting additional investigation.
Our ex vivo studies further highlighted the ability of HIV proteins to downregulate spines. Using the non-infectious HIV-1 Tg rats, we observed a significant reduction in dendritic spine density and branching in neurons of the PFC compared with control animals. Additionally, levels of FHC were increased in brain tissue lysates from the frontal cortex of these HIV-1 Tg rats, confirming the correlation between FHC and spine numbers. To our knowledge, this is the first study demonstrating spine reduction in the PFC of HIV-1 Tg rat, a crucial finding because dendritic spine density is one of the pathophysiological changes well correlated with cognitive impairment in HIV patients (Masliah et al., 1997; Everall et al., 1999). These animals also show behavioral changes consistent with such a deficit. Despite the physical limitations of the HIV-1 Tg rat (i.e., congenital cataracts), past studies using modified behavioral tests have revealed neurological deficits reminiscent of the human condition in the HIV Tg rats (Vigorito et al., 2007; Lashomb et al., 2009). More recent studies focused on different brain areas have also confirmed the value of this model to HAND research (Moran et al., 2014; Repunte-Canonigo et al., 2014; Roscoe et al., 2014). A limitation of the HIV Tg rats in this specific context is that expression of the viral proteins occurs throughout their life, and this could potentially interfere with normal CNS development, thus possibly adding a confounding factor in the evaluation of spine density. Although we cannot exclude this possibility, it is unlikely that this has created a significant bias in this study, because the findings from the gp120-treated animals are almost identical.
Our investigations in rats treated with gp120 further characterized the factors involved in the neurocognitive impairment. The behavioral analyses performed indicate that specific aspects of cognitive flexibility, including reversal learning and extra-dimensional shift, are altered in animals whose brain tissue was exposed to low concentrations of gp120 for a finite period of time. Changes in motivation, discrimination, or perseveration could also be influencing the performance of gp120-treated animals. However, these results are consistent with previous studies showing deficits in flexible attention, a cognitive ability dependent on intact prefrontal cortical function (Birrell and Brown, 2000; McAlonan and Brown, 2003). As might have been predicted, layer II/III pyramidal neurons from gp120-treated rats that failed behavioral testing also had fewer spines compared with control animals. Although these results do not exclude the possible contribution of other viral proteins—namely Tat, which can also alter the excitatory and inhibitory synapses ratio and cause dendritic pruning in other brain regions (Hargus and Thayer, 2013; Fitting et al., 2014)—they provide evidence about the specific ability of gp120 treatment to reduce spine and branch number in vivo and affect executive function.
Outside of spine numbers, additional analysis of dendritic spines must be considered, including spine type and location. We are currently investigating whether HIV proteins may preferentially affect specific spine types.
In conclusion, this study bears important implications for understanding mechanisms of dendritic spine loss in the brain of HIV patients by focusing on potential contributors to neurocognitive impairment in the post-cART era, in which productive viral replication is limited. The lack of the infectious component in both the in vitro and in vivo models used here allowed us to identify specific viral (i.e., gp120 and possibly tat) and host (IL1β/TNF-α) factors with intrinsic ability to alter FHC independently of other comorbid factors (i.e., drug abuse, aging, altered iron metabolism) or overt inflammation. The data presented here suggest that targeting the IL-1β-dependent FHC increase may represent a valid strategy for neuroprotective adjuvant therapies in HAND; importantly, IL-1β receptor antagonists are already used in the clinic for other purposes. Furthermore, the ability of FHC to modulate CXCR4 signaling is outside of its critical iron-sequestration and storage functions (Pitcher et al., 2014), suggesting that its association to CXCR4 can be targeted specifically without detrimental effects to the cell.
Footnotes
This work was supported by National Institutes of Health Grants DA15014 and DA32444 (O.M.) and MH097623 (B.D.W., O.M.). L.F. is a fellow of the “Interdisciplinary and Translational Research Training in neuroAIDS” (National Institutes of Health Grant T32-MH078795). We thank Matilde Hoffmann and Maricar Palalay for assistance with the dendritic spine studies and David Sulzer (Columbia University) and James Russell (National Institutes of Health/National Institute of Child Health and Human Development) for helpful discussion and review of this manuscript.
References
- Bałkowiec-Iskra E, Vermehren-Schmaedick A, Balkowiec A. Tumor necrosis factor-alpha increases brain-derived neurotrophic factor expression in trigeminal ganglion neurons in an activity-dependent manner. Neuroscience. 2011;180:322–333. doi: 10.1016/j.neuroscience.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardi G, Sengupta R, Khan MZ, Patel JP, Meucci O. Human immunodeficiency virus gp120-induced apoptosis of human neuroblastoma cells in the absence of CXCR4 internalization. J Neurovirol. 2006;12:211–218. doi: 10.1080/13550280600848373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Birrell JM, Brown VJ. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci. 2000;20:4320–4324. doi: 10.1523/JNEUROSCI.20-11-04320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloss EB, Janssen WG, Ohm DT, Yuk FJ, Wadsworth S, Saardi KM, McEwen BS, Morrison JH. Evidence for reduced experience-dependent dendritic spine plasticity in the aging prefrontal cortex. J Neurosci. 2011;31:7831–7839. doi: 10.1523/JNEUROSCI.0839-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhari SM, Hegde R, Callen S, Yao H, Adany I, Li Q, Li Z, Pinson D, Yeh HW, Cheney PD, Buch S. Morphine potentiates neuropathogenesis of SIV infection in rhesus macaques. J Neuroimmune Pharmacol. 2011;6:626–639. doi: 10.1007/s11481-011-9272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook A, Hippensteel R, Shimizu S, Nicolai J, Fatatis A, Meucci O. Interactions between chemokines: regulation of fractalkine/CX3CL1 homeostasis by SDF/CXCL12 in cortical neurons. J Biol Chem. 2010;285:10563–10571. doi: 10.1074/jbc.M109.035477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, Pasciuto E, Li KW, Fernández E, Di Marino D, Buzzi A, Ostroff LE, Klann E, Zwartkruis FJ, Komiyama NH, Grant SG, Poujol C, Choquet D, Achsel T, Posthuma D, Smit AB, Bagni C. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron. 2013;79:1169–1182. doi: 10.1016/j.neuron.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias R, Robbins TW, Roberts AC. Primate analogue of the Wisconsin Card Sorting Test: effects of excitotoxic lesions of the prefrontal cortex in the marmoset. Behav Neurosci. 1996;110:872–886. doi: 10.1037/0735-7044.110.5.872. [DOI] [PubMed] [Google Scholar]
- Dias R, Robbins TW, Roberts AC. Dissociable forms of inhibitory control within prefrontal cortex with an analog of the Wisconsin Card Sort Test: restriction to novel situations and independence from “on-line“ processing. J Neurosci. 1997;17:9285–9297. doi: 10.1523/JNEUROSCI.17-23-09285.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu D, Hao J, Hara Y, Kaufmann J, Janssen WG, Lou W, Rapp PR, Morrison JH. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J Neurosci. 2010;30:7507–7515. doi: 10.1523/JNEUROSCI.6410-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M, Masliah E. Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol. 1999;9:209–217. doi: 10.1111/j.1750-3639.1999.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Knapp PE, Zou S, Marks WD, Bowers MS, Akbarali HI, Hauser KF. Interactive HIV-1 Tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na(+) influx, mitochondrial instability, and Ca(2)(+) overload. J Neurosci. 2014;34:12850–12864. doi: 10.1523/JNEUROSCI.5351-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetghebeur PJ, Lerdrup L, Sylvest A, Dias R. Erythropoietin reverses the attentional set-shifting impairment in a rodent schizophrenia disease-like model. Psychopharmacology. 2010;212:635–642. doi: 10.1007/s00213-010-1990-9. [DOI] [PubMed] [Google Scholar]
- Hargus NJ, Thayer SA. Human immunodeficiency virus-1 Tat protein increases the number of inhibitory synapses between hippocampal neurons in culture. J Neurosci. 2013;33:17908–17920. doi: 10.1523/JNEUROSCI.1312-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser KF, El-Hage N, Buch S, Nath A, Tyor WR, Bruce-Keller AJ, Knapp PE. Impact of opiate-HIV-1 interactions on neurotoxic signaling. J Neuroimmune Pharmacol. 2006;1:98–105. doi: 10.1007/s11481-005-9000-4. [DOI] [PubMed] [Google Scholar]
- Herndon JG, Moss MB, Rosene DL, Killiany RJ. Patterns of cognitive decline in aged rhesus monkeys. Behav Brain Res. 1997;87:25–34. doi: 10.1016/S0166-4328(96)02256-5. [DOI] [PubMed] [Google Scholar]
- Impey S, Davare M, Lesiek A, Fortin D, Ando H, Varlamova O, Obrietan K, Soderling TR, Goodman RH, Wayman GA. An activity-induced microRNA controls dendritic spine formation by regulating Rac1-PAK signaling. Mol Cell Neurosci. 2010;43:146–156. doi: 10.1016/j.mcn.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Khan MZ, Brandimarti R, Musser BJ, Resue DM, Fatatis A, Meucci O. The chemokine receptor CXCR4 regulates cell-cycle proteins in neurons. J Neurovirol. 2003;9:300–314. doi: 10.1080/13550280390201010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ, Brandimarti R, Shimizu S, Nicolai J, Crowe E, Meucci O. The chemokine CXCL12 promotes survival of postmitotic neurons by regulating Rb protein. Cell Death Differ. 2008;15:1663–1672. doi: 10.1038/cdd.2008.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft-Terry SD, Buch SJ, Fox HS, Gendelman HE. A coat of many colors: neuroimmune crosstalk in human immunodeficiency virus infection. Neuron. 2009;64:133–145. doi: 10.1016/j.neuron.2009.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashomb AL, Vigorito M, Chang SL. Further characterization of the spatial learning deficit in the human immunodeficiency virus-1 transgenic rat. J Neurovirol. 2009;15:14–24. doi: 10.1080/13550280802232996. [DOI] [PubMed] [Google Scholar]
- Li M, Ransohoff RM. Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Prog Neurobiol. 2008;84:116–131. doi: 10.1016/j.pneurobio.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Luo C, Mines M, Zhang J, Fan GH. Chemokine CXCL12 induces binding of ferritin heavy chain to the chemokine receptor CXCR4, alters CXCR4 signaling, and induces phosphorylation and nuclear translocation of ferritin heavy chain. J Biol Chem. 2006;281:37616–37627. doi: 10.1074/jbc.M607266200. [DOI] [PubMed] [Google Scholar]
- Lindl KA, Marks DR, Kolson DL, Jordan-Sciutto KL. HIV-associated neurocognitive disorder: pathogenesis and therapeutic opportunities. J Neuroimmune Pharmacol. 2010;5:294–309. doi: 10.1007/s11481-010-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysko DE, Putt M, Golden JA. SDF1 reduces interneuron leading process branching through dual regulation of actin and microtubules. J Neurosci. 2014;34:4941–4962. doi: 10.1523/JNEUROSCI.4351-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar D, Nebhan CA, Hu L, Anderson B, Webb DJ. An APPL1/Akt signaling complex regulates dendritic spine and synapse formation in hippocampal neurons. Mol Cell Neurosci. 2011;46:633–644. doi: 10.1016/j.mcn.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42:963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- McAlonan K, Brown VJ. Orbital prefrontal cortex mediates reversal learning and not attentional set shifting in the rat. Behav Brain Res. 2003;146:97–103. doi: 10.1016/j.bbr.2003.09.019. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Steiner J, Sacktor N, Nath A. Human immunodeficiency virus-associated neurocognitive disorders: mind the gap. Ann Neurol. 2010;67:699–714. doi: 10.1002/ana.22053. [DOI] [PubMed] [Google Scholar]
- Meucci O, Miller RJ. gp120-induced neurotoxicity in hippocampal pyramidal neuron cultures: protective action of TGF-beta1. J Neurosci. 1996;16:4080–4088. doi: 10.1523/JNEUROSCI.16-13-04080.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LL, Miller SC, Torti SV, Tsuji Y, Torti FM. Iron-independent induction of ferritin H chain by tumor necrosis factor. Proc Natl Acad Sci U S A. 1991;88:4946–4950. doi: 10.1073/pnas.88.11.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Kim HJ, Shin AH, Thayer SA. Synapse loss induced by interleukin-1beta requires pre- and post-synaptic mechanisms. J Neuroimmune Pharmacol. 2012;7:571–578. doi: 10.1007/s11481-012-9342-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran LM, Booze RM, Mactutus CF. Modeling deficits in attention, inhibition, and flexibility in HAND. J Neuroimmune Pharmacol. 2014;9:508–521. doi: 10.1007/s11481-014-9539-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13:240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, Cass W, Turchan JT. Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S62–S69. doi: 10.1097/00126334-200210012-00006. [DOI] [PubMed] [Google Scholar]
- Nicolai J, Burbassi S, Rubin J, Meucci O. CXCL12 inhibits expression of the NMDA receptor's NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell Death Disease. 2010;1:e33. doi: 10.1038/cddis.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale S, Winston A, Letendre S, Michael BD, McArthur JC, Khoo S, Solomon T. Controversies in HIV-associated neurocognitive disorders. Lancet Neurol. 2014;13:1139–1151. doi: 10.1016/S1474-4422(14)70137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Yasuda R, Oertner TG, Svoboda K. The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J Neurosci. 2004;24:2054–2064. doi: 10.1523/JNEUROSCI.5066-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JP, Sengupta R, Bardi G, Khan MZ, Mullen-Przeworski A, Meucci O. Modulation of neuronal CXCR4 by the micro-opioid agonist DAMGO. J Neurovirol. 2006;12:492–500. doi: 10.1080/13550280601064798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Vigorito M, Liu X, Zhou D, Wu X, Chang SL. The HIV-1 transgenic rat as a model for HIV-1 infected individuals on HAART. J Neuroimmunol. 2010;218:94–101. doi: 10.1016/j.jneuroim.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Pitcher J, Abt A, Myers J, Han R, Snyder M, Graziano A, Festa L, Kutzler M, Garcia F, Gao WJ, Fischer-Smith T, Rappaport J, Meucci O. Neuronal ferritin heavy chain and drug abuse affect HIV-associated cognitive dysfunction. J Clin Invest. 2014;124:656–669. doi: 10.1172/JCI70090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulliam L, Zhou M, Stubblebine M, Bitler CM. Differential modulation of cell death proteins in human brain cells by tumor necrosis factor alpha and platelet activating factor. J Neurosci Res. 1998;54:530–538. doi: 10.1002/(SICI)1097-4547(19981115)54:4<530::AID-JNR10>3.0.CO%3B2-1. [DOI] [PubMed] [Google Scholar]
- Reid W, Sadowska M, Denaro F, Rao S, Foulke J, Jr, Hayes N, Jones O, Doodnauth D, Davis H, Sill A, O'Driscoll P, Huso D, Fouts T, Lewis G, Hill M, Kamin-Lewis R, Wei C, Ray P, Gallo RC, Reitz M, Bryant J. An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc Natl Acad Sci U S A. 2001;98:9271–9276. doi: 10.1073/pnas.161290298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repunte-Canonigo V, Lefebvre C, George O, Kawamura T, Morales M, Koob GF, Califano A, Masliah E, Sanna PP. Gene expression changes consistent with neuroAIDS and impaired working memory in HIV-1 transgenic rats. Mol Neurodegener. 2014;9:26. doi: 10.1186/1750-1326-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT. Ferritin translation by interleukin-1and interleukin-6: the role of sequences upstream of the start codons of the heavy and light subunit genes. Blood. 1996;87:2525–2537. [PubMed] [Google Scholar]
- Rogers JT, Andriotakis JL, Lacroix L, Durmowicz GP, Kasschau KD, Bridges KR. Translational enhancement of H-ferritin mRNA by interleukin-1 beta acts through 5′ leader sequences distinct from the iron responsive element. Nucleic Acids Res. 1994;22:2678–2686. doi: 10.1093/nar/22.13.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscoe RF, Jr, Mactutus CF, Booze RM. HIV-1 transgenic female rat: synaptodendritic alterations of medium spiny neurons in the nucleus accumbens. J Neuroimmune Pharmacol. 2014;9:642–653. doi: 10.1007/s11481-014-9555-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal W, 3rd, Zhang L, Guo M, Jones O, Davis H, Bryant JL. Immune activation, viral gene product expression and neurotoxicity in the HIV-1 transgenic rat. J Neuroimmunol. 2012;247:16–24. doi: 10.1016/j.jneuroim.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta R, Burbassi S, Shimizu S, Cappello S, Vallee RB, Rubin JB, Meucci O. Morphine increases brain levels of ferritin heavy chain leading to inhibition of CXCR4-mediated survival signaling in neurons. J Neurosci. 2009;29:2534–2544. doi: 10.1523/JNEUROSCI.5865-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Abt A, Meucci O. Bilaminar co-culture of primary rat cortical neurons and glia. J Vis Exp. 2011;pii:3257. doi: 10.3791/3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh IN, El-Hage N, Campbell ME, Lutz SE, Knapp PE, Nath A, Hauser KF. Differential involvement of p38 and JNK MAP kinases in HIV-1 Tat and gp120-induced apoptosis and neurite degeneration in striatal neurons. Neuroscience. 2005;135:781–790. doi: 10.1016/j.neuroscience.2005.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticozzi C, Belmonte G, Meini A, Carbotti P, Grasso G, Palmi M. IL-1beta induces GFAP expression in vitro and in vivo and protects neurons from traumatic injury-associated apoptosis in rat brain striatum via NFkappaB/Ca(2)(+)-calmodulin/ERK mitogen-activated protein kinase signaling pathway. Neuroscience. 2013;252:367–383. doi: 10.1016/j.neuroscience.2013.07.061. [DOI] [PubMed] [Google Scholar]
- Thomson AM, Cahill CM, Cho HH, Kassachau KD, Epis MR, Bridges KR, Leedman PJ, Rogers JT. The acute box cis-element in human heavy ferritin mRNA 5′-untranslated region is a unique translation enhancer that binds poly(C)-binding proteins. J Biol Chem. 2005;280:30032–30045. doi: 10.1074/jbc.M502951200. [DOI] [PubMed] [Google Scholar]
- Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood. 2002;99:3505–3516. doi: 10.1182/blood.V99.10.3505. [DOI] [PubMed] [Google Scholar]
- Torti SV, Kwak EL, Miller SC, Miller LL, Ringold GM, Myambo KB, Young AP, Torti FM. The molecular cloning and characterization of murine ferritin heavy chain, a tumor necrosis factor-inducible gene. J Biol Chem. 1988;263:12638–12644. [PubMed] [Google Scholar]
- Tyor WR, Glass JD, Griffin JW, Becker PS, McArthur JC, Bezman L, Griffin DE. Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol. 1992;31:349–360. doi: 10.1002/ana.410310402. [DOI] [PubMed] [Google Scholar]
- Vigorito M, LaShomb AL, Chang SL. Spatial learning and memory in HIV-1 transgenic rats. J Neuroimmune Pharmacol. 2007;2:319–328. doi: 10.1007/s11481-007-9078-y. [DOI] [PubMed] [Google Scholar]
- Viviani B, Gardoni F, Bartesaghi S, Corsini E, Facchi A, Galli CL, Di Luca M, Marinovich M. Interleukin-1 beta released by gp120 drives neural death through tyrosine phosphorylation and trafficking of NMDA receptors. J Biol Chem. 2006;281:30212–30222. doi: 10.1074/jbc.M602156200. [DOI] [PubMed] [Google Scholar]
- Volinsky N, Gantman A, Yablonski D. A Pak- and Pix-dependent branch of the SDF-1alpha signalling pathway mediates T cell chemotaxis across restrictive barriers. Biochem J. 2006;397:213–222. doi: 10.1042/BJ20051655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Miller SC, Tsuji Y, Torti SV, Torti FM. Interleukin 1 induces ferritin heavy chain in human muscle cells. Biochem Biophys Res Commun. 1990;169:289–296. doi: 10.1016/0006-291X(90)91466-6. [DOI] [PubMed] [Google Scholar]
- Yang G, Parkhurst CN, Hayes S, Gan WB. Peripheral elevation of TNF-alpha leads to early synaptic abnormalities in the mouse somatosensory cortex in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2013;110:10306–10311. doi: 10.1073/pnas.1222895110. [DOI] [PMC free article] [PubMed] [Google Scholar]