

Abstract
Vacuolar protein sorting-35 (VPS35) is essential for endosome-to-Golgi retrieval of membrane proteins. Mutations in the VPS35 gene have been identified in patients with autosomal dominant PD. However, it remains poorly understood if and how VPS35 deficiency or mutation contributes to PD pathogenesis. Here we provide evidence that links VPS35 deficiency to PD-like neuropathology. VPS35 was expressed in mouse dopamine (DA) neurons in substantia nigra pars compacta (SNpc) and STR (striatum)—regions that are PD vulnerable. VPS35-deficient mice exhibited PD-relevant deficits including accumulation of α-synuclein in SNpc-DA neurons, loss of DA transmitter and DA neurons in SNpc and STR, and impairment of locomotor behavior. Further mechanical studies showed that VPS35-deficient DA neurons or DA neurons expressing PD-linked VPS35 mutant (D620N) had impaired endosome-to-Golgi retrieval of lysosome-associated membrane glycoprotein 2a (Lamp2a) and accelerated Lamp2a degradation. Expression of Lamp2a in VPS35-deficient DA neurons reduced α-synuclein, supporting the view for Lamp2a as a receptor of chaperone-mediated autophagy to be critical for α-synuclein degradation. These results suggest that VPS35 deficiency or mutation promotes PD pathogenesis and reveals a crucial pathway, VPS35-Lamp2a-α-synuclein, to prevent PD pathogenesis.
SIGNIFICANCE STATEMENT VPS35 is a key component of the retromer complex that is essential for endosome-to-Golgi retrieval of membrane proteins. Mutations in the VPS35 gene have been identified in patients with PD. However, if and how VPS35 deficiency or mutation contributes to PD pathogenesis remains unclear. We demonstrated that VPS35 deficiency or mutation (D620N) in mice leads to α-synuclein accumulation and aggregation in the substantia nigra, accompanied with DA neurodegeneration. VPS35-deficient DA neurons exhibit impaired endosome-to-Golgi retrieval of Lamp2a, which may contribute to the reduced α-synuclein degradation through chaperone-mediated autophagy. These results suggest that VPS35 deficiency or mutation promotes PD pathogenesis, and reveals a crucial pathway, VPS35-Lamp2a-α-synuclein, to prevent PD pathogenesis.
Keywords: α-synuclein, autophagy, LAMP2a, Parkinson's disease, VPS35
Introduction
PD, the second most common progressive neurodegenerative disorder, is pathologically characterized by DA neuronal loss and accumulation of α-synuclein-containing Lewy bodies. Mutations in numerous genes have been identified in PD patients. For example, mutations in α-synuclein (also called Park1) and leucine-rich repeat kinase 2 (LRRK2, or Park8) cause autosomal dominant PD, and mutations in parkin (Park2), DJ-1 (Park7), Pink1 (Park6), and ATP13A2 have been linked to autosomal recessive PD (Savitt et al., 2006; von Coelln et al., 2006; Gasser, 2007). Investigation of the function of these genes and mutant proteins has revealed pathophysiological mechanisms for both familial and sporadic PD (Savitt et al., 2006; Gasser, 2007; Lee et al., 2012).
Vacuolar protein sorting-35 (VPS35; also called Park17) is a key component of the retromer complex. The retromer, initially identified in yeast, is evolutionally conserved across different species, including Caenorhabditis elegans, Drosophila, mouse, and human. It contains two subprotein complexes: the cargo-selective subcomplex that consists of a trimer of VPS35, VPS29, and VPS26, and the membrane deformation subcomplex that consists of sortin nexin dimers (Seaman, 2005; Bonifacino and Hurley, 2008; van Weering et al., 2010). The retromer plays an essential role in endosome-to-Golgi retrieval of membrane proteins. Numerous transmembrane proteins/receptors have been identified as retromer cargos, which include VPS10/sortilin family proteins (Seaman, 2005), cation-independent mannose 6-phosphate receptor (CI-M6PR; Seaman, 2004), mammalian iron transporter DMT1 (Tabuchi et al., 2010), C. elegans phagocytosis receptor Ced1 (Chen et al., 2010), APP (Vieira et al., 2010), APP processing β1 secretase (Wen et al., 2011), Wntless (Belenkaya et al., 2008; Pan et al., 2008; Yang et al., 2008), β2-adrenergic receptor (Temkin et al., 2011), type 1 receptor for parathyroid hormone (Feinstein et al., 2011), and AMPA-type glutamate receptors (Munsie et al., 2015). Intriguingly, mutations in the VPS35 gene are identified in autosomal dominant PD patients (Vilarino-Guell et al., 2011; Zimprich et al., 2011). VPS35 protein is reduced in the substantia nigra of PD patients (MacLeod et al., 2013) and in the hippocampus of AD patients (Small et al., 2005). These clinical investigations demonstrate VPS35/retromer abnormality in both PD and AD, suggesting that VPS35/retromer dysfunction or deficiency may be a risk factor for the pathogenesis of both PD and AD. Whereas the in vivo evidence for VPS35 deficiency in mice to promote AD pathogenesis has been shown (Wen et al., 2011), it lacks evidence for VPS35 deficiency to contribute to PD pathogenesis in mice.
In this paper, we provide evidence that links VPS35 deficiency to PD-relevant neuropathology. Hemizygous deletion of the VPS35 gene results in PD-relevant deficits including accumulation of α-synuclein protein in substantia nigra pars compacta (SNpc)-DA neurons; reduction of DA transmitter, TH protein, and DA neurons in SNpc and STR (striatum); and impairment of locomotor behavior. We also found that VPS35-deficient DA neurons exhibit alterations in lysosome morphology with enlarged lysosome-associated membrane glycoprotein 1 (Lamp1+), but reduced Lamp2a+ vesicles. The Lamp2a reduction was associated with the α-synuclein increase in VPS35-deficient DA neurons. Expression of Lamp2a into VPS35-deficient DA neurons reduced α-synuclein, supporting the view for Lamp2a as a receptor of chaperone-mediated autophagy (CMA) essential for α-synuclein degradation. Intriguingly, VPS35/retromer deficiency or mutation (D620N) impaired the endosome-to-Golgi retrieval of Lamp2a, thus accelerating its lysosomal degradation. These results suggest that VPS35 is a critical regulator of Lamp2a in DA neurons, which may underlie VPS35 deficiency-promoted PD pathology.
Materials and Methods
Animals and behavior tests.
VPS35 mutant mice have been described previously (Wen et al., 2011). In brief, the mutant mice were generated by injection of mutant ES cells obtained from Bay Genomics and backcrossed with C57BL/6 mice for >10 generations. The homozygotes (VPS35−/−, also called VPS35m/m) die early during embryonic development, but heterozygotes (VPS35+/− or VPS35+/m) have a normal life span (Wen et al., 2011). Mice were housed in a room with a 12 h light/dark cycle with water and rodent chow diet. Control mice were VPS35+/+ littermates for each experiment. All experimental procedures were approved by the Animal Subjects Committee at the Georgia Regents University according to NIH guidelines.
Behavioral tests were conducted during the active phase with 2-, 12-, and 18-month-old mice. Mice were transferred to the testing room in advance and were allowed at least 1 h to acclimate. The open-field test was performed in an arena of 50 × 50 cm at 20 cm of height. Mice, placed in the center of the open field, were allowed to explore the arena undisturbed for 10 min after which they were removed. The arena was cleaned with 70% ethanol between each animal. Video analysis and data acquisition were obtained with Noldus tracking software (EthoVision XT, 7.0) to analyze total distance and mean velocity. Rearing frequency was scored manually by an investigator blind to genotype. To evaluate sensorimotor coordination, mice were placed on an accelerating rotarod (Rotor-Rod System; San Diego Instruments) and assessed for ability to maintain balance on the rotating bar that accelerated from 4 to 40 rpm over a 5 min period. Mouse latency to fall from the rod was recorded and mean time spent on the rotarod over a period of three successive trails was scored. To evaluate a mouse's motor function and muscle strength, the mouse was placed on its forelimbs on a tension bar and pulled back gently until it released the bar. Grip strength was determined by a digital grip-strength meter (Animal Grip Strength System; San Diego Instruments).
Cloning, RNAi, and mutagenesis.
The VPS35 cDNA was subcloned into mammalian expression construct (pEGFP, in which GFP is fused with the VPS35-C terminus). The VPS35 mutant plasmids were constructed through site-directed mutagenesis to convert Asp620 to Asn by the method described previously (Xie et al., 2005). The miRNA-VPS35-EmGFP expression vector was described previously (Wen et al., 2011; Wang et al., 2012; Xia et al., 2013). The miRNA-VPS35-eBFP was constructed by replacing the EmGFP with the eBFP sequence from pBad-eBFP2 (Addgene). Lamp2a mammalian expression construct was obtained from DNASU. All the constructs were confirmed by DNA sequencing and Western analysis.
Cell culture and transfection.
NLT cells were cultured at 37°C in DMEM (Hyclone) supplemented with 5% FBS (Invitrogen) and 100 U/ml penicillin G and streptomycin (Invitrogen). For transient transfection, indicated cells were transfected using polyethylenimine (PEI; Sigma-Aldrich) as described previously (Yin et al., 2013). In brief, cells were cultured in 6-well plates at 70% confluence and were incubated with precipitates formed by 3 μg of plasmid DNA and 1 μl of PEI (5 mg/ml). After 8–12 h of incubation with PEI/DNA polyplex, cells were replaced with the fresh serum-containing medium. At 72 h after initial transfection, cells were fixed with 4% PFA for immunofluorescence staining analysis or lysed with SDS-loading buffer for immunoblotting analysis.
Dopaminergic neuron culture and transfection.
For the midbrain DA neuronal culture, the ventral midbrain was dissected from P0 VPS35+/+ or VPS35+/− mice and placed in prechilled HBSS. The brain pieces were then digested in papain (28 U/ml) for 25 min at 35°C under humidified oxygen chamber. Cells were dissociated, washed twice, resuspended in the plating medium (60% Neurobasal-A, 30% Basal Media Eagle, 10% FBS), and plated onto polyornithine-coated coverslips at a density of 130,000 cells/cm2. The growth media supplemented with 1× B27, and 2 mm GlutaMAX-1 and 10 ng/ml glial cell line-derived neurotrophic factor (Sigma) were replaced 4 h later. Approximately 40% DA neurons were obtained in this culture based on TH-staining analysis. At DIV 5 of culture, neurons were transfected with the indicated plasmids by calcium phosphate-mediated gene transfer, as described previously (Mani and Ryan, 2009). In brief, 3 μg of plasmid DNA was mixed with calcium chloride and then added into the phosphate buffer, pH 7.04, at room temperature. This step generates a precipitate that is dispersed onto the cultured neurons (24-well plate). After 90 min of incubation with precipitates, neurons were replaced with the fresh medium and cultured for another 2 d for further analysis. We were able to get ∼10% of DA neurons expressing the plasmid of interest.
β-gal detection, immunofluorescence staining, and confocal imaging analyses.
VPS35+/+ or VPS35+/− mice were killed and perfused with 2% glutaraldehyde and 2% formaldehyde immediately. Brain sections (45 μm) cut by the Leica vibratome cutting system were mounted onto Superfrost Plus slides (Fisher Scientific). For β-gal activity detection, the slides were fixed with 0.5% glutaraldehyde and incubated with X-gal solution (2 mm MgCl2, 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, and 0.1% X-gal) in the dark at 37°C for 12 h. The slides were washed, mounted in Permount (Thermo Fisher Scientific), and imaged using microscope with a high-sensitivity camera and equipped with a Plan-Neofluar 5× and Plan-Neofluar 10×/0.30 NA objective lens (Axioplan 2; Carl Zeiss).
For immunofluorescence staining analysis, sections or cells on coverslips were fixed with 4% PFA and 4% sucrose at room temperature for 15 min, permeabilized with 0.2% saponin, and blocked with 10% donkey serum and 0.1% saponin. Antibodies used were mouse anti-TH (Sigma; 1:10,000), mouse anti-NeuN (Millipore; 1:2000), rat anti-Lamp1 [Developmental Studies Hybridoma Bank (DHSB); 1:1000], rat anti-Lamp2 (DHSB; 1:1000), rabbit anti-Lamp2a (Abcam; 1:500), rabbit anti-VPS35 (1:1000), rabbit anti-α-synuclein (Cell Signaling Technology; 1:100), rabbit anti-EEA1 (Cell Signaling Technology; 1:500), and rabbit anti-Golgin-97 (Abcam; 1:500). Sections or cells were incubated with appropriate fluorescent secondary antibodies (Jackson ImmunoResearch) at room temperature for 1 h. Slides were mounted using Vectashield mounting medium with and without DAPI. Confocal images were obtained using a Zeiss LSM 510 oil-immersion 40× or 63× objective with sequential-acquisition setting. The acquired 8-bit RGB images were exported to TIFF files with the LSM 510 viewer software. Photoshop software CC (Adobe) was used to adjust the brightness. Figures were constructed in Illustrator version CC (Adobe). For fluorescent quantification, morphometric measurements of images were performed using Image-Pro Plus software (MediaCybernetics).
Stereological neuron counts.
The unbiased stereological estimation of the total number of TH+, NeuN+, or Nissl+ cells in midbrain was used as previously described (Yin et al., 2013). In brief, serial coronal sections across the midbrain (every fourth section from the bregma −2.92 to −4.16 mm) were stereologically counted by using Stereo Investigator software. Investigators were blinded to the genotype of each mouse.
Western blotting analysis.
Cells and tissue were lysed/homogenized in RIPA buffer (50 mm Tris, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% SDS, and 0.5% sodium deoxycholate) supplemented with protease inhibitors (Roche Applied Science). After a 20 min incubation on ice, protein extracts were clarified by centrifugation at 12,000 × g for 20 min at 4°C and protein concentrations were determined by BCA Protein Assay Kit (Pierce Biotechnology). For Western blots analysis, 20 μg of lysate per lane was separated by Bis-Tris SDS-PAGE gel. After transfer to nitrocellulose membranes, the membranes were immunoblotted with the following antibodies: rabbit anti-VPS35 (1:10,000; gift from Dr. K.-W. Kim, Columbia University, New York), rabbit anti-VPS26 (Abcam; 1:1000), mouse anti-TH (Sigma; 1:40,000), mouse anti-β-actin (Sigma; 1:10,000), mouse anti α-synuclein (BD Bio; 1:4000), rabbit anti-phosphor (S129)-α-synuclein (Abcam; 1:2000), rabbit anti-Lamp1 (Millipore; 1:2000), rat anti-Lamp2 (DHSB; 1:4000), rabbit anti-EEA1 (Cell Signaling Technology;1:5000), rabbit anti-Lamp2a (Abcam; 1:5000), rabbit anti-Lamp2b (Abcam; 1:1000), rabbit anti-LC3 (Abcam; 1:2000), mouse anti p62/SQSTM1 (Novus Biologicals; 1:5000), and rabbit anti-GFP (Life Technologies; 1:5000). For semiquantitative analysis, protein bands were detected by the Odyssey Infrared Imaging System (Li-Cor) and analyzed using ImageJ software.
Soluble and insoluble brain fractions for α-synuclein protein analysis.
Brain tissues were homogenized with ice-cold Triton X-100 lysis buffer (175 mm NaCl, 30 mm Tris, 5 mm EDTA, and 1% Triton X-100, plus protease and phosphatase inhibitor cocktails) and incubated on ice for 25 min. After sonication, the brain lysate was centrifuged at 14,000 g for 15 min and the supernatant from this step was the “TX-100 soluble fraction.” The remaining pellet was extracted in 2% SDS and 8 m urea and present as “TX-100 insoluble, SDS-urea soluble fraction.”
Monoamine analysis in tissue homogenates.
Striatum and ventral midbrain were isolated to detect levels of DA and its metabolite by HPLC with electrochemical detection (Eicom) according to the manufacturer's instructions. In brief, tissues were weighed and then homogenized in 0.2 m ice-cold perchloric acid (0.5 ml/100 mg wet tissue) and incubated on ice bath for 60 min. One hundred nanograms of isoproterenol (ISO) per 100 mg of wet tissue was added as an internal control. After centrifuging at 20,000 g at 4°C for 20 min, the supernatant was filtered with a 0.44 μm Millipore filter and injected into the HPLC column (Eicompak SC3ODS; 3.0* 100 mm) for analysis. The mobile phase contained 110 mm citrate buffer/100 mm EDTA/70 mm sodium octane sulfonate and 20% (v/v) methanol. Flow rate was at 340 μl/min.
Real-time analysis of mRNA expression.
The total RNA was isolated with the RNeasy Plus Mini Kit (Qiagen) and 5 μg of RNA was reverse transcribed into cDNA using Maxima First Strand cDNA Synthesis Kit (Life Technologies) according to the manufacturer's directions. The SYBR Green Real-Time PCR method was used to compare gene expression. The primers for Vps35 were 5′-CAGCTGGCTGCTATCACCTT and 5′-TCTTCCCCATTTTTGTCTGTG; for Lamp1 5′-TGCCAGCTCTAGCCTGTTTT and 5′-CTGGACCTGCACACTGAAGA; for Lamp2a 5′-TTGGCTAATGGCTCAGCTTT and 5′-ATGGGCACAAGGAAGTTGTC; for Lamp2b 5′-TTGGCTAATGGCTCAGCTTT and 5′-GTCAGCAGCACATTCCTCAG; and for GAPDH 5′-CACCAACTGCTTAGC and 5′-GCATGGACTGTGGTCATGAG.
The rAAV5 virus production and injection.
The full length of mouse VPS35-D620N cDNA was subcloned into the pAAV-cDNA-EGFP-T2A-V5 vector (in which the GFP is not fused with the VPS35-D620N). We chose AAV serotype 5 for the virus transduction because it provides a better efficiency than other AAV serotypes, based on a previous publication (McFarland et al., 2009) and our pilot studies. The AAV5 of VPS35-D620N was generated by the core facility at Emory University, according to a previous publication (Huang et al., 2013). The AAV5 viruses were purified by using the iodixanol gradient centrifugation procedure, and the titer of the viruses was determined by real-time PCR. The viruses were diluted to a final concentration of 1 × 1012 vg/ml, and the aliquots (10 μl each) were kept at −80°C.
Stereotaxic injections were performed under ketamine/xylazine anesthesia (100 mg/kg and 12 mg/kg, i.p.) using a 5 μl Hamilton syringe with a 33 gauge blunt-tip needle. The anesthetic male C57BL/6 mouse was placed into a stereotactic device (Kopf Instruments) and the skull was exposed. The injection coordinates were calculated from bregma with AP −2.8 mm, lateral −1.3 mm, and ventrodorsal (VD) −4.2. Subsequently, the needle was lowered into the brain 0.1 mm below the VD coordinate and retracted back up to the correct level. A total of 1 μl of virus was loaded for each mouse at the speed of ∼0.2 μl/min. After the injection, the needle was left in place for 5 min before retraction. Mice were killed at 2 weeks post surgery.
Statistics.
Statistical analyses were performed by using Prism 5 (GraphPad Software) using Student's t test and one-way ANOVA. All values were expressed as mean ± SEM. The test was considered significant when p < 0.05. For all analyses, the following apply: *p < 0.05; ns, not significant p > 0.05. Normally distributed data were analyzed by ANOVA followed by Bonferroni or Fisher's LSD post hoc tests as necessary. Student's t test was used to compare pairs of means.
Results
Expression of VPS35 in mouse DA neurons in SNpc and STR
To understand how VPS35 is involved in PD pathogenesis, we first asked whether VPS35 is expressed in PD-vulnerable brain regions or DA neurons in mouse. To this end, we stained for β-gal activity in VPS35 heterozygous mice (VPS35+/−), where the lacZ gene is “knocked-in” into the VPS35 gene and could serve as a reporter of the expression of endogenous VPS35 (Wen et al., 2011; Xia et al., 2013; Liu et al., 2014). Indeed, β-gal activity was detected in the SNpc and STR—two regions associated with PD (Fig. 1A). The activity in the SNpc was age dependent, detectable at P10, peaking in adult age (P70), and remaining positive in 1-year-old or older mice (Fig. 1B; data not shown). VPS35 expression in both SNpc and STR was confirmed by Western blot analysis that showed a single band (∼90 kDa) in various brain regions. The band's intensity was reduced to ∼50% in VPS35+/− samples (Fig. 1C), demonstrating the antibody specificity. VPS35 expression was comparable in STR, ventral midbrain (VM), hippocampus (Hip), and cortex (Ctx; Fig. 1C). VPS26, another component of the retromer, was also expressed in the same brain regions and decreased in VPS35+/− mice (Fig. 1C). Immunohistochemical analysis demonstrated that VPS35 costained with TH, a marker for DA neurons, in the soma and processes of SNpc-DA neurons (Fig. 1D, left). Notice that the VPS35 signal was specific because it was diminished in TH+ or DA neurons of VPS35+/− mice (Fig. 1D, right). These results thus demonstrate that VPS35 is expressed in mouse DA neurons.
Figure 1.

Expression of VPS35 in DA neurons. A, Detection of the β-gal activity (blue color) in VPS35+/− SNpc, VTA, red nucleus (RN), STR, and hippocampus CA3 at P45. cc, Corpus callosum. The β-gal activity is used as a reporter for VPS35's expression, because the LacZ gene is inserted in the intron of the VPS35 gene in VPS35+/− mouse. Scale bar, 200 μm. B, Detection of the β-gal activity in VPS35+/− VM from different aged mice (E18.5, P10, P70, and P360). In young adult brain (e.g., P70), β-gal activity reached the peak level in VTA and SNpc, which remained positive in 1-year-old or older mice. Scale bars, 200 μm. C, Western blot analysis of retromer complex VPS35 (∼90 kDa) and VPS26 (∼36 kDa) protein levels in homogenates of Hip, Ctx, VM, and STR of 8-month-old VPS35+/+ and VPS35+/− brains. Note that ∼50% reductions in both VPS35 and VPS26 protein levels were detected in homogenates from VPS35+/− brain samples. D, Coimmunofluorescence labeling of TH (green) and VPS35 (red) in the SNpc brain sections from 2-month-old (2 m) VPS35+/+ and VPS35+/− mice. The VPS35 immunoreactivity is reduced in TH+ neurons from VPS35+/− SNpc (right). Insets marked by yellow dashed rectangles are amplified images. Scale bars: 80 μm; inset, 10 μm. PFC, prefrontal cortex; IB, immunoblot.
Degenerative-like DA neurons in aged VPS35-deficient SNpc and STR
Homozygous VPS35−/− die early during embryonic development, whereas the life span of VPS35+/− is normal (Wen et al., 2011; Xia et al., 2013). To investigate whether VPS35-deficient mice show PD-relevant neuropathology, we analyzed VPS35+/+ and VPS35+/− mice at different ages. In young adult (e.g., 2 months old), VPS35+/+ and VPS35+/− brain sections showed no obvious difference in the number of TH+ neurons and their processes (Fig. 2A–D). However, TH+ somas were fewer with disturbed and decreased fibers/processes in SNpcs of aged (∼12 months or older) mutant mice (Fig. 2A–D). There was a ∼20% (n = 4, p = 0.029) loss of TH+ somas in the SNpcs of mutant mice at 12 months age and the reduction of SNpc neuron number was confirmed by NeuN and Nissl staining analyses (Figs. 2A,B, 3A–C). Consistently, TH protein was lower in aged (12 months or older) VPS35+/− VMs (Fig. 2E,F) and STRs (Figs. 2G,H, 3D,E). Note that neurons in aged VPS35+/− hippocampus appeared to be normal (Fig. 3F,G). These results reveal an age-dependent and brain region-specific reduction of TH proteins and DA neurons in VPS35+/− mice.
Figure 2.
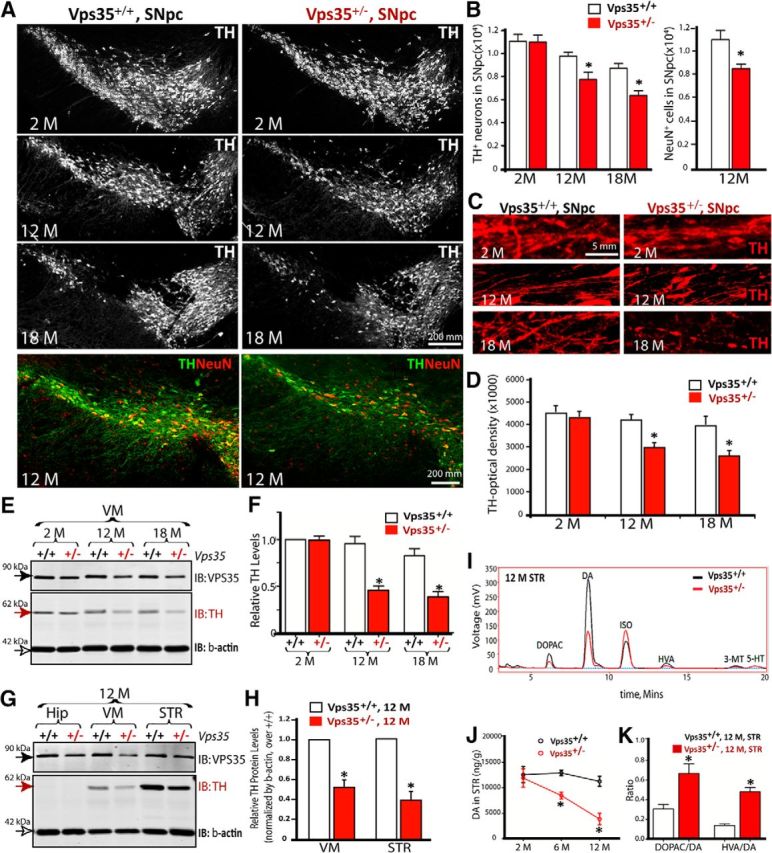
Progressive degeneration of DA neurons in aged VPS35+/− mice. A, Reduced number of TH+ neurons in VPS35+/− SNpc. Shown were representative images of midbrain coronal sections of age-matched VPS35+/+ and VPS35+/− littermates. B, Quantification of data in A. Data were quantified by stereological analysis and shown as mean ± SEM (n = 4; *p <0.05). C, D, Degenerated processes of SNpc TH+ neurons. Representative images were shown in C; quantification of TH optical density were presented in D as mean ± SEM (n = 4; *p <0.05). E, F, TH reduction in VPS35+/− VM. VM homogenates of indicted mice were subjected to Western analysis for TH and VPS35. Representative blots were shown in E. Quantification of TH levels (normalized by 2-M VPS35+/+ control) was presented in F. G, H, Western blot analysis of TH and VPS35 protein levels in homogenates of the Hip, VM, and STR of 12-month-old VPS35+/+ and VPS35+/− mice. Quantification analysis of the relative TH levels (normalized by the VPS35+/+ controls) was presented in H. F, H, Data presented were mean ± SEM from three independent experiments; *p < 0.01. I–K, HPLC analysis of DA, its metabolite DOPAC, and HVA in the STR of indicated age groups of VPS35+/+ and VPS35+/− mice (n = 4 for each genotype). ISO is included as an internal control. Quantification analysis of DA levels in STR was presented in J. The ratios of DOPAC/DA and HVA/DA were presented in K. Data presented in J and K were mean ± SEM; *p < 0.01. IB, immunoblot.
Figure 3.

Decreased DA neurons and TH+ fibers in aged VPS35+/− SNpc and STR. A, Immunostaining indicating reduced TH+ neurons and neuronal processes in 12-month-old Vps35+/− SNpc. B, C, Nissl staining indicating decreased neurons in 12-month-old Vps35+/− SNpc. B, Representative images. C, Stereological analysis of cell numbers in SNpc. Data were presented as mean ± SEM (n = 5; *p < 0.05). D, E, Immunostaining showing reduced TH+ neuronal processes in 12-month-old Vps35+/− STR. D, Representative images. E, Quantification of TH optical density (mean ± SEM; n = 4; *p < 0.05). F, G, Immunostaining indicating normal NeuN+ neuronal density in 12-month-old VPS35+/− Hip. F, Representative images. G, Quantification of densities of CA1 neurons (mean ± SEM; n = 4; *p < 0.05).
To determine whether the activity of DA neurons or TH was altered, we measured DA and its metabolites by HPLC in STR and VM regions in VPS35+/+ and VPS35+/− littermates. DA levels were significantly reduced in 6 months (34.4%, n = 3, p = 0.0068) or older (65.7%, n = 3, p = 0.0085) mutant STRs and VMs (Fig. 2I,J; data not shown). The ratios of 3,4-dihydroxyphenylacetic acid (DOPAC)/DA or 3,4-dihydroxyphenylacetic acid (HVA)/DA were much higher in 12-month-old VPS35+/− STRs, compared with that in age-matched VPS35+/+ controls (Fig. 2K). DA reduction was specific because no change in 5-HT levels was detected between VPS35+/− and controls (data not shown). These results suggest that young mutant mice may have functional deficit of DA neurons or decrease of DA neurotransmitter in advance of DA neurodegeneration in aged mice.
Accumulation of α-synuclein in VPS35-deficient SNpc-DA neurons
Another pathological feature of PD is accumulation of α-synuclein in the Lewy body (von Coelln et al., 2006). We thus compared α-synuclein levels in VPS35+/+ and VPS35+/− VMs (Fig. 4). α-Synuclein exhibits monomeric (∼18 kDa), intermediate (20–75 kDa), and oligomeric (75–150 kDa) species (Fig. 4A,C). Interestingly, both monomeric and oligomeric, or phosphorylated and unphosphorylated, species of α-synuclein were increased in VPS35+/− VMs (Fig. 4A–D). The increase was age dependent, as it was detectable in 6-month-old and prominent in aged (18-month-old) mutant mice (Fig. 4A–D). The increase was detected only in VM, but not in the Hip, even in aged mutant mice (Fig. 4E,F). Immunostaining analysis showed an age-dependent increase in intracellular α-synuclein signals in TH+ neurons in SNpcs (Fig. 4G,H).These results demonstrate another PD-relevant neuropathological feature in aged VPS35+/− brain.
Figure 4.

Accumulation of α-synuclein proteins in VPS35+/− SNpc-DA neurons. A–D, Western blot analysis of soluble and insoluble α-synuclein levels from VM of various aged (6-, 12-, and 18-month-old) VPS35+/+ and VPS35+/− mice. TX-100 soluble (A, B) and insoluble (SDS/Urea soluble; C, D) fractions were isolated and analyzed via Western blotting using indicated antibodies. VPS35 was used to confirm VPS35+/− mice, and β-actin was used as a loading control. Data were quantified and presented in B and D as mean ± SEM; n = 3; *p < 0.01. E, F, Western blot analysis of soluble homogenates of α-synuclein levels from various brain regions (VM, STR, and Hip) of 12-month-old VPS35+/+ and VPS35+/− mice. Data in E were quantified and presented in F as mean ± SEM; n = 3; *p < 0.01. G, H, Coimmunofluorescence staining analyses of α-synuclein and TH in SNpcs from 6-, 12-, and 18-month-old VPS35+/+ and VPS35+/− mice. Coronal midbrain sections from VPS35+/+ and VPS35+/− mice at indicated ages were stained with α-synuclein and TH antibodies. Asterisk, Indicates the cell with somatic accumulation of α-synuclein. Scale bar, 10 μm. Quantitative analysis of the DA (TH+) neurons with somatic accumulation of α-synuclein in VMs at indicated ages was presented in H (mean ± SEM; n = 3; *p < 0.01). IB, immunoblot.
Impairment of locomotor activity in aged VPS35-deficient mice
Given that PD patients usually have impaired locomotion activity, we examined motor functions of VPS35+/− mice. No obvious difference was detected between the two groups in young adult age (data not shown). Even in aged animals, no difference was observed in the rotarod and gait tests (Fig. 5A,B). However, in the open-field test, total distance and velocity of 12- and 18-month-old VPS35+/− mice were reduced, compared with WT controls (Fig. 5C,D). The rearing frequency (vertical movement) of aged mutant mice was notably decreased (35.6% decreased in 12-month-old and 82.2% decreased in 18-month-old mutant mice; n = 4–5, p < 0.01; Fig. 5E). Notice that the movement reduction occurred in both central and periphery areas of the arena (Fig. 5F) and the amount of time mice spent in the central versus periphery area was similar between two genotypes (data not shown), suggesting no anxiety-related deficit. These results indicate that aged VPS35+/− mice exhibited reduced locomotor activity.
Figure 5.
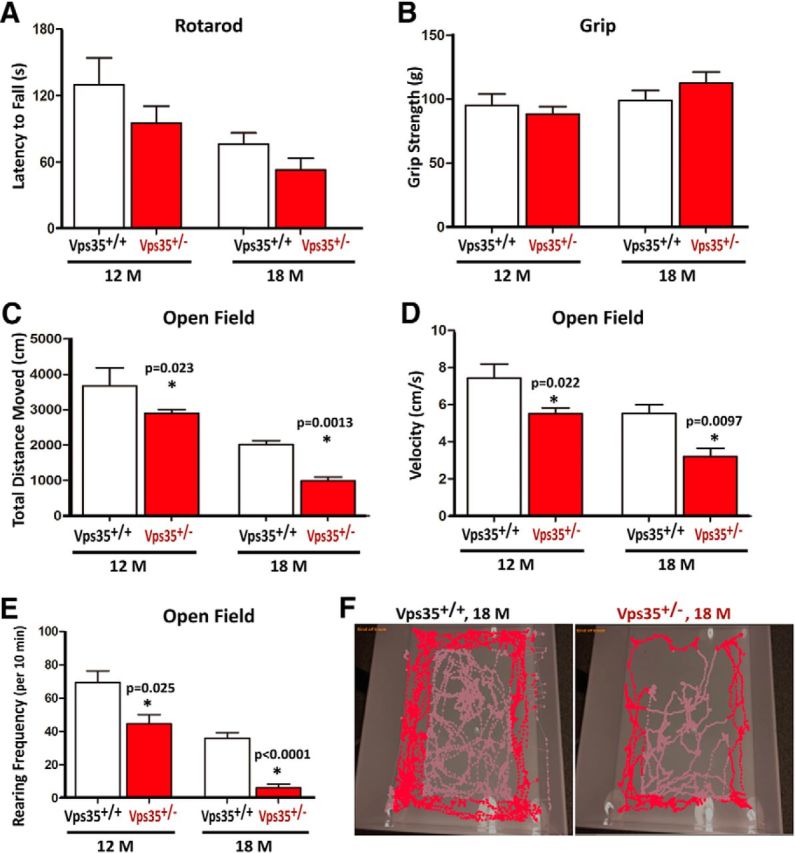
Intact motor coordination, but attenuated exploratory motility, in aged VPS35+/− mice. A, B, Motor coordination and grip performance were determined by rotarod test (A) and grip strength measurement (B) in 12- and 18-month-old VPS35+/+ and VPS35+/− mice. Rotarod latency to fall in A was quantified by the accelerating rotarod test. Forelimb grip strength in B was determined by a digital weight gauge. C–E, Open-field exploratory behavior was attenuated in VPS35+/− mice. Total distance traveled (C), mean velocity (D), and rearing frequency (E) in open-field test were decreased in VPS35+/− mice as compared with that of controls. F, Path of open-field exploration. A–E, N = 5 (for 18-month-old) and 12–14 for (12-month-old) VPS35+/+ and VPS35+/− mice. Data were presented as mean ± SEM; *p < 0.05.
Reduction of Lamp2, but not Lamp1, in VPS35-deficient DA neurons in culture
To understand how VPS35 deficiency contributes to PD-like neuropathology, we examined neuronal morphology in primary cultured DA neurons from neonatal VPS35+/+ and VPS35+/− midbrains. Immunostaining analysis using antibodies against TH or Tuj1 (to label neuronal processes) showed no obvious defect in DA neuronal processes derived from the VPS35+/− mouse (Fig. 6A; data not shown). We then examined subcellular organelles by coimmunostaining analyses. A relatively normal morphology of EEA1-labeled early endosomes and GRASP-marked trans-Golgi network (TGN) was detected in VPS35+/− DA neurons (Fig. 6A–F). However, the Lamp1+ late endosomes/early lysosomes appeared enlarged in the mutant DA neurons (Fig. 6E,G), suggesting that the late endosome to lysosome pathway may be altered because of VPS35 deficiency. We further examined Lamp2-associated lysosomes in VPS35+/+ and VPS35+/− DA neurons. In contrast to Lamp1, the Lamp2+ vesicles appeared smaller in size with reduced intensity in VPS35+/− DA neurons, compared withVPS35+/+ controls (Fig. 6H,I). These results suggest that VPS35 in DA neurons may be required for morphogenesis of lysosomes, possibly by regulating Lamp1 and/or Lamp2.
Figure 6.

Reduced Lamp2+ and enlarged Lamp1+ vesicles in primary cultured VPS35+/− DA neurons. A–C, EEA1+ early endosomes in TH+ DA neurons from VPS35+/+ and VPS35+/− neonatal (P0) midbrains (DIV 7). Representative confocal images were shown in A, and quantification analyses of EEA1+ vesicle size and distribution (in soma vs dendrite) were presented in B and C, respectively (n = 100 vesicles from 6 different cells of each group). D, F, GRASP+ Golgi structure in TH+ DA neurons from VPS35+/+ and VPS35+/− neonatal midbrains. Representative images were shown in D, and quantification analysis of GRASP+ Golgi size was presented in F. Mean ± SEM, n = 6 neurons. E, G, Lamp1+ late endosomes/early lysosomes in TH+ DA neurons from VPS35+/+ and VPS35+/− neonatal midbrains. Representative images were shown in E, and quantification analysis of Lamp1+ vesicle size was presented in G (n = 100 vesicles from 6 different cells of each group); *p < 0.05. H, I, Lamp2+ lysosomes in TH+ DA neurons from VPS35+/+ and VPS35+/− neonatal midbrains. Representative images were shown in H, and quantification analysis of Lamp2+ vesicle size was presented in I (n = 100 vesicles from 6 different cells of each group); *p < 0.05.
Reduction of Lamp2a accompanied with accumulation of α-synuclein in VPS35-deficient SNpc-DA neurons
Lamp2a is known to be critical for CMA-mediated α-synuclein degradation (Vogiatzi et al., 2008; Xilouri et al., 2013a; Murphy et al., 2015). We thus asked whether Lamp2/Lamp2a is reduced in VPS35+/− brain in vivo, and if so, whether Lamp2a reduction is associated with the α-synuclein increase in VPS35+/− DA neurons. Homogenates were prepared from the VM region of VPS35+/+ and VPS35+/− mice at different ages and subjected to Western blotting. As shown in Figure 7A, Lamp2-reduction was detected in the mutant VM at 6 months of age by using the antibody against the N terminus of Lamp2 (Fig. 7A,B). Lamp2 has three isoforms (a–c) with different C termini, and only Lamp2a is associated with CMA-mediated α-synuclein degradation (Xilouri et al., 2013a; Murphy et al., 2015). We thus assessed whether Lamp2a or Lamp2b is reduced in VPS35+/−. By using specific antibodies against the C terminus of Lamp2a/Lamp2b, we found that Lamp2a, but not Lamp2b, was reduced in VPS35+/− VM (Fig. 7A,B). Note that no obvious change in protein levels of Lamp1 and EEA1 was detected even in aged mutant mice (Fig. 7A,B), providing additional evidence for the specificity of the Lamp2a reduction. Moreover, as the α-synuclein increases in Vps35+/− mice, the Lamp2a reduction was detected in the mutant VM at 6 months of age, but it was not detected in VPS35+/− Hip or STR, even at 12 months of age (Fig. 7A,B). These results suggest a temporal and brain-regional association between Lamp2a reduction and the α-synuclein increase in VPS35+/− mice. We next asked whether the Lamp2a reduction is also associated with the α-synuclein increase in VPS35+/− DA neurons. Indeed, immunostaining analysis showed that Lamp2a was reduced in VPS35+/− SNpc-DA neurons (marked by TH), which was accompanied with the increase of α-synuclein (Fig. 7C,D). Finally, we examined if the Lamp2a reduction is responsible for the increase of α-synuclein. VPS35-deficient DA neurons were transfected with the plasmid encoding Lamp2a. Expressing exogenous Lamp2a indeed abolished the α-synuclein accumulation (Fig. 7E,F). These results suggest that VPS35 deficiency in DA neurons results in a decrease in Lamp2a, which leads to an increase in α-synuclein, supporting the view for Lamp2a, a lysosome-associated transmembrane receptor for CMA, to mediate α-synuclein degradation (Vogiatzi et al., 2008).
Figure 7.
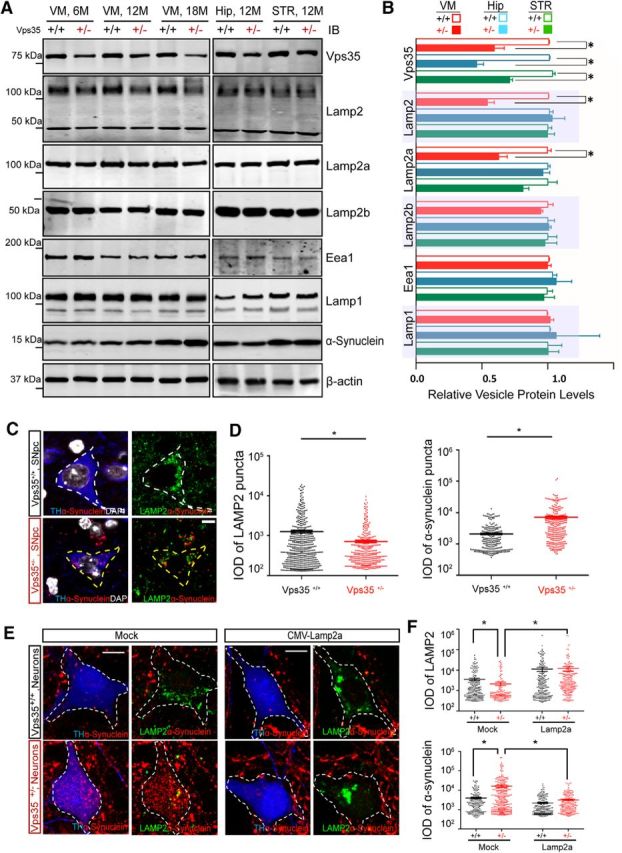
Association of Lamp2 reduction with α-synuclein increase in VPS35-deficient DA neurons. A, B, Reduced Lamp2 in aged VPS35+/− VM, but not VPS35+/− Hip or STR. Soluble extracts from VM, Hip, and STR of indicated mice were subjected to Western bot analysis. A, Representative blots. B, Quantification of protein levels (normalized by VPS35+/+ controls). Data presented were mean ± SEM (n = 3 mice/genotype); *p < 0.05. C, D, Reduced Lamp2 level in VPS35+/− DA neurons. C, Representative images of TH+ DA neurons. D, Quantitation analysis (normalized by VPS35+/+) of Lamp2+ vesicle size and α-synuclein puncta size (mean ± SEM; n = 10 neurons; *p < 0.05). IOD, Integrated optical density. Scale bar, 5 μm. E, F, Reduced α-synuclein in VPS35+/− DA neurons expressing Lamp2a. Primary cultured VPS35+/+ and VPS35+/− DA neurons were transiently transfected with MOCK control and Lamp2a plasmids. The transfected DA neurons were fixed and subjected to coimmunostaining analysis using antibodies against TH (blue), Lamp2 (green), and α-synuclein (red). E, Representative images. F, Quantification analysis Lamp2+ vesicle size and α-synuclein puncta size (mean ± SEM; n = 10 neurons; *p < 0.05). Scale bar, 10 μm.
Accelerated Lamp2a degradation and impaired Lamp2a endosome-to-Golgi retrieval in VPS35-deficient neurons
We then investigated the underlying mechanisms for Lamp2a reduction in VPS35-deficient neurons. The Lamp2a reduction was not caused by a problem in transcription because a comparable level of Lamp2a mRNA was detected between VPS35+/+ and VPS35+/− VM at 12 months of age (Fig. 8A). We next examined if the Lamp2a reduction was from an accelerated Lamp2a degradation. Indeed, the half-life of Lamp2a in VPS35+/− DA neurons was shorter (∼3 h) than that in VPS35+/+ controls (∼8 h; Fig. 8B,C). In contrast, the α-synuclein protein was more stable in VPS35+/− DA neurons than that in VPS35+/+ controls (Fig. 8B,C), providing additional evidence for their reverse relationship. To determine whether the lysosome-degradation pathway is responsible for the Lamp2a reduction, we treated Vps35+/− DA neurons with or without bafilomycin A1 (BafA1; 200 nm for 24 h), an inhibitor of lysosomal acidification and function (Yoshimori et al., 1991). The Lamp2a in Vps35+/− DA neurons was restored to a nearly normal level (Fig. 8D,E), indicating that VPS35 deficiency may result in accelerated lysosome-mediated Lamp2a degradation, leading to the Lamp2a reduction.
Figure 8.
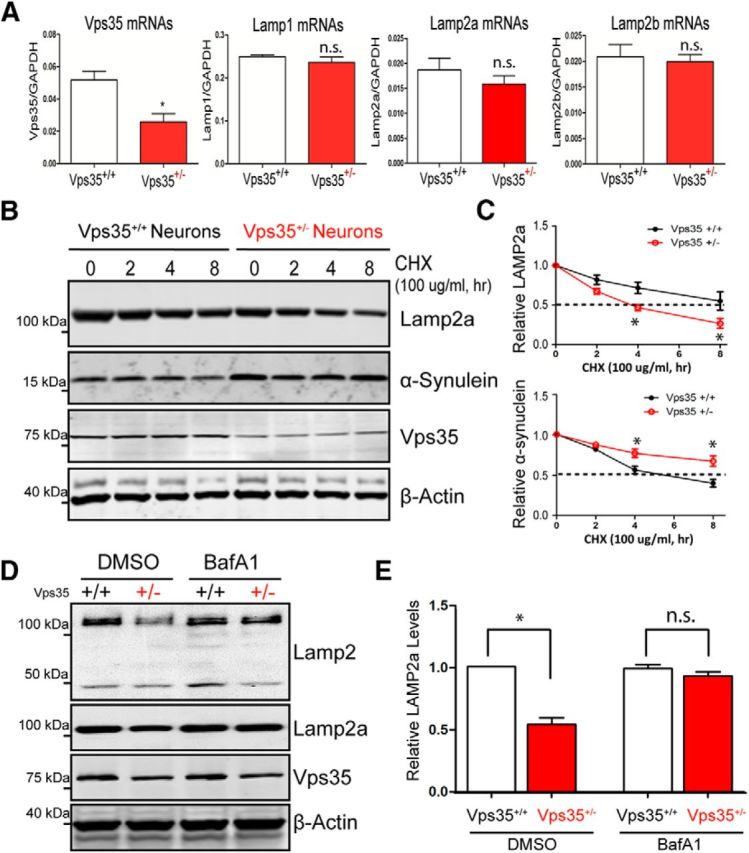
Accelerated lysosomal degradation of Lamp2a in VPS35-deficient DA neurons. A, No difference of Lamp2a mRNA levels was detected between VPS35+/+ and VPS35+/− VM. The mRNAs of 6-month-old mice were analyzed by real-time PCR analysis. The values of mean ± SEM (n = 3 samples /genotype) were presented; *p < 0.05). B, C, Lamp2a protein degradation was accelerated, but α-synuclein degradation was impaired, in VPS35+/− DA neurons. DA neurons were incubated with CHX (100 μg/ml) for indicated times. Cell lysates were subjected to Western blotting analyses using indicated antibodies. B, Representative blots. C, Quantification of Lamp2a and α-synuclein protein levels (normalized with VPS35+/+ at time 0), respectively; *p < 0.05. D, E, Lamp2a reduction was restored in VPS35+/− DA neurons treated with BafA1. VPS35+/+ and VPS35+/− DA neurons were treated with BafA1 (200 nm for 24 h). The resulting lysates were subjected to Western blotting analyses. D, Representative blots. E, Quantification of Lamp2a levels (normalized with VPS35+/+); *p < 0.05.
We further asked whether the increased Lamp2a degradation is from impaired Lamp2a trafficking in VPS35-deficient neurons. To address this issue, Lamp2a subcellular distribution in NLT cells expressing control and miRNA-VPS35 was examined. NLT, a GnRH neuroblastoma cell line, was chosen because both VPS35 and Lamp2a were highly expressed in this cell line (data not shown); NLT cells have a spread morphology with relative large subcellular organelles; and NLT cells expressing miRNA-VPS35 could efficiently (>80%) knock down endogenous VPS35 expression (Wen et al., 2011; Xia et al., 2013). Consistent with the results of VPS35+/− DA neurons (Fig. 6), Lamp2/Lamp2a was reduced, and Lamp1 was increased, in miRNA-VPS35 (VPS35-KD) NLT cells (Fig. 9A,B). Intriguingly, a portion of Lamp2 as well as Lamp1 was colocalized with VPS35 (Fig. 9A,C), and the colocalization of VPS35 with Lamp2, but not Lamp1, was largely distributed in the perinuclear area (Fig. 9A,D), where the Golgi compartment is located in. Indeed, a fraction of Lamp2a was associated with the GM130-marked TGN in control cells, and such a Lamp2a distribution in TGN was abolished in VPS35-KD cells (Fig. 9E,F). Instead, Lamp2a distribution in Lamp1+ late endosomes/early lysosomes was increased in VPS35-KD NLT cells (Fig. 9E,F). The Lamp2-VPS35 colocalization was detected not only in NLT cells, but also in SNpc-DA neurons of WT mouse brain, which was reduced in VPS35+/− SNpc-DA neurons (Fig. 9G,H). These results thus support the hypothesis that Lamp2a may be a cargo of VPS35/retromer, undergoing endosome-to-Golgi retrieval, an event critical to prevent Lamp2a distribution in Lamp1+ lysosomes and to suppress Lamp2a degradation.
Figure 9.
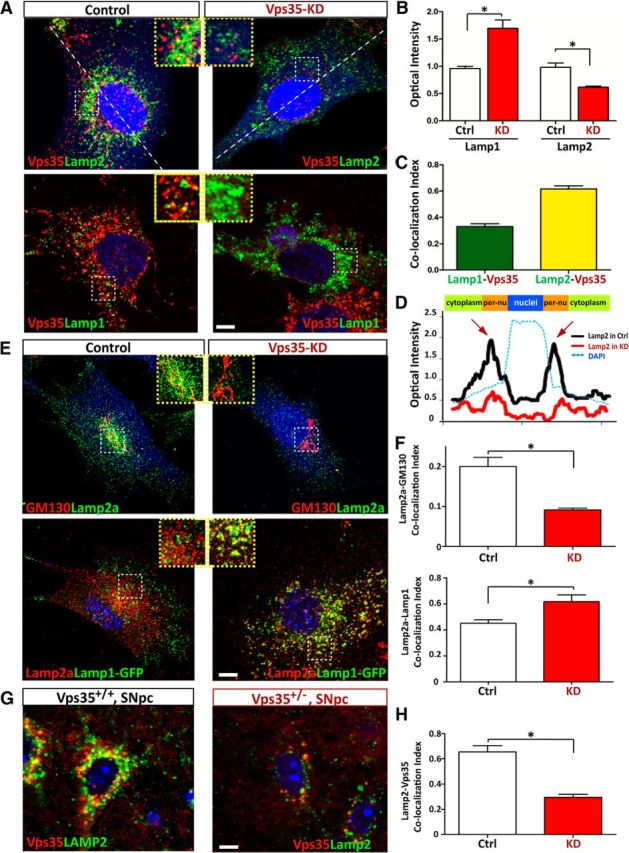
Impaired endosome-to-Golgi retrieval of Lamp2a in VPS35-deficient cells. A–C, Reduction of Lamp2, but increase of Lamp1, in VPS35-deficient NLT cells. NLT cells were transfected with miRNA-VPS35 or scramble control (marked by eBFP) and examined for the distribution of Lamp1/2 and endogenous VPS35. A, Representative images. Scale bar, 10 μm. B, Quantification of Lamp1/2 optical density was presented as mean ± SEM (n = 10; *p <0.05). C, Colocalization index of Lamp1 with VPS35 or Lamp2 with VPS35. D, Altered Lamp2 distribution in VPS35-deficient NLT cells. Line profile intensity of Lamp2a from the dotted line in A was presented. E, F, Coimmunostaining analysis of Lamp2a with endogenous GM130 and exogenous Lamp1-GFP in NLT cells transfected with control or miRNA-VPS35. E, Representative images. Scale bar, 10 μm. F, The colocalization index of Lamp2a with the indicated markers (GM130 and Lamp1) was determined by the measurement of overlapped signal (yellow fluorescence) over total Lamp2a. Data were shown as mean ± SEM (n = 10); *p < 0.01 compared with control cells. G, H, Decreased Lamp2a in DA neurons from 12-month-old VPS35+/− mice. Middle brain sections were immunostained with VPS35 (red) and Lamp2a (green). Scale bar, 5 μm. G, Representative images. Scale bar, 10 μm. H, Colocalization index of Lamp2 with VPS35. Ctrl, Control; KD, knockdown.
Decreased Lamp2a distribution in Golgi apparatus, reduced Lamp2a, and increased α-synuclein in mouse DA neurons expressing PD-linked VPS35 mutant (D620N)
VPS35-D620N mutation is identified in autosomal dominant PD patients (Vilarino-Guell et al., 2011; Zimprich et al., 2011). We thus determined whether this mutant alters functions of VPS35/retromer, such as endosome-to-Golgi retrieval of membrane proteins. We generated GFP-fused VPS35-D620N plasmids, which had a similar distribution pattern as endogenous VPS35 or exogenous GFP-fused VPS35-WT when expressed in HEK293 cells (Fig. 10A). We first asked whether VPS35-D620N alters the distribution of CI-M6PR, which is a well characterized retromer/VPS35 cargo and regulates lysosome function (Seaman, 2005). Expression of VPS35-D620N in HEK293 cells did not alter the distribution of endogenous CI-M6PR to the TGN compartments (Fig. 10B,D). As control, VPS35 deficiency by its miRNA resulted in the disappearance of CI-M6PR (Fig. 10C,E). However, CI-M6PR's intensity in the TGN was reduced in cells expressing VPS35-D620N compared with that of untransfected cells (Fig. 10B,E). These results suggest that VPS35-D620N may have a weak inhibitory effect on the endosome-to-Golgi retrieval of endogenous CI-M6PR, in line with a previous report (MacLeod et al., 2013).
Figure 10.
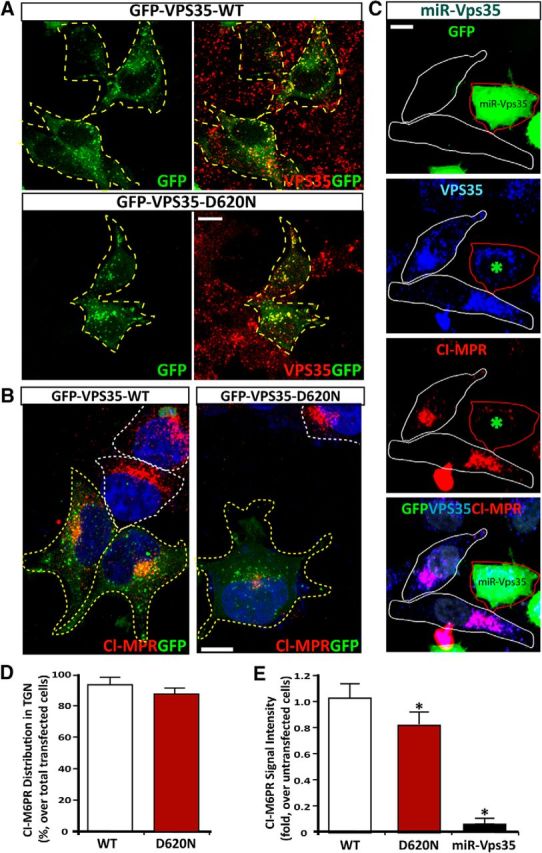
Expression of PD-linked VPS35 mutant (D620N) and its effect on CI-MPR distribution. A, Colocalization of VPS35-WT and D620N mutant (marked by GFP) with endogenous VPS35 (by anti-VPS35 antibodies) in HeLa cells. B, C, CI-MPR distribution in HEK293 (B) and HeLa cells (C) expressing indicated plasmids. Transfected cells were marked by yellow dashed lines, and untransfected/control cells were marked by white dashed lines. Scale bars: B, C, 10 μm. D, E, Quantitative analysis of data in B and C (mean ± SEM; n = 20 cells). CI-M6PR intensity in TGN was quantified (over untransfected cells) and shown in D. Compact juxtanuclear distribution of CI-M6PR is shown in E (*p < 0.05).
Next, we determined whether VPS35-D620N alters Lamp2a distribution in NLT cells. As shown in Figure 11, A and B, the endogenous Lamp2a was partially colocalized with exogenous GFP-VPS35-WT and GFP-VPS35-D620N. However, the colocalization of GFP-VPS35-D620N with Lamp2a was reduced, compared with that of GFP-VPS35-WT with Lamp2a (Fig. 11A,B). Interestingly, as in VPS35-KD cells, Lamp2a distribution in the perinuclear area and the Lamp2a signal were reduced in VPS35-D620N-expressing cells (Fig. 11A,C,D). These results suggest that the D620N mutant may serve as a dominant-negative inhibitor for endosome-to-Golgi retrieval of Lamp2a.
Figure 11.

Impaired endosome-to-Golgi retrieval of Lamp2a in NLT cells expressing VPS35-D620N. Decreased Lamp2a in NLT cells expressing PD-linked VPS35 mutant (D620N). Transfected NLT cells were fixed and immunostained using anti-Lamp2a antibody. A, Representative images. Scale bar, 10 μm. B, Pearson's correlation coefficient of VPS35/VPS35-D620N-GFP with Lamp2a. C, Line profile intensity of Lamp2a from the dotted line in A. D, Quantification of Lamp2a optical density. The values of mean ± SEM (n = 10; *p <0.05) were presented.
To determine whether VPS35-D620N impairs Lamp2a trafficking in vivo, we generated AAV5 encoding VPS35-D620N (with a T2A-GFP), in which the GFP is not fused with VPS35-D620N protein. The AAV5 viruses were stereotactically injected into the VM of 4-month-old WT mice (C57BL/6). Two weeks after injection, mouse SNpcs were examined by immunostaining analyses. GFP was detected in large TH+ DA neurons (Fig. 12A), and VPS35 was elevated because of the exogenous VPS35-D620N expression (Fig. 12B). Interestingly, in VPS35-D620N-expressing neurons, the Lamp2a was reduced, compared with that of uninfected or AAV-GFP-infected control neurons (Fig. 12C,D). Coimmunostaining analysis of Lamp2a with Golgin-97 (a marker for Golgi) also showed a slight reduction of Lamp2a in the Golgi compartment of D620N-expressing DA neurons, compared with that of uninfected DA neurons (Fig. 12E,F). Western blot analysis provided additional support for the Lamp2a reduction and α-synuclein increase selectively in VPS35-D620N-expressing VM (Fig. 12G,H). These results, in agreement with the data in VPS35-D620N-expressing NLT cells, suggest that D620N mutation, acting as VPS35 deficiency, impairs endosome-to-Golgi retrieval of Lamp2a and decreases Lamp2a protein.
Figure 12.
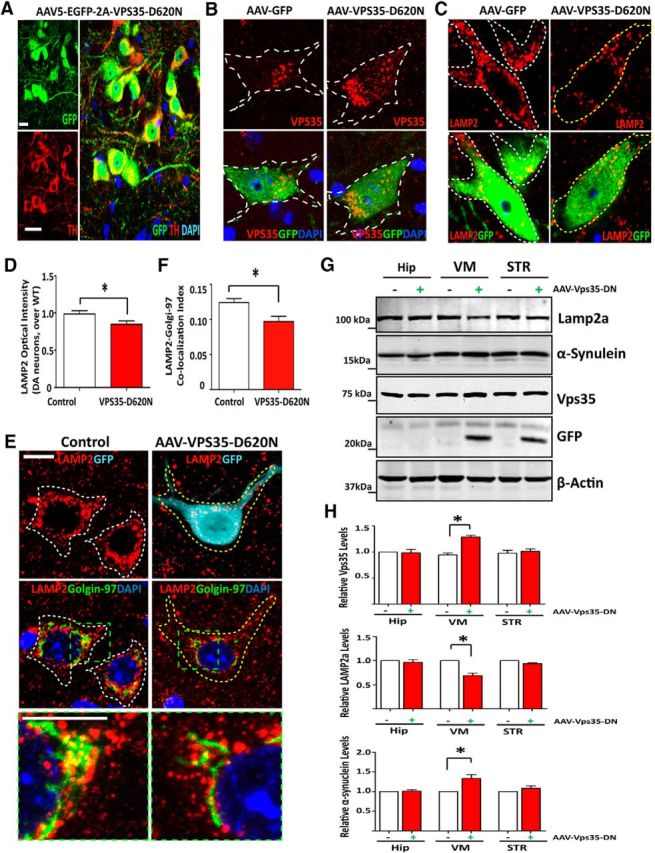
Reduced Lamp2a and impaired Lamp2 endosome-to-Golgi retrieval in mouse SNpc-DA neurons infected with AAV of EGFP-2A-VPS35-D620N. A, The AAV5 infected GFP-positive cells in the SNpc region were colocalized with TH-positive dopamine neurons. Sections were stained GFP (green) and TH (red). Scale bar, 10 μm. B, GFP-infected cells expressing more VPS35 punctae (red) compared with the uninfected cells. Sections were coimmunostained GFP (turquoise) with VPS35 (red) and TH (green). Scale bar, 10 μm. C, D, Lamp2 was reduced in AAV5-VPS35-D620N-infected DA neurons by immunostaining analysis. C, Representative images were from the injected SNpc. Scale bar, 10 μm. D, Quantification analysis of Lamp2 optical density was presented as mean ± SEM (n = 10; *p < 0.05). E, F, Lamp2 distribution was altered in AAV5-VPS35-D620N-infected DA neurons. E, Coimmunostaining analysis of Lamp2 with endogenous Golgi marker (Golgin-97) in TH+ DA neurons with/without AAV-GFP-VPS35-D620N infection. Scale bar, 10 μm. F, Quantification analysis of the colocalization index of Lamp2 with Golgin-97 in D. Data were shown as mean ± SEM (n = 10); *p < 0.01 compared with uninfection side. G, H, Reduced LAMP2a and increased α-synuclein were detected in AAV5-VPS35-D620N-infected VM by Western blot analysis. Soluble extracts from Hip, VM, and STR from the virus-injected (+) and contralateral (−) hemispheres were subjected to Western bot analysis. G, Representative blots. H, Quantification analysis of data in F (normalized by VPS35+/+ controls). Data were presented as mean ± SEM (n = 3 mice/genotype); *p < 0.05, compared with uninfection controls.
Discussion
Major findings of this study are as follows. First, VPS35+/− mice exhibit PD-like deficits, such as α-synuclein accumulation in SNpc-DA neurons, decreased TH proteins and DA transmitter in STRs and SNpcs, dystrophic TH+ neurites/axons, and impaired motor behavior. Second, VPS35 expression in DA neurons is necessary for proper lysosome morphogenesis and function. Third, VPS35 regulates Lamp2a trafficking and prevents its degradation likely by endosome-to-Golgi retrieval of Lamp2a. Fourth, VPS35/retromer-mediated endosome-to-Golgi retrieval of Lamp2a is impaired by the PD-linked VPS35 mutation (D620N). Finally, Lamp2a is critical for α-synuclein degradation, and the impairment in VPS35-Lamp2a-α-synuclein degradation pathway may underlie VPS35 deficiency or mutation-induced PD pathogenesis. Our work establishes a working model (Fig. 13) in which VPS35/retromer deficiency or mutation facilitates PD disease progression in an age-dependent manner, which is likely in part due to the impairment of VPS35/retromer regulation of Lamp2 trafficking and function.
Figure 13.

Illustration of a working model for VPS35 in Lamp2a endosomal-to-Golgi retrieval, where VPS35 assists in preventing Lamp2a degradation, promoting α-synuclein degradation, and suppressing PD pathogenesis.
Taking advantage of the VPS35+/− mouse, we have previously demonstrated that VPS35 haploinsufficiency enhances Aβ-associated neuropathology in the Tg2576 mouse model of AD (Wen et al., 2011). Young VPS35+/− mice also exhibit hyperactivation of osteoclasts and bone loss, features of Paget's disease (Xia et al., 2013). Here, we present evidence that aged VPS35+/− mice display PD-relevant phenotypes, such as age-dependent decrease of TH protein, increase of α-synuclein accumulation, and impairment of motor behavior. These observations support the view for VPS35/retromer deficiency as a risk factor in the pathogenesis of PD, in addition to AD pathogenesis, in line with human clinical investigations (Small et al., 2005; Rogaeva et al., 2007; Vilarino-Guell et al., 2011; Zimprich et al., 2011).
In viewing the PD-relevant phenotypes in VPS35+/− mice, the increase of α-synuclein in SNpc-DA neurons appears to be a prominent early step in comparison with other phenotypes (Figs. 2–4). This observation supports VPS35 deficiency or mutation as a critical mechanism underlying α-synuclein increase. This view is in agreement with reports that α-synuclein mutant identified in PD patients impairs its degradation, resulting in its accumulation (Cuervo et al., 2004), and overexpression of WT α-synuclein is sufficient to cause a PD-like deficit in mice (Lin et al., 2012). While it remains unclear exactly how α-synuclein accumulation promotes PD pathogenesis, multiple cellular deficits are induced by overexpression of α-synuclein in DA neurons, including impairment of DA transmitter release (Perez et al., 2002), mitochondrial fission deficit (Kamp et al., 2010; Lim et al., 2012), and altered autophagosome formation (Winslow et al., 2010). All of these cellular defects may contribute to DA neurodegeneration and PD pathogenesis.
How does VPS35 deficiency result in α-synuclein-accumulation? Our results have pointed to impaired α-synuclein degradation as a prime mechanism. WT α-synuclein is degraded by both pathways: CMA and ubiquitin-proteasomal (Webb et al., 2003). Lamp2a is a receptor of CMA crucial for α-synuclein degradation (Vogiatzi et al., 2008; Mak et al., 2010; Xilouri et al., 2013b). Interestingly, Lamp2a is reduced in VPS35-deficient or D620N-expressing DA neurons in the midbrain (Figs. 6, 12). The Lamp2a reduction is associated with the α-synuclein increase in VPS35-deficient DA neurons (Fig. 7C). Expression of Lamp2a in VPS35-deficient DA neurons abolished α-synuclein accumulation (Fig. 7E,F7). Thus, it is conceivable that the impaired Lamp2a-mediated α-synuclein degradation underlies the increase of α-synuclein protein levels in VPS35-deficient or mutant DA neurons. This view is in line with the report that Lamp2a reduction by its RNAi in culture results in accumulation of both soluble and detergent-insoluble species of α-synuclein (Vogiatzi et al., 2008). While we believe that this is a crucial step for α-synuclein degradation, it may not be the only one. For CMA-mediated α-synuclein degradation, in addition to Lamp2a-mediated recognition of its cargo protein, α-synuclein, it requires lysosome proteases, such as cathepsin D (Qiao et al., 2008). It is of interest to note that VPS35 deficiency also disrupts the trafficking of cathepsin D, though this event is not confirmed in DA neurons yet (Xia et al., 2013; Follett et al., 2014; Miura et al., 2014). We speculate that the α-synuclein increase may result from the dysfunctional CMA and lysosomes in VPS35-deficient DA neurons.
How does VPS35 regulate CMA and lysosome function? As we know, VPS35 is a key component of the retromer that is essential for endosome-to-Golgi retrieval of transmembrane proteins/cargos (Bonifacino and Hurley, 2008; van Weering et al., 2010). A well characterized cargo of VPS35/retromer is CI-M6PR, an M6P receptor crucial for delivery of numerous lysosome proteases, including cathepsin D (Seaman, 2004). Thus, it is possible that VPS35/retromer deficiency or D620N mutation in DA neurons results in a loss of endosome-to-Golgi retrieval of CI-M6PR and an impairment of CI-M6PR-regulated lysosome function. In addition to CI-M6PR, our results suggest that Lamp2a is another cargo of VPS35/retromer in DA neurons. VPSS35 is necessary for endosome-to-Golgi retrieval of Lamp2a and for maintenance of Lamp2a protein level in DA neurons (Figs. 8, 9). This function is impaired by PD-linked VPS35-D620N mutation (Fig. 11). These observations lead to the speculation that by retrieval of both Lamp2a and CI-M6PR lysosome membrane proteins, VPS35/retromer plays a critical role in the maintenance of proper functions of CMA-lysosome degradation pathway. Insights may be gained by future work to further investigate how the VPS35/retromer selectively regulates Lamp2a trafficking in DA neurons and CMA/lysosome functions.
It is noteworthy that dysfunction in CMA has been reported in both familial and sporadic PD (Cuervo and Wong, 2014). The two most commonly mutated proteins in PD patients, α-synuclein and LRRK2, undergo degradation in lysosomes via CMA (Cuervo and Wong, 2014). Mutations in both α-synuclein and LRRK2 proteins increased their binding to Lamp2a, which preclude not only their own degradation, but also inhibit the degradation of other CMA substrates (Martinez-Vicente et al., 2008; Orenstein et al., 2013; Cuervo and Wong, 2014). For example, mutant LRRK2 impairs CMA-mediated clearance of α-synuclein, exacerbating the intracellular accumulation of α-synuclein (Orenstein et al., 2013). The persistence of α-synuclein bound to the lysosomal membrane further compromises CMA activity, forming a vicious cycle (Martinez-Vicente et al., 2008; Cuervo and Wong, 2014). In addition, CMA malfunction is found in PD patients' brain samples (Alvarez-Erviti et al., 2010). A good correlation is detected between brain-regional deficiency in Lamp2a and the selective vulnerability of the brain regions to α-synuclein aggregation (Malkus and Ischiropoulos, 2012; Murphy et al., 2015). Our results presented in this paper are in line with these observations, revealing a link between VPS35 deficiency/mutation with Lamp2a/CMA impairment, and providing additional evidence for dysfunction in CMA to contribute to PD pathogenesis.
While we believe that the impaired CMA-mediated α-synuclein degradation is a crucial mechanism underlying VPS35 deficiency or mutation-associated PD pathogenesis, it does not exclude other possibilities. For example, VPS25-D620N is found to impair glutamatergic neurotransmission likely due to the altered AMPA-type glutamate receptor trafficking (Munsie et al., 2015). VPS35 was recently reported to interact with EIF4G1, and to be involved in protein translation (Dhungel et al., 2015). The VPS35-EIF4G1 complex may be impaired by VPS35 deficiency and/or mutation, which may also contribute to the increase of α-synuclein. It is possible that VPS35 deficiency or mutation leads to deficits in multiple molecular/cellular pathways, and a combination of these deficits promotes PD pathogenesis.
Footnotes
This study was supported in part by grants from the National Institutes of Health (AG045781 to W.C.X.) and the Department of Veterans Affairs (BX00838 to W.C.X.). We also thank Shan Xiong for VPS35 mutant mouse maintenance and genotyping and members of the Xiong and Mei laboratories for helpful discussions.
The authors declare no competing financial interests.
References
- Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010;67:1464–1472. doi: 10.1001/archneurol.2010.198. [DOI] [PubMed] [Google Scholar]
- Belenkaya TY, Wu Y, Tang X, Zhou B, Cheng L, Sharma YV, Yan D, Selva EM, Lin X. The retromer complex influences Wnt secretion by recycling wntless from endosomes to the trans-Golgi network. Dev Cell. 2008;14:120–131. doi: 10.1016/j.devcel.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. 2008;20:427–436. doi: 10.1016/j.ceb.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Xiao H, Zhang K, Wang B, Gao Z, Jian Y, Qi X, Sun J, Miao L, Yang C. Retromer is required for apoptotic cell clearance by phagocytic receptor recycling. Science. 2010;327:1261–1264. doi: 10.1126/science.1184840. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24:92–104. doi: 10.1038/cr.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Dhungel N, Eleuteri S, Li LB, Kramer NJ, Chartron JW, Spencer B, Kosberg K, Fields JA, Stafa K, Adame A, Lashuel H, Frydman J, Shen K, Masliah E, Gitler AD. Parkinson's disease genes VPS35 and EIF4G1 interact genetically and converge on alpha-synuclein. Neuron. 2015;85:76–87. doi: 10.1016/j.neuron.2014.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein TN, Wehbi VL, Ardura JA, Wheeler DS, Ferrandon S, Gardella TJ, Vilardaga JP. Retromer terminates the generation of cAMP by internalized PTH receptors. Nat Chem Biol. 2011;7:278–284. doi: 10.1038/nchembio.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, Tay S, Zhe Y, Wood SA, Mellick GD, Silburn PA, Collins BM, Bugarcic A, Teasdale RD. The Vps35 D620N mutation linked to Parkinson's disease disrupts the cargo sorting function of retromer. Traffic. 2014;15:230–244. doi: 10.1111/tra.12136. [DOI] [PubMed] [Google Scholar]
- Gasser T. Update on the genetics of Parkinson's disease. Mov Disord. 2007;22(Suppl 17):S343–S350. doi: 10.1002/mds.21676. [DOI] [PubMed] [Google Scholar]
- Huang X, Hartley AV, Yin Y, Herskowitz JH, Lah JJ, Ressler KJ. AAV2 production with optimized N/P ratio and PEI-mediated transfection results in low toxicity and high titer for in vitro and in vivo applications. J Virol Methods. 2013;193:270–277. doi: 10.1016/j.jviromet.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, Eimer S, Winklhofer KF, Haass C. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29:3571–3589. doi: 10.1038/emboj.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Dawson VL, Dawson TM. Animal models of Parkinson's disease: vertebrate genetics. Cold Spring Harb Perspect Med. 2012;2:a009324. doi: 10.1101/cshperspect.a009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KL, Ng XH, Grace LG, Yao TP. Mitochondrial dynamics and Parkinson's disease: focus on parkin. Antioxid Redox Signal. 2012;16:935–949. doi: 10.1089/ars.2011.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Sgobio C, Liu G, Yu J, Sun L, Shim H, Gu XL, Luo J, Long CX, Ding J, Mateo Y, Sullivan PH, Wu LG, Goldstein DS, Lovinger D, Cai H. Conditional expression of Parkinson's disease-related mutant alpha-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci. 2012;32:9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Tang FL, Erion J, Xiao H, Ye J, Xiong WC. Vps35 haploinsufficiency results in degenerative-like deficit in mouse retinal ganglion neurons and impairment of optic nerve injury-induced gliosis. Mol Brain. 2014;7:10. doi: 10.1186/1756-6606-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem. 2010;285:13621–13629. doi: 10.1074/jbc.M109.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkus KA, Ischiropoulos H. Regional deficiencies in chaperone-mediated autophagy underlie alpha-synuclein aggregation and neurodegeneration. Neurobiol Dis. 2012;46:732–744. doi: 10.1016/j.nbd.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani M, Ryan TA. Live imaging of synaptic vesicle release and retrieval in dopaminergic neurons. Front Neural Circuits. 2009;3:3. doi: 10.3389/neuro.04.003.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland NR, Lee JS, Hyman BT, McLean PJ. Comparison of transduction efficiency of recombinant AAV serotypes 1, 2, 5, and 8 in the rat nigrostriatal system. J Neurochem. 2009;109:838–845. doi: 10.1111/j.1471-4159.2009.06010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura E, Hasegawa T, Konno M, Suzuki M, Sugeno N, Fujikake N, Geisler S, Tabuchi M, Oshima R, Kikuchi A, Baba T, Wada K, Nagai Y, Takeda A, Aoki M. VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson's disease. Neurobiol Dis. 2014;71:1–13. doi: 10.1016/j.nbd.2014.07.014. [DOI] [PubMed] [Google Scholar]
- Munsie LN, Milnerwood AJ, Seibler P, Beccano-Kelly DA, Tatarnikov I, Khinda J, Volta M, Kadgien C, Cao LP, Tapia L, Klein C, Farrer MJ. Retromer-dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson's disease VPS35 mutation p.D620N. Hum Mol Genet. 2015;24:1691–1703. doi: 10.1093/hmg/ddu582. [DOI] [PubMed] [Google Scholar]
- Murphy KE, Gysbers AM, Abbott SK, Spiro AS, Furuta A, Cooper A, Garner B, Kabuta T, Halliday GM. Lysosomal-associated membrane protein 2 Isoforms are differentially affected in early Parkinson's disease. Mov Disord. 2015 doi: 10.1002/mds.26141. doi: 10.1002/mds.26141. Advance online publication. Retrieved January 16, 2015. [DOI] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16:394–406. doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan CL, Baum PD, Gu M, Jorgensen EM, Clark SG, Garriga G. C. elegans AP-2 and retromer control Wnt signaling by regulating mig-14/Wntless. Dev Cell. 2008;14:132–139. doi: 10.1016/j.devcel.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci. 2002;22:3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao LY, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S, Xie ZL, Speake LD, Parks R, Crabtree D, Liang Q, Crimmins S, Schneider L, Uchiyama Y, Iwatsubo T, Zhou Y, Peng L, Lu Y, Standaert DG, Walls KC, et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain. 2008;1:17. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. 2004;165:111–122. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN. Recycle your receptors with retromer. Trends Cell Biol. 2005;15:68–75. doi: 10.1016/j.tcb.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, Kim TW. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- Tabuchi M, Yanatori I, Kawai Y, Kishi F. Retromer-mediated direct sorting is required for proper endosomal recycling of the mammalian iron transporter DMT1. J Cell Sci. 2010;123:756–766. doi: 10.1242/jcs.060574. [DOI] [PubMed] [Google Scholar]
- Temkin P, Lauffer B, Jäger S, Cimermancic P, Krogan NJ, von Zastrow M. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat Cell Biol. 2011;13:715–721. doi: 10.1038/ncb2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weering JR, Verkade P, Cullen PJ. SNX-BAR proteins in phosphoinositide-mediated, tubular-based endosomal sorting. Semin Cell Dev Biol. 2010;21:371–380. doi: 10.1016/j.semcdb.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira SI, Rebelo S, Esselmann H, Wiltfang J, Lah J, Lane R, Small SA, Gandy S, da Cruz E Silva EF, da Cruz E Silva OA. Retrieval of the Alzheimer's amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol Neurodegener. 2010;5:40. doi: 10.1186/1750-1326-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, et al. VPS35 Mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Coelln R, Thomas B, Andrabi SA, Lim KL, Savitt JM, Saffary R, Stirling W, Bruno K, Hess EJ, Lee MK, Dawson VL, Dawson TM. Inclusion body formation and neurodegeneration are parkin independent in a mouse model of alpha-synucleinopathy. J Neurosci. 2006;26:3685–3696. doi: 10.1523/JNEUROSCI.0414-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CL, Tang FL, Peng Y, Shen CY, Mei L, Xiong WC. VPS35 regulates developing mouse hippocampal neuronal morphogenesis by promoting retrograde trafficking of BACE1. Biol Open. 2012;1:1248–1257. doi: 10.1242/bio.20122451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Wen L, Tang FL, Hong Y, Luo SW, Wang CL, He W, Shen C, Jung JU, Xiong F, Lee DH, Zhang QG, Brann D, Kim TW, Yan R, Mei L, Xiong WC. VPS35 haploinsufficiency increases Alzheimer's disease neuropathology. J Cell Biol. 2011;195:765–779. doi: 10.1083/jcb.201105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC. alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia WF, Tang FL, Xiong L, Xiong S, Jung JU, Lee DH, Li XS, Feng X, Mei L, Xiong WC. Vps35 loss promotes hyperresorptive osteoclastogenesis and osteoporosis via sustained RANKL signaling. J Cell Biol. 2013;200:821–837. doi: 10.1083/jcb.201207154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Ding YQ, Hong Y, Feng Z, Navarre S, Xi CX, Zhu XJ, Wang CL, Ackerman SL, Kozlowski D, Mei L, Xiong WC. Phosphatidylinositol transfer protein-alpha in netrin-1-induced PLC signalling and neurite outgrowth. Nat Cell Biol. 2005;7:1124–1132. doi: 10.1038/ncb1321. [DOI] [PubMed] [Google Scholar]
- Xilouri M, Brekk OR, Stefanis L. alpha-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol. 2013a;47:537–551. doi: 10.1007/s12035-012-8341-2. [DOI] [PubMed] [Google Scholar]
- Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, Kirik D, Stefanis L. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain. 2013b;136:2130–2146. doi: 10.1093/brain/awt131. [DOI] [PubMed] [Google Scholar]
- Yang PT, Lorenowicz MJ, Silhankova M, Coudreuse DY, Betist MC, Korswagen HC. Wnt signaling requires retromer-dependent recycling of MIG-14/Wntless in Wnt-producing cells. Dev Cell. 2008;14:140–147. doi: 10.1016/j.devcel.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Yin DM, Chen YJ, Lu YS, Bean JC, Sathyamurthy A, Shen C, Liu X, Lin TW, Smith CA, Xiong WC, Mei L. Reversal of behavioral deficits and synaptic dysfunction in mice overexpressing neuregulin 1. Neuron. 2013;78:644–657. doi: 10.1016/j.neuron.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]