

ABSTRACT
Pyruvate dehydrogenase kinases (PDKs) modulate energy homeostasis in multiple tissues and cell types, under various nutrient conditions, through phosphorylation of the α subunit (PDHE1α, also known as PDHA1) of the pyruvate dehydrogenase (PDH) complex. However, the roles of PDKs in meiotic maturation are currently unknown. Here, by undertaking knockdown and overexpression analysis of PDK paralogs (PDK1–PDK4) in mouse oocytes, we established the site-specificity of PDKs towards the phosphorylation of three serine residues (Ser232, Ser293 and Ser300) on PDHE1α. We found that PDK3-mediated phosphorylation of Ser293-PDHE1α results in disruption of meiotic spindle morphology and chromosome alignment and decreased total ATP levels, probably through inhibition of PDH activity. Unexpectedly, we discovered that PDK1 and PDK2 promote meiotic maturation, as their knockdown disturbs the assembly of the meiotic apparatus, without significantly altering ATP content. Moreover, phosphorylation of Ser232-PDHE1α was demonstrated to mediate PDK1 and PDK2 action in meiotic maturation, possibly through a mechanism that is distinct from PDH inactivation. These findings reveal that there are divergent roles of PDKs during oocyte maturation and indicate a new mechanism controlling meiotic structure.
KEY WORDS: Pyruvate dehydrogenase kinase, Oocyte, Meiosis, Metabolism, Spindle
Highlighted Article: In mouse ooctytes, different isoforms of pyruvate dehydrogenase kinase have differing effects on the assembly of the spindle, chromosome alignment and ATP content.
INTRODUCTION
Oocyte development is key in establishing female fertility. At around the time of birth, mammalian oocytes are arrested in prophase of the first meiotic division, which is also called the germinal vesicle stage. Following a stimulation by luteinizing hormone at puberty, immature fully-grown oocytes reinitiate meiosis, characterized by germinal vesicle breakdown (GVBD). With the chromatin condensation and microtubule organization, the oocytes proceed through the meiosis I (MI) division, extruding the first polar body (Pb1). Oocytes then become arrested at metaphase II (MII), waiting for fertilization (Jones and Lane, 2013; Wang and Sun, 2007). Accurate control of spindle assembly and chromosome alignment during meiotic maturation is necessary to produce a healthy oocyte. Errors in this process cause aneuploid eggs, which is a major cause of miscarriage and birth defects in humans (Hassold and Hunt, 2001).
Balanced and efficient metabolism is crucial for the execution of oocyte and subsequent embryo development. Throughout the period of oocyte growth, pyruvate and oxygen consumption is markedly increased (Harris et al., 2009). Fully-grown oocytes have limited capacity to utilize glucose (Biggers et al., 1967; Saito et al., 1994; Zuelke and Brackett, 1992); instead, glucose first needs to be transferred to granulosa cells and then metabolized into pyruvate to support oocyte maturation (Eppig, 1976; Fagbohun and Downs, 1992). Oocyte-specific deletion of mouse PDHA1, the gene encoding the PDHE1α subunit of the pyruvate dehydrogenase (PDH) complex, results in compromised energetic status and meiotic defects in mouse oocytes (Johnson et al., 2007), further highlighting the importance of pyruvate metabolism during oocyte development.
PDH is able to convert pyruvate into acetyl coenzyme A (acetyl-CoA) and thereby modulates the entry of glucose-derived carbons into the tricarboxylic acid (TCA) cycle. The TCA cycle provides an efficient means of producing ATP and many biosynthetic intermediates. The PDH complex is composed of three catalytic enzymes (E1, E2 and E3) and their regulatory proteins. The activity of PDH complex is under the control of two classes of enzymes, pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP), through a reversible phosphorylation–dephosphorylation cycle (McFate et al., 2008; Sugden and Holness, 2006). Studies in somatic cells indicate that PDH activity is inhibited by site-specific phosphorylation at three serine residues of the E1 α subunit (PDHE1α): Ser232, Ser293 and Ser300 (Hitosugi et al., 2011; Korotchkina and Patel, 2001; Rardin et al., 2009). To date, four paralogous PDK genes (PDK1–PDK4) have been identified in mammals (Gudi et al., 1995; Tso et al., 2014). PDK1 expression is limited to heart and PDK3 is mainly found in testis, whereas PDK2 can be detected in many tissues. PDK4 is the most abundant of the PDKs in heart and skeletal muscle (Bowker-Kinley et al., 1998; Gudi et al., 1995; Korotchkina and Patel, 2001). These findings suggest that different PDK isoforms are utilized to meet the tissue- or cell-type-specific requirements for proper tuning of PDH activity. Some information is known about how PDK activity is regulated from biochemical or structural analysis and expression studies. For example, through binding to the inner lipoyl domain of the PDH E2 catalytic subunit, dihydrolipoyl acetyltransferase, PDKs have been showed to access their PDH-E2-bound PDH substrate. In the case of PDK2, ADP dissociation limits the PDH-E2-enhanced activity of PDK2, and pyruvate binding to the regulatory domain could further inhibit this process. NADH and acetyl-CoA stimulates PDK2 activity through a decrease in acetylation of lipoyl group and the resultant increase in ADP dissociation rate (for a review see Roche and Hiromasa, 2007). PDK4 is regulated, in part, through differences in gene transcription, as in a number of cell types the gene is transcriptionally inactive, through histones on the PDK4 gene being in the non-acetylated state (Kwon and Harris, 2004; Kwon et al., 2006).
The pivotal roles of PDKs in energy homeostasis in diverse tissues and under various nutrient conditions have been extensively reported (Hitosugi et al., 2011; Jeoung and Harris, 2008; Kim et al., 2006; Michelakis et al., 2010; Takubo et al., 2013; Zhang et al., 2014). For example, PDK4 is involved in the fatty acid utilization in muscle by interacting with cluster of differentiation 36 (CD36) and FoxO1 (Nahle et al., 2008). PDK2 is essential for the maintenance of cell cycle quiescence and glycolytic metabolic status in hematopoietic stem cells (Takubo et al., 2013). It has recently been found that metabolic perturbations result in significant meiotic defects in mouse oocytes (Luzzo et al., 2012; Wang et al., 2009; Wu et al., 2015), which might be through the action of PDKs on PDH. However, up to now, it is not known which PDKs are expressed and what are their functions in the maturation of the meiotic oocyte, even in the unperturbed metabolic state.
In this study, by employing a morpholino (MO) knockdown screen and overexpression analysis, we discovered distinct roles for PDK paralogs during mouse oocyte maturation, particularly in controlling meiotic spindle structure and metabolic activity, and report our findings below.
RESULTS
Expression of PDKs in mouse oocytes
PDH links glycolysis to the TCA cycle by catalyzing the irreversible oxidative decarboxylation of pyruvate, leading to the generation of CO2, NADH and acetyl-CoA. Given the crucial role PDH plays in cellular energy metabolism and biosynthetic pathways, multiple levels of regulation are applied so that the demands of specific cell types are balanced. Pyruvate dehydrogenase kinases (PDKs) are important inhibitors of PDH activity in numerous cell types, through phosphorylation of the PDHE1α subunit (Fig. 1A). As a first step to investigate the potential involvement of PDKs during oocyte maturation, we examined the expression of PDK genes in mouse oocytes by quantitative real-time PCR (qRT-PCR). As shown in Fig. 1B, expression of all the PDK members was detected, with PDK3 mRNA being most abundant and PDK4 mRNA being least abundant, suggesting that they might play different roles.
Fig. 1.
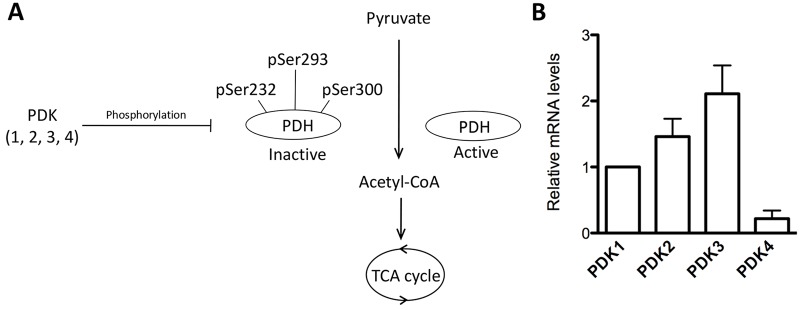
PDK gene expression in mouse oocyte. (A) Schematic illustrating a model for control of PDH activity and energy metabolism by PDKs in somatic cells. pSer293-PDHE1α has been reported as the major site of downregulation of PDH activity. (B) Fully-grown germinal vesicle oocytes were collected for RNA sampling, and relative quantification of PDK mRNA levels was conducted by real-time RT-PCR. GAPDH expression served as an internal control. Data are expressed as the mean±s.d. from three independent experiments.
PDK paralogs preferentially phosphorylate specific sites on PDHE1α in oocytes
PDKs phosphorylate PDHE1α on three sites, Ser232, Ser293 and Ser300 (Rardin et al., 2009; Sugden and Holness, 2003). There are four PDK paralogs (PDK1–PDK4) and each isoform has specific activities and different affinities for these serine residues on PDHE1α, which allows for individual responses to altering metabolic demands (Bowker-Kinley et al., 1998; Korotchkina and Patel, 1995, 2001; Roche and Hiromasa, 2007) (Fig. 1A). Thus, to understand the underlying pathways mediating the action of PDKs in oocyte maturation, we wished to systematically evaluate the specificity of PDK1–PDK4 towards the three phosphorylation sites of PDHE1α in mouse oocytes. MO knockdown screening combined with antibodies specific for the three phosphorylation sites has provided us a route to address this question.
Fully-grown oocytes were microinjected with PDK-paralog-targeting MOs and then stained with a panel of antibodies against phosphorylated Ser232, Ser293 or Ser300 of the PDHE1α subunit (hereafter denoted pSer232-PDHE1α, pSer293-PDHE1α and pSer300-PDHE1α, respectively). The specificity of these three antibodies to the phosphorylated PDHE1α sites has been previously assessed in cell lines (Rardin et al., 2009), and they have also been employed in other studies (Jaswal et al., 2010; Miquel et al., 2012). Partial knockdown of each PDK isoform protein level after injection of individual paralogous gene MOs was confirmed by immunoblotting (supplementary material Fig. S1). As shown in Fig. 2A, pSer293-PDH resides in the cytoplasm of control germinal vesicle and metaphase II oocytes. Confocal scanning and quantitative analysis further demonstrated that knockdown of PDK3 or PDK4 led to a marked reduction of pSer293-PDHE1α. In comparison, knockdown of PDK1 or PDK2 had little or no effects on the phosphorylation of this site (Fig. 2B). For Ser300-PDHE1α site, its phosphorylation was consistently detected in the cytoplasm during oocyte maturation, and was weakly inhibited only when PDK4 was knocked down in oocytes (Fig. 2C,D). It is possible that there is redundancy among PDKs for the Ser300-PDHE1α site such that knockdown of any single paralog has a small or no effect.
Fig. 2.

Differing effects of PDK knockdown on phosphorylation of serine residues on PDHE1α. (A) Control and PDK knockdown (KD) oocytes were stained with anti-pSer293-PDHE1α antibody (green) and counterstained with propidium iodide to label chromosomes (red). (B) Quantification of pSer293-PDHE1α fluorescence shown in A. (C) Control and PDK-KD oocytes were labeled with anti-pSer300-PDHE1α antibody (green) and counterstained with propidium iodide (red). (D) Quantification of pSer300-PDHE1α fluorescence shown in C. (E) Control and PDK-KD oocytes were labeled with anti-pSer232-PDHE1α antibody (green) and counterstained with propidium iodide (red). (F) Quantification of pSer232-PDHE1α fluorescence in E. Arrows in A, C and E point to chromosome congression failure in PDK1-KD and PDK2-KD oocytes. Yellow arrowheads in E show the pSer232-PDH signal in control metaphase oocytes. Representative confocal sections are shown in A, C and E. For B, D and F, results are mean±s.d. At least 60 oocytes for each group were analyzed, and the experiments were conducted three times. *P<0.05 versus controls. (G) Schematic representation of the PDK paralog phosphorylation site specificity for oocyte PDHE1α. Scale bars: 20 µm.
Phosphorylated Ser232-PDHE1α exhibited an unexpected subcellular localization during oocyte maturation – it was distributed in the cytoplasm of control germinal vesicle oocytes, but as oocytes enter into metaphase it became concentrated on the spindle region and its poles (Fig. 2E, arrowheads). Moreover, we found that knockdown of PDK1 or PDK2 resulted in a significant decrease in pSer232-PDHE1α, whereas the pSer232-PDHE1α signal in PDK3- or PDK4-depleted oocytes remained unchanged (Fig. 2E,F). To assess whether the anti-pSer232-PDHE1α antibody staining pattern is specific, we also stained oocytes following PDHE1α knockdown. The results showed that the pSer232-PDHE1α signal was reduced in oocytes depleted of PDHE1α when compared to controls (supplementary material Fig. S2), suggesting that at least some of the spindle and pole staining is specific. Surprisingly, in the PDK1 or PDK2 knockdown, we consistently observed abnormal chromosome alignment in metaphase oocytes (Fig. 2, arrows), whereas knockdown of PDK3 or PDK4 did not result in such defects. Thus PDK1 and PDK2 might promote the assembly of meiotic apparatus.
Collectively, these data reveal that in mouse oocytes, PDK3 shows preference for specific phosphorylation of Ser293-PDHE1α, PDK4 seems to be able to slightly affect both pSer293-PDHE1α and pSer300-PDHE1α, and PDK1 and PDK2 appear to primarily modulate the phosphorylation status of Ser232-PDHE1α (Fig. 2G). The specificity of the PDK isoforms for individual phosphorylation sites indicates that different PDKs might control different cellular events during oocyte meiosis.
PDK3 overexpression induces meiotic defects in oocytes
Studies of the oocyte specific knockout of PDHA1 (Johnson et al., 2007) have demonstrated that PDHE1α functions in meiotic maturation and spindle or chromosome organization, likely through generation of ATP and NAD(P)H. Phosphorylation of PDHE1α by PDKs inactivates the PDH complex to modulate energy homeostasis (Lu et al., 2008; Peters et al., 2001; Stacpoole, 2012). Therefore, we sought to determine whether up-regulation of PDK in oocytes, which is predicted to inactivate PDHE1α through increased phosphorylation, would affect meiotic maturation in ways that are similar to the PDHA1 knockout. Exogenous Myc–PDK-encoding mRNA for each paralog was separately injected into immature oocytes, which were arrested for 20 h with milrinone to allow synthesis of new PDK protein. The oocytes were then washed and matured in vitro to first check their maturational progression. After 3 h culture, control and all PDK-overexpressing oocytes resumed meiosis normally, as indicated by the similar GVBD rate (supplementary material Fig. S3A). Elevated PDK3 expression significantly reduced the rate of Pb1 extrusion in oocytes at 14 h (supplementary material Fig. S3B), consistent with the proposal that PDHE1α activity is decreased upon PDK3 overexpression (also see below). In contrast, overexpression of PDK1, PDK2 or PDK4 had no apparent effect on meiotic progression. Efficient protein overexpression for each of the exogenous Myc–PDK paralogs was confirmed by immunoblotting (supplementary material Fig. S3C–F).
To examine whether increased PDK levels in oocytes influences the phosphorylation status of their corresponding target sites on PDHE1α, oocytes injected with PDK mRNAs were stained with a panel of antibodies against pSer232-PDHE1α, pSer293-PDHE1α or pSer300-PDHE1α. In line with the knockdown experiments, overexpression of PDK3 enhanced the pSer293-PDHE1α signal in the cytoplasm (Fig. 3A,B). Similarly, overexpression of PDK1 or PDK2 in oocytes increased the fluorescence intensity of pSer232-PDHE1α (Fig. 3C,D, data only shown for PDK1). Furthermore, in rescue experiments, we found that overexpression of PDK3 significantly restored pSer293-PDHE1α staining in PDK3-MO-injected oocytes, and correspondingly PDK2 overexpression restored pSer232-PDHE1α staining in PDK2 MO-injected oocytes (supplementary material Fig. S4A–D). These results further support the idea that the signals from anti-pSer232-PDHE1α and anti-pSer293-PDHE1α antibody staining are specific to the activity of PDK2 and PDK3, respectively. However, overexpression of PDK4 seems incapable of markedly boosting the signal for phosphorylation of Ser232-PDHE1α, Ser293-PDHE1α or Ser300-PDHE1α (data not shown).
Fig. 3.

PDK3 overexpression induces meiotic defects in oocytes. (A) Control and PDK3-overexpressing oocytes were stained with anti-pSer293-PDHE1α antibody (green) and with propidium iodide to label chromosomes (red). (B) Quantification of pSer293-PDHE1α fluorescence in A. Data are expressed as the mean±s.d. from three independent experiments in which at least 60 oocytes were analyzed. (C) Control and PDK1-overexpressing oocytes were stained with pSer232-PDHE1α antibody (green) and with propidium iodide (red). (D) Quantification of pSer232-PDHE1α fluorescence in C. Data are expressed as the mean±s.d. from three independent experiments in which at least 60 oocytes were analyzed. (E) Control and PDK3-overexpressing oocytes were stained with α-tubulin antibody to visualize the spindle (green) and counterstained with propidium iodide (red). Spindle defects and chromosome misalignment are indicated by arrowheads and arrows, respectively. Representative confocal sections are shown. (F) Quantitative analysis of control and PDK-overexpressing oocytes with abnormal spindle and chromosomes. Data are expressed as the mean±s.d. percentage from three independent experiments in which at least 120 oocytes were analyzed. *P<0.05 versus controls. Scale bars: 20 µm.
Importantly, when PDK3 is overexpressed, misaligned chromosomes are readily observed in metaphase oocytes (Fig. 3A, arrow) with high levels of phosphorylation on Ser293-PDHE1α, not on other serine residues. This correlation suggests that hyperphosphorylation of Ser293-PDHE1α induced by PDK3 overexpression might be linked to the meiotic abnormalities. We thus extended our analysis to examine spindle and chromosome organization in PDK-overexpressing oocytes. Cells were labeled with an anti-tubulin antibody to visualize the spindle and co-stained with propidium iodide for chromosomes. PDK3 overexpression (Fig. 3E,F), in comparison to controls, significantly elevated the incidence of disorganized spindle (arrowheads) and misaligned chromosomes (arrows), whereas PDK1, PDK2 or PDK4 overexpression did not confer an adverse effect on these structures (Fig. 3F). These results suggest that PDK3 overexpression, concomitant with increased phosphorylation levels of Ser293-PDHE1α, are likely to decrease PDHE1a activity and induce meiotic defects in oocytes.
Metabolic dysfunction in PDK3-overexpressing oocytes
Having shown that overexpression of PDK3 disrupts oocyte maturation, we assessed whether PDK overexpression also affects metabolic activity in oocytes. Pyruvate metabolism by the PDH complex is a major route for energy production in mammalian oocytes, with the product of pyruvate oxidation, acetyl-CoA, the starting point for the TCA cycle that provides the major energy currency molecule of the cell, ATP. Thus, we measured the bulk intracellular ATP levels, using a bioluminescence assay, as a proxy for affects of manipulations of PDK activity and PDHE1α phosphorylation state. As shown in Fig. 4A, overexpression of PDK1, PDK2 or PDK4 had little effects on the ATP content. However, PDK3 overexpression resulted in an ∼50% reduction in bulk ATP levels compared to control oocytes, indicating impairment of mitochondrial function. Mitochondria are the major reactive oxygen species (ROS) generator, as well as one of the main targets of ROS-induced oxidative damage (Ramalho-Santos et al., 2009). Next, we asked whether ROS generation is also affected when PDK3 is overexpressed in oocytes. To address this question, live oocytes were stained with CM-H2DCFDA, a mitochondria-specific dye that fluoresces when oxidized by ROS, and then examined under a fluorescence microscope. PDK3-overexpressing oocytes showed significantly more mitochondrial ROS, as determined by mean fluorescence (Fig. 4B,C), whereas overexpression of PDK1, PDK2 or PDK4 had little effects on ROS generation (data not shown). These results suggest that PDK3 can function to control metabolic activity during oocyte maturation.
Fig. 4.
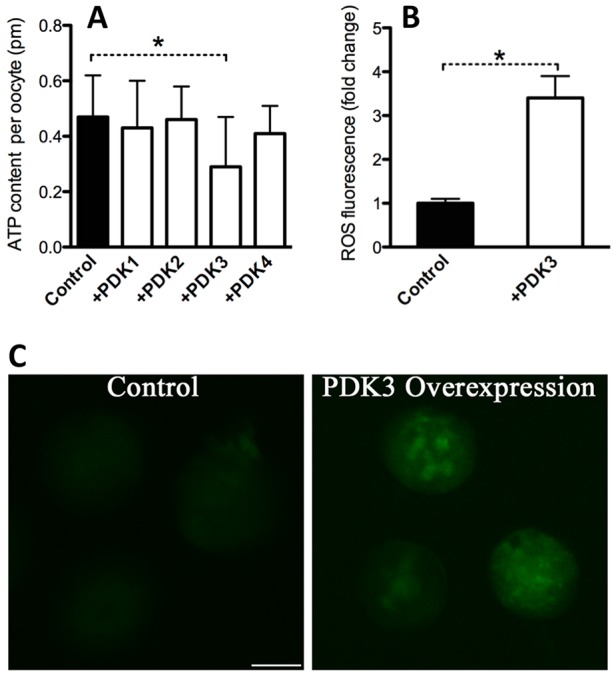
PDK3 overexpression causes metabolic dysfunction in oocytes. (A) Histogram showing the total ATP content in control and PDK-overexpressing oocytes (n=50 for each group). (B) Effects of PDK overexpression on intracellular ROS levels, as estimated by CM-H2DCFDA fluorescence intensity. (C) Representative images of CM-H2DCFDA fluorescence in oocytes analyzed in B. For A and B, at least 50 oocytes were examined for each group, data are expressed as the mean±s.d. from three independent experiments, *P<0.05 versus controls. Scale bar: 50 µm.
Phosphorylation of Ser293-PDHE1α is important for PDK3-controlled oocyte metabolism and meiosis
Given that PDK3 primarily modulates the phosphorylation status of Ser293-PDHE1α (Fig. 2), we asked whether Ser293-PDHE1α phosphorylation mediates the affects of PDK3 on oocyte metabolism and meiotic apparatus. Towards this goal, we first generated Ser293-PDHE1α mutants and investigated their roles during oocyte maturation. Ser293 was mutated to an alanine residue (S293A), to preclude phosphorylation, or to an aspartate residue (S293D), to mimic permanent phosphorylation (Vagnoni et al., 2011). Wild-type (WT), S293A or S293D mutant mRNA was then injected into fully-grown oocytes for total ATP measurement and spindle and chromosome analysis. In comparison to WT controls, oocytes expressing the S293D-PDHE1α mutant exhibited lower levels of ATP (Fig. 5A) and a higher proportion of meiotic defects (Fig. 5B), consistent with the phenotypes of PDK3-overexpressing oocytes. By contrast, oocytes expressing the S293A-PDHE1α mutant had little effects on both cellular metabolism and meiosis.
Fig. 5.
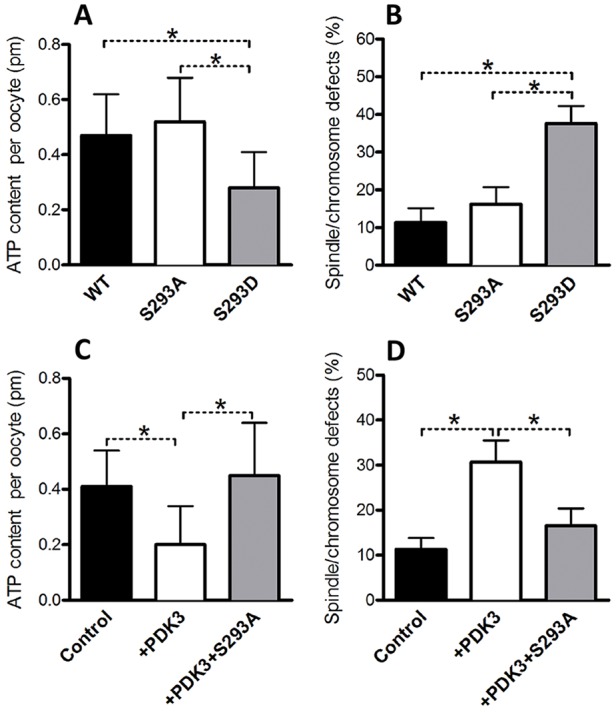
Phosphorylation of Ser293-PDHE1α is a major target mediating the effects of PDK3 on oocyte metabolism and meiosis. WT, S293A-PDHE1α or S293D-PDHE1α mutant cRNA was microinjected into fully grown oocytes to evaluate their effects on ATP levels and the meiotic apparatus. The histograms show (A) a reduced total ATP content and (B) an increased frequency of spindle and chromosome defects in oocytes expressing phosphorylation mimetic mutant S293D as compared to WT controls (n=90 for each group). In rescue experiments, PDK3 and S293A-PDHE1α mutant cRNAs were simultaneously injected into oocytes, and then metabolic and meiotic phenotypes were examined. The histograms show that non-phosphorylated S293A-PDHE1α mutant can (C) suppress the total ATP reduction and (D) ameliorate spindle and chromosome defects in oocytes overexpressing PDK3 (n=75 for each group). Results are mean±s.d. from three replicates. *P<0.05.
Furthermore, to test whether the meiotic defects observed upon PDK3 overexpression are mediated by phosphorylation of Ser293-PDHE1α, a functional rescue experiment was conducted. PDK3 mRNA and S293A-PDHE1α mutant mRNAs were simultaneously injected into oocytes, and then meiotic and metabolic phenotypes were examined. As shown in Fig. 5C,D, we found that the phenotypic defects of oocytes overexpressing PDK3 were partially suppressed by the co-expression of the non-phosphorylatable S293A-PDHE1α mutant. Owing to the limitation of oocyte number, we have not yet been able to analyze the relationship between PDHE1α phosphorylation and PDH complex activity in mouse oocytes, although we assume that Ser293 phosphorylation inactivates PDH, as observed in somatic cells. Taken together, these data indicate that Ser293 is a major, if not unique, phosphorylation site on PDHE1α, mediating the action of PDK3 on oocyte metabolism and meiotic maturation.
PDK1 and PDK2 promote spindle organization and chromosome congression in oocytes by phosphorylating Ser232-PDHE1α
Two of our observations would not be expected if PDK1- and PDK2-mediated Ser232-PDHE1α phosphorylation inhibited the activity of the PDH complex: (1) that from the initial analysis of PDK1 or PDK2 MO knockdown showing meiotic maturation defects and (2) that overexpression of PDK1 or PDK2 did not influence oocyte maturation (in contrast to overexpression of PDK3 described above). Therefore we explored the function of PDK1 and PDK2-mediated phosphorylation of Ser232-PDHE1α further.
First, we individually knocked down PDK1–PDK4 by MO injection, with a sham MO standard injected as control, and examined meiotic maturation phenotypes. Our results showed that loss of any PDKs barely affected the fraction of oocytes that underwent GVBD (Fig. 6A). However, Pb1 extrusion was significantly decreased in PDK1-MO (38.4±3.9%) and PDK2-MO (47.3±4.7%) oocytes compared to controls (90.6±2.8%) (mean±s.d., Fig. 6B), indicating that PDK1 and PDK2 are involved in meiotic progression. Furthermore, confocal microscopy revealed a significantly higher percentage of meiotic defects in PDK1-MO (33.2±5.7%) and PDK2-MO (27.6±5.3%) oocytes relative to controls (8.9±2.8%) (Fig. 6C,D), displaying diverse malformed spindles, multipolar spindles (arrowheads) and chromosome congression failures (arrows). These phenotypes differed sharply from metaphase spindles in control oocytes, which presented a typical barrel-shaped spindle and well-aligned chromosomes at the equator. By contrast, knockdown of PDK3 or PDK4 in oocytes had no evident effects on these meiotic structures (Fig. 6C,D). We also evaluated the effects of PDK knockdown on total intracellular ATP levels. Of note, total ATP content was not significantly altered in oocytes depleted of any PDKs compared to control cells, although there was a slight increase in PDK3-MO oocytes (Fig. 6E).
Fig. 6.

Effects of PDK knockdown on spindle formation and chromosome alignment during oocyte meiosis. (A,B) Quantitative analysis of GVBD and Pb1 extrusion in control (n=130), PDK1-knockdown (KD) (n=140), PDK2-KD (n=136), PDK3-KD (n=138) and PDK4-KD (n=120) oocytes. Results are mean±s.d. from three replicates. (C) Control and PDK-KD oocytes were stained with anti-α-tubulin antibody to visualize spindles (green) and counterstained with propidium iodide to visualize chromosomes (red). Spindle defects and chromosome misalignment are indicated by arrowheads and arrows, respectively. Representative confocal sections are shown. (D) Quantification of control and PDK-KD oocytes with abnormal spindle and chromosomes. Data are expressed as the mean±s.d. from three independent experiments in which at least 100 oocytes were analyzed. (E) Measurement of total ATP levels in control and PDK-KD oocytes. Data are expressed as the mean±s.d. from three independent experiments in which at least 50 oocytes were analyzed. *P<0.05 versus controls. Scale bar: 20 µm.
To further confirm the specificity of PDK1 and PDK2 MO knockdown, we performed an mRNA rescue experiment, where we expressed PDK2-encoding cRNA that contains a 5′UTR that is not recognized by the MO (see Materials and Methods). As shown in supplementary material Fig. S4E–G, PDK2 overexpression could efficiently rescue the effects of MO knockdown. The majority of oocytes were rescued, progressed to metaphase II and displayed the normal meiotic structures observed in controls. These results indicate that PDK1 and PDK2 are required for normal spindle formation and meiotic chromosome congression.
Given that PDK1 and PDK2 preferentially regulates the phosphorylation state of Ser232 on PDHE1α in oocytes, we next examined whether Ser232-PDHE1α phosphorylation is a major target mediating the effects of PDK1 and PDK2 on meiotic apparatus. The non-phosphorylatable S232A-PDHE1α and phospho-mimetic S232D-PDHE1α mutants were constructed and then the corresponding mRNA individually microinjected into oocytes to evaluate the meiotic and metabolic phenotypes. As shown in Fig. 7A, the non-phosphorylatable mutant S232A-PDHE1α resulted in an almost threefold increase in spindle defects and chromosome misalignment compared to the WT control, whereas the S232D-PDHE1α phospho-mimetic form had little effect on the meiotic spindle and meiotic chromosomes. We next found that the PDK1-knockdown meiotic phenotypes could be rescued by overexpression of the phospho-mimetic S232D-PDHE1α (Fig. 7B). Additionally, similar to PDK1 or PDK2 knockdown, overexpression of either S232A-PDHE1α or S232D-PDHE1α mutants did not significantly alter bulk ATP levels in oocytes (Fig. 7C). Taken together, these results indicate that PDK1 and PDK2 promote spindle assembly and chromosome alignment in oocytes by regulating the phosphorylation status of Ser232-PDHE1α, possibly through a mechanism that is independent of the PDHE1α activity that controls ATP production.
Fig. 7.
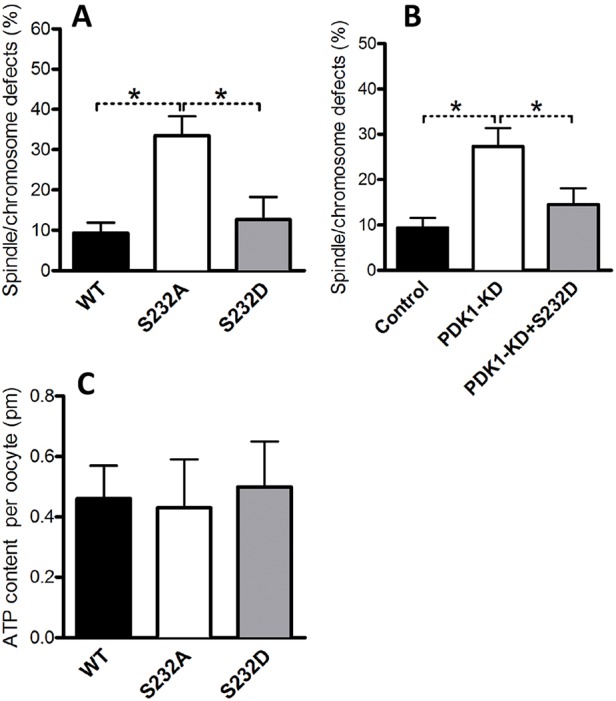
Phosphorylation of Ser232-PDHE1α is important for PDK1/2-controlled meiotic regulation. WT, S232A-PDHE1α or S232D-PDHE1α mutant cRNA was injected into oocytes to evaluate their effects on the meiotic apparatus and ATP levels. (A) The histogram shows that the non-phosphorylatable mutant S232A resulted in an ∼threefold increase in spindle and chromosome defects in oocytes compared to WT control (n=90 for each group). (B) The histogram shows that phosphorylation mimetic mutant S232D rescues the spindle and chromosome defects in PDK1-knockdown oocytes (n=75 for each group). (C) Histogram showing that neither S232A-PDHE1α nor S232D-PDHE1α mutants altered total ATP levels in oocytes (n=90 for each group). Results are mean±s.d. from three replicates. *P<0.05.
DISCUSSION
This study was designed to uncover the functions of PDKs during oocyte maturation. We first examined the site-specificity of the four PDK paralogs towards the phosphorylation of three serine residues on PDHE1α. We found, through screening the entire family, that PDK3 is the primary PDK that functions in metabolic control during oocyte maturation, which is mediated by phosphorylation of Ser293-PDHE1α, which inactivates the PDH complex. Additionally, we unexpectedly discovered that PDK1 and PDK2 promote spindle formation and chromosome congression in mouse oocytes, likely by phosphorylating Ser232-PDHE1α, and which appears not to inactivate PDH (Fig. 8).
Fig. 8.

Diagram illustrating how experimental perturbations of PDK paralog activity affect site-specific phosphorylation for PDHE1α and oocyte functions. Overexpression of PDK3 (upward arrow), but not knockdown, results in increased pSer293-PDHE1α, which leads to decreased PDH activity resulting in decreased total ATP, increased ROS and spindle assembly defects and chromosome misalignment in oocytes. Knockdown of PDK1 or PDK2 (downward arrow), but not overexpression, results in decreased pSer232-PDHE1α, which leads to spindle assembly defects and chromosome misalignment in metaphase oocytes. See text for details.
Phosphorylation site-specific control of PDHE1α by PDKs
It has been widely reported that mammalian PDK paralogs are expressed in a tissue- and cell-type-specific manner, with PDK1 present mostly in the heart, PDK2 in a number of tissues, PDK3 in testis and PDK4 having elevated amounts in heart and skeletal muscle (Bowker-Kinley et al., 1998; Patel and Korotchkina, 2001). Here, qRT-PCR data showed that all PDKs could be detected in mouse oocytes, and that PDK3 appears most abundant (Fig. 1). The activity of the PDH complex in animals is regulated in large part by reversible phosphorylation of Ser232, Ser293 and Ser300 on the PDHE1α subunit. Analysis of the phosphorylation and inactivation of the purified heart PDH has revealed that the relative initial rates of phosphorylation of individual sites are in the order Ser293>Ser300>Ser232. Early experiments indicated that Ser293 is the predominant inactivating site (Kolobova et al., 2001; Sale and Randle, 1981), and later in vitro biochemical assays found that phosphorylation at any site can inhibit enzymatic activity, although much higher inhibition is observed for the phosphorylation of Ser293 than for Ser300 and Ser232 (Gray et al., 2014; Korotchkina and Patel, 1995). On the basis of these data, it has been proposed that phosphorylation of Ser293 is responsible for PDH inactivation, whereas phosphorylation of Ser300 and Ser232 are perhaps not primarily concerned with cellular metabolism.
Employing in vitro kinase assays with recombinant PDHE1α proteins, Korotchkina and Patel reported that all four PDKs can phosphorylate Ser293 and Ser300, whereas PDK1 uniquely phosphorylates Ser232 (Korotchkina and Patel, 2001). To assess PDHE1α phosphorylation in vivo in mouse oocytes, we genetically manipulated PDK activity and then used phospho-site-specific antibodies to detect phosphorylation. We found that, in mouse oocytes, both PDK1 and PDK2 control the phosphorylation of Ser232-PDHE1α, whereas PDK3 primarily controls phosphorylation of Ser293-PDHE1α (Figs 2 and 3). Although PDK4 seems to be able to slightly regulate the phosphorylation state of both Ser293 and Ser300 sites, neither knockdown nor overexpression of PDK4 was demonstrated to have obvious effects on oocyte maturation (supplementary material Fig. S3 and Fig. 6) and is not discussed further.
Diverging roles of PDKs in oocyte meiosis
There is mounting evidence of crosstalk between PDKs and metabolic control in diverse cell types and tissues. For example, PDK1 and PDK3 have been reported to promote metabolic switching and drug resistance in cancer cells (Lu et al., 2008; McFate et al., 2008). Consistent with the metabolic function of PDKs in other cell types, we found that overexpression of PDK3, through specifically increasing the phosphorylation of Ser293-PDHE1α, resulted in a significant reduction of total ATP levels in mouse oocytes (Figs 4 and 5), as well as increased ROS production, indicative of mitochondrial dysfunction. Abnormal meiotic spindles and chromosome alignment was observed in PDK3-overexpressing oocytes, which was rescued by overexpression of phosphorylation blocking mutant S293A-PDHE1α (Figs 3 and 5). PDHA1−/− (the PDHE1α subunit) mouse oocytes display similar metabolic and meiotic phenotypes (Johnson et al., 2007). Chromatin condensation during meiosis is an ATP-dependent process. Numerous studies have suggested that insufficient ATP availability and increased oxidative stress are tied to spindle defects and chromsomal abnormalities in oocytes (Eichenlaub-Ritter et al., 2004; Tilly and Sinclair, 2013; Zeng et al., 2007). Our results are therefore consistent with PDK3 overexpression inducing Ser293-PDHE1α hyperphosphorylation, leading to inactivation of PDH and resulting in spindle and chromosome disorganization. We propose that under normal conditions, PDK3 activity is low in oocytes, allowing optimal PDH complex activity to produce sufficient ATP and metabolites for meiotic maturation.
Unexpectedly, PDK1 and PDK2 function to promote meiotic spindle organization and chromosome alignment (Fig. 6), which appears to be acting through phosphorylation of Ser232-PDHE1α as: (1) the knockdown of PDK1 or PDK2 causes a decrease in Ser232-PDHE1α staining, (2) the PDK1 or PDK2 knockdown phenotypes are rescued by overexpression of the phospho-mimetic mutant Ser232Asp-PDHE1α, and (3) overexpression of the phosphorylation-defective mutant S232A-PDHE1α results in similar phenotypes to those seen upon PDK1 and PDK2 knockdown (Figs 2, 6 and 7). These results are not readily consistent with a model where the PDK1 and PDK2 phosphorylation of Ser232-PDHE1α results in inactivation of the PDH complex. pSer232-PDHE1α localizes to the cytoplasm in germinal-vesicle-stage oocytes but accumulates at the spindle and pole area at metaphase, which is not where the vast majority of mitochondria reside. We suggest that oocyte PDK1 and PDK2 preferentially phosphorylate Ser232-PDHE1α in a subpopulation of mitochondria that are spindle associated. Total ATP levels in oocytes with PDK1 or PDK2 knockdown were slightly lower, but not statistically significantly different from the levels in controls. One possibility is that the PDK1- or PDK2-regulated Ser232-PDHE1α phosphorylation that promotes spindle formation and chromosome alignment is independent of PDH complex function to produce ATP. pSer232-PDHE1α might interact with the proteins needed for the assembly of meiotic apparatus in oocytes, which remains to be determined. It should be noted that the ATP levels measured were bulk (i.e. total) ATP per oocyte, not ATP levels in the vicinity of the spindle and poles where pSer232-PDHE1α is localized. Thus an alternative hypothesis is that, in metaphase II oocytes, pSer232-PDHE1α localized to mitochondria in the region of the spindle promotes PDH complex activity that is necessary for the meiotic divisions. However, we cannot rule out the PDK1 and PDK2 might be acting on a distinct substrate to promote spindle and chromosome organization. Additional experiments will be required to understand PDK1 and PDK2 function in mouse oocyte meiosis.
Accurate control of spindle assembly and chromosome dynamics and well-balanced energy metabolism are the crucial determinants for oocyte quality. This study uncovers the different roles of PDK isoforms in these processes, providing a new mechanism controlling oocyte maturation. As any errors during meiotic division could lead to pregnancy loss and infertility, our findings are not only of fundamental scientific interest but also vital for prevention of birth defects and treatment of reproductive disease in women.
MATERIALS AND METHODS
All chemicals and culture media were purchased from Sigma (St Louis, MO) unless stated otherwise.
Mice
ICR mice were used in all experiments. All experiments were approved by the Animal Care and Use Committee of Nanjing Medical University and were performed in accordance with institutional guidelines.
Antibodies
Rabbit polyclonal anti-PDK1 and rabbit polyclonal anti-Myc antibodies were purchased from Abcam (Cambridge, MA; catalog numbers ab92959 and ab9106); rabbit polyclonal anti-PDK2, rabbit polyclonal anti-PDK3 and rabbit polyclonal anti-PDK4 antibodies were purchased from Novus Biologicals (Concord, MA; catalog numbers NBP1-87307, NBP1-32581 and NBP1-07047); rabbit polyclonal anti-PDHE1α (pSer232), rabbit polyclonal anti-PDHE1α (pSer293), and rabbit polyclonal anti-PDHE1α (pSer300) antibodies were purchased from EMD Chemicals (Beverly, MA; catalog numbers AP1063, AP1062 and AP1064); mouse monoclonal anti-β-actin and mouse monoclonal anti-α-tubulin antibodies conjugated to FITC were purchased from Sigma (catalog numbers A5441 and F2168). FITC-conjugated goat anti-rabbit-IgG antibody was purchased from Thermo Fisher Scientific (Rockford, IL).
Oocyte collection and culture
To collect fully-grown germinal vesicle oocytes, mice were superovulated with 5 IU pregnant mares serum gonadotropin (PMSG) by intraperitoneal injection, and 48 h later, cumulus-enclosed oocytes were obtained by manual rupturing of antral ovarian follicles. To collect denuded oocytes, cumulus cells were removed by repeatedly pipetting. For in vitro maturation, oocytes were cultured in M2 medium under mineral oil at 37°C in a 5% CO2 incubator.
Plasmid construction and mRNA synthesis
Total RNA was extracted from 100 denuded oocytes using an Arcturus PicoPure RNA isolation kit (Applied Biosystems, CA), and the cDNA was generated with QIAquick PCR purification kit (Qiagen, Germany). PCR products were purified, digested with FseI and AscI (NEB Inc, MA), and then cloned into the pCS2+ vector with six Myc tags. The pCS2+ vectors encoding the Myc–PDHE1α substitution mutants S232A, S232D, S293A and S293D, were generated with the use of a QuikChange site-directed mutagenesis kit (Stratagene). The related primer sequences can be found in supplementary material Tables S2, S3.
For the synthesis of mRNA, the PDK and PDHE1α-pCS2+ plasmids were linearized by NotI. Capped cRNAs were made using in vitro transcription with SP6 mMESSAGE mMACHINE (Ambion, CA) according to the manufacturer's instructions, and then purified with an RNeasy Micro kit (Qiagen). Synthesized RNA was aliquoted and stored at −80°C.
PDK knockdown and overexpression
Microinjections of MO or mRNA, with a Narishige microinjector, were used to knock down or overexpress proteins in mouse oocytes, respectively. For overexpression experiments, 10 pl cRNA solution (10 ng/µl) was injected into cytoplasm of germinal vesicle oocytes. The same amount of RNase-free PBS was injected as control. For knockdown experiments, MOs against each PDK were designed by Gene Tools (Philomath, OR) through their site selection procedure, to target the initiation of translation and were thus complementary to the 5′UTR or N-terminal coding sequences; PDK2-MO and PDK3-MO targeted the 5′UTR of mRNA, whereas PDK1-MO and PDK4-MO targeted coding sequence because there were not appropriate oligonucleotides that were specific for their 5′UTR. MOs were diluted with water to give a stock concentration of 1 mM, and then 2.5 pl MO solution was injected into oocytes. A non-targeting MO was injected as a control.
For rescue experiments using PDK2 or PDK3 cRNA, 10 pl cRNA solution (10 ng/µl) was microinjected into oocytes 2 h after MO injection. The PDK2 and PDK3 cRNAs contain a 5′UTR that is not recognized by the MO: PDK1-MO (CDS), 5′-TTGTTGCTCCTCGATTAGCCATGTC-3′; PDK2-MO (UTR), 5′-GGCTTGGATCTCGCTGCTGTCCC-3′; PDK3-MO (UTR), 5′-CCTCACTAGAGCAGTGTGCAGATCC-3′; PDK4-MO (CDS), 5′-CATCACGAAGCGGGCTGCCTTCATC-3′; CTRL-MO, 5′-CCTCTTACCTCAGTTACAATTTATA-3′.
After injections, oocytes were arrested at germinal vesicle stage in M2 medium supplemented with 2.5 µM milrinone, a phosphodiesterase inhibitor that blocks meiotic maturation, for 20 h, to allow time for either MO-mediated knockdown of gene-specific mRNA translation or to permit overexpression. Then oocytes were washed three times in milrinone-free M2 medium and cultured for in vitro maturation.
Western blotting
A pool of ∼100–150 denuded oocytes was lysed in Laemmli sample buffer containing protease inhibitor and then subjected to SDS-PAGE. The separated proteins were transferred onto a PVDF membrane. Membranes were blocked in TBS containing 0.1% Tween 20 and 5% low-fat dried milk powder for 1 h and then incubated with primary antibodies. After multiple washes in TBS containing 0.1% Tween 20 and incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies, the protein bands were visualized using an ECL Plus Western Blotting Detection System. The membrane was then washed and reblotted with anti-β-actin (1:5000) antibody as a loading control. All western blot experiments were repeated at least three times.
Quantitative real-time PCR
qRT-PCR was performed as described previously (Ma et al., 2013). Total RNA was isolated from 50 oocytes using an RNAqueous-Micro Kit (Ambion, TX) following the manufacturer's protocols. The cDNA was quantified by qRT-PCR using an ABI Stepone Plus Real-time PCR system (Applied Biosystems, CA). The fold change in gene expression was calculated using the ΔΔCt method with the house keeping gene, glyceraldehydes-3-phosphate dehydrogenase (GAPDH), as the internal control. The result was expressed as fold change relative to PDK1. Primer sequences are listed in supplementary material Table S1.
Determination of total ATP content
Total ATP content in pools of 10–20 oocytes was determined using the bioluminescent somatic cell assay kit (Sigma), following the procedure described by Combelles and Albertini (2003) and the manufacturer's recommendations. A six-point standard curve (0, 0. 1, 0.5, 1.0, 10, and 50 pmol of ATP) was generated in each assay and the ATP content was calculated by using the formula derived from the linear regression of the standard curve.
Evaluation of intracellular ROS
To detect intercellular ROS in living oocytes, CM-H2DCFDA from Invitrogen was used. CM-H2DCFDA was prepared in DMSO prior to loading. Oocytes were incubated with 5 μM CM-H2DCFDA for 30 min, and then immediately observed under a fluorescence microscope (AxioVert, Zeiss, Germany).
Immunofluorescence and confocal microscopy
Oocytes were fixed with 4% paraformaldehyde for 30 min and then permeabilized with 0.5% Triton X-100 for 20 min. Following blocking in 1% BSA-supplemented PBS for 1 h, samples were incubated overnight at 4°C with primary antibody, and then at room temperature for 1 h with secondary antibody. Chromosomes were evaluated by staining with propidium iodide for 10 min. After washes in PBS, oocyte samples were mounted on anti-fade medium (Vectashield, Burlingame, CA, USA) and examined under a Laser Scanning Confocal Microscope (LSM 710, Zeiss, Germany) equipped with 40× or 63× oil objectives.
To quantify the intensity of fluorescence, Image J software (NIH) was used as previously described (Wang et al., 2012). For quantification of oocytes with meiotic defects, the gross morphology of spindle and chromosomes was assessed. The metaphase II spindle was classified as normal if it displayed the typical barrel shape. The spindle was scored as abnormal if it displayed defects, such as single or multiple poles, prominent asters and other malformations. The alignment of chromosomes was defined as normal if they tightly lay on the metaphase plate or abnormal if one or more bivalent was displaced from the metaphase plate.
Statistical analysis
Data are presented as mean±s.d., unless otherwise indicated. Differences between two groups were analyzed by Student's t-test. Multiple comparisons between more than two groups were analyzed by one-way ANOVA test using Prism 5.0. P<0.05 was considered to be significant.
Supplementary Material
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
X.H., X.Z., T.S. and Q.W. designed research; X.H., L.Z., L.H., J.G. and R.M. performed research; X.H., K.M., T.S. and Q.W. analyzed data; K.M., T.S. and Q.W. wrote the paper.
Funding
This work was supported by National Key Scientific Research Projects [grant number 2014CB943200]; and National Natural Science Foundation of China [grant number 31271541 and 31301181]. K.M. was supported by the American Diabetes Association [grant number 76274]. T.S. was supported by the National Institutes of Health [grant number R01 GM100756]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.167049/-/DC1
References
- Biggers J. D., Whittingham D. G. and Donahue R. P. (1967). The pattern of energy metabolism in the mouse oocyte and zygote. Proc. Natl. Acad. Sci. USA 58, 560-567. 10.1073/pnas.58.2.560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowker-Kinley M. M., Davis W. I., Wu P., Harris R. A. and Popov K. M. (1998). Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329, 191-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combelles C. M. H. and Albertini D. F. (2003). Assessment of oocyte quality following repeated gonadotropin stimulation in the mouse. Biol. Reprod. 68, 812-821. 10.1095/biolreprod.102.008656 [DOI] [PubMed] [Google Scholar]
- Eichenlaub-Ritter U., Vogt E., Yin H. and Gosden R. (2004). Spindles, mitochondria and redox potential in ageing oocytes. Reprod. Biomed. Online 8, 45-58. 10.1016/S1472-6483(10)60497-X [DOI] [PubMed] [Google Scholar]
- Eppig J. J. (1976). Analysis of mouse oogenesis in vitro. Oocyte isolation and the utilization of exogenous energy sources by growing oocytes. J. Exp. Zool. 198, 375-381. 10.1002/jez.1401980311 [DOI] [PubMed] [Google Scholar]
- Fagbohun C. F. and Downs S. M. (1992). Requirement for glucose in ligand-stimulated meiotic maturation of cumulus cell-enclosed mouse oocytes. J. Reprod. Fertil. 96, 681-697. 10.1530/jrf.0.0960681 [DOI] [PubMed] [Google Scholar]
- Gray L. R., Tompkins S. C. and Taylor E. B. (2014). Regulation of pyruvate metabolism and human disease. Cell. Mol. Life. Sci. 71, 2577-2604. 10.1007/s00018-013-1539-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudi R., Melissa M. B.-K., Kedishvili N. Y., Zhao Y. and Popov K. M. (1995). Diversity of the pyruvate dehydrogenase kinase gene family in humans. J. Biol. Chem. 270, 28989-28994. 10.1074/jbc.270.48.28989 [DOI] [PubMed] [Google Scholar]
- Harris S. E., Leese H. J., Gosden R. G. and Picton H. M. (2009). Pyruvate and oxygen consumption throughout the growth and development of murine oocytes. Mol. Reprod. Dev. 76, 231-238. 10.1002/mrd.20945 [DOI] [PubMed] [Google Scholar]
- Hassold T. and Hunt P. (2001). To err (meiotically) is human: the genesis of human aneuploidy. Nat. Rev. Genet. 2, 280-291. 10.1038/35066065 [DOI] [PubMed] [Google Scholar]
- Hitosugi T., Fan J., Chung T.-W., Lythgoe K., Wang X., Xie J., Ge Q., Gu T.-L., Polakiewicz R. D., Roesel J. L. et al. (2011). Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 44, 864-877. 10.1016/j.molcel.2011.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaswal J. S., Lund C. R., Keung W., Beker D. L., Rebeyka I. M. and Lopaschuk G. D. (2010). Isoproterenol stimulates 5′-AMP-activated protein kinase and fatty acid oxidation in neonatal hearts. Am. J. Physiol. Heart Circ. Physiol. 299, H1135-H1145. 10.1152/ajpheart.00186.2010 [DOI] [PubMed] [Google Scholar]
- Jeoung N. H. and Harris R. A. (2008). Pyruvate dehydrogenase kinase-4 deficiency lowers blood glucose and improves glucose tolerance in diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 295, E46-E54. 10.1152/ajpendo.00536.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. T., Freeman E. A., Gardner D. K. and Hunt P. A. (2007). Oxidative metabolism of pyruvate is required for meiotic maturation of murine oocytes in vivo. Biol. Reprod. 77, 2-8. 10.1095/biolreprod.106.059899 [DOI] [PubMed] [Google Scholar]
- Jones K. T. and Lane S. I. R. (2013). Molecular causes of aneuploidy in mammalian eggs. Development 140, 3719-3730. 10.1242/dev.090589 [DOI] [PubMed] [Google Scholar]
- Kim J.-W., Tchernyshyov I., Semenza G. L. and Dang C. V. (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177-185. 10.1016/j.cmet.2006.02.002 [DOI] [PubMed] [Google Scholar]
- Kolobova E., Tuganova A., Boulatnikov I. and Popov K. M. (2001). Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem. J. 358, 69-77. 10.1042/0264-6021:3580069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotchkina L. G. and Patel M. S. (1995). Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase. Site-specific regulation. J. Biol. Chem. 270, 14297-14304. 10.1074/jbc.270.24.14297 [DOI] [PubMed] [Google Scholar]
- Korotchkina L. G. and Patel M. S. (2001). Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J. Biol. Chem. 276, 37223-37229. 10.1074/jbc.M103069200 [DOI] [PubMed] [Google Scholar]
- Kwon H.-S. and Harris R. A. (2004). Mechanisms responsible for regulation of pyruvate dehydrogenase kinase 4 gene expression. Adv. Enzyme Regul. 44, 109-121. 10.1016/j.advenzreg.2003.11.020 [DOI] [PubMed] [Google Scholar]
- Kwon H.-S., Huang B., Ho Jeoung N., Wu P., Steussy C. N. and Harris R. A. (2006). Retinoic acids and trichostatin A (TSA), a histone deacetylase inhibitor, induce human pyruvate dehydrogenase kinase 4 (PDK4) gene expression. Biochim. Biophys. Acta 1759, 141-151. 10.1016/j.bbaexp.2006.04.005 [DOI] [PubMed] [Google Scholar]
- Lu C.-W., Lin S.-C., Chen K.-F., Lai Y.-Y. and Tsai S.-J. (2008). Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 283, 28106-28114. 10.1074/jbc.M803508200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzo K. M., Wang Q., Purcell S. H., Chi M., Jimenez P. T., Grindler N., Schedl T. and Moley K. H. (2012). High fat diet induced developmental defects in the mouse: oocyte meiotic aneuploidy and fetal growth retardation/brain defects. PLoS ONE 7, e49217 10.1371/journal.pone.0049217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Flemr M., Strnad H., Svoboda P. and Schultz R. M. (2013). Maternally recruited DCP1A and DCP2 contribute to messenger RNA degradation during oocyte maturation and genome activation in mouse. Biol. Reprod. 88, 11 10.1095/biolreprod.112.105312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFate T., Mohyeldin A., Lu H., Thakar J., Henriques J., Halim N. D., Wu H., Schell M. J., Tsang T. M., Teahan O. et al. (2008). Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem. 283, 22700-22708. 10.1074/jbc.M801765200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelakis E. D., Sutendra G., Dromparis P., Webster L., Haromy A., Niven E., Maguire C., Gammer T. L., Mackey J. R., Fulton D. et al. (2010). Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2, 31ra34 10.1126/scitranslmed.3000677 [DOI] [PubMed] [Google Scholar]
- Miquel E., Cassina A., Martínez-Palma L., Bolatto C., Trías E., Gandelman M., Radi R., Barbeito L. and Cassina P. (2012). Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis. PLoS ONE 7, e34776 10.1371/journal.pone.0034776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahle Z., Hsieh M., Pietka T., Coburn C. T., Grimaldi P. A., Zhang M. Q., Das D. and Abumrad N. A. (2008). CD36-dependent regulation of muscle FoxO1 and PDK4 in the PPAR delta/beta-mediated adaptation to metabolic stress. J. Biol. Chem. 283, 14317-14326. 10.1074/jbc.M706478200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M. S. and Korotchkina L. G. (2001). Regulation of mammalian pyruvate dehydrogenase complex by phosphorylation: complexity of multiple phosphorylation sites and kinases. Exp. Mol. Med. 33, 191-197. 10.1038/emm.2001.32 [DOI] [PubMed] [Google Scholar]
- Peters S. J., Harris R. A., Wu P., Pehleman T. L., Heigenhauser G. J. and Spriet L. L. (2001). Human skeletal muscle PDH kinase activity and isoform expression during a 3-day high-fat/low-carbohydrate diet. Am. J. Physiol. Endocrinol. Metab. 281, E1151-E1158. [DOI] [PubMed] [Google Scholar]
- Ramalho-Santos J., Varum S., Amaral S., Mota P. C., Sousa A. P. and Amaral A. (2009). Mitochondrial functionality in reproduction: from gonads and gametes to embryos and embryonic stem cells. Hum. Reprod. Update 15, 553-572. 10.1093/humupd/dmp016 [DOI] [PubMed] [Google Scholar]
- Rardin M. J., Wiley S. E., Naviaux R. K., Murphy A. N. and Dixon J. E. (2009). Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal. Biochem. 389, 157-164. 10.1016/j.ab.2009.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche T. E. and Hiromasa Y. (2007). Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell. Mol. Life Sci. 64, 830-849. 10.1007/s00018-007-6380-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T., Hiroi M. and Kato T. (1994). Development of glucose utilization studied in single oocytes and preimplantation embryos from mice. Biol. Reprod. 50, 266-270. 10.1095/biolreprod50.2.266 [DOI] [PubMed] [Google Scholar]
- Sale G. J. and Randle P. J. (1981). Analysis of site occupancies in [32P] phosphorylated pyruvate dehydrogenase complexes by aspartyl-prolyl cleavage of tryptic phosphopeptides. Eur. J. Biochem. 120, 535-540. 10.1111/j.1432-1033.1981.tb05733.x [DOI] [PubMed] [Google Scholar]
- Stacpoole P. W. (2012). The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell 11, 371-377. 10.1111/j.1474-9726.2012.00805.x [DOI] [PubMed] [Google Scholar]
- Sugden M. C. and Holness M. J. (2003). Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 284, E855-E862. 10.1152/ajpendo.00526.2002 [DOI] [PubMed] [Google Scholar]
- Sugden M. C. and Holness M. J. (2006). Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch. Physiol. Biochem. 112, 139-149. 10.1080/13813450600935263 [DOI] [PubMed] [Google Scholar]
- Takubo K., Nagamatsu G., Kobayashi C. I., Nakamura-Ishizu A., Kobayashi H., Ikeda E., Goda N., Rahimi Y., Johnson R. S., Soga T. et al. (2013). Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49-61. 10.1016/j.stem.2012.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly J. L. and Sinclair D. A. (2013). Germline energetics, aging, and female infertility. Cell Metab. 17, 838-850. 10.1016/j.cmet.2013.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso S.-C., Qi X., Gui W.-J., Wu C.-Y., Chuang J. L., Wernstedt-Asterholm I., Morlock L. K., Owens K. R., Scherer P. E., Williams N. S. et al. (2014). Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J. Biol. Chem. 289, 4432-4443. 10.1074/jbc.M113.533885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnoni A., Rodriguez L., Manser C., De Vos K. J. and Miller C. C. J. (2011). Phosphorylation of kinesin light chain 1 at serine 460 modulates binding and trafficking of calsyntenin-1. J. Cell Sci. 124, 1032-1042. 10.1242/jcs.075168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. and Sun Q.-Y. (2007). Evaluation of oocyte quality: morphological, cellular and molecular predictors. Reprod. Fertil. Dev. 19, 1-12. 10.1071/RD06103 [DOI] [PubMed] [Google Scholar]
- Wang Q., Ratchford A. M., Chi M. M.-Y., Schoeller E., Frolova A., Schedl T. and Moley K. H. (2009). Maternal diabetes causes mitochondrial dysfunction and meiotic defects in murine oocytes. Mol. Endocrinol. 23, 1603-1612. 10.1210/me.2009-0033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Chi M. M. and Moley K. H. (2012). Live imaging reveals the link between decreased glucose uptake in ovarian cumulus cells and impaired oocyte quality in female diabetic mice. Endocrinology 153, 1984-1989. 10.1210/en.2011-1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L. L., Russell D. L., Wong S. L., Chen M., Tsai T.-S., St John J. C., Norman R. J., Febbraio M. A., Carroll J. and Robker R. L. (2015). Mitochondrial dysfunction in oocytes of obese mothers: transmission to offspring and reversal by pharmacological endoplasmic reticulum stress inhibitors. Development 142, 681-691. 10.1242/dev.114850 [DOI] [PubMed] [Google Scholar]
- Zeng H.-t., Ren Z., Yeung W. S. B., Shu Y.-m., Xu Y.-w., Zhuang G.-l. and Liang X.-y. (2007). Low mitochondrial DNA and ATP contents contribute to the absence of birefringent spindle imaged with PolScope in in vitro matured human oocytes. Hum. Reprod. 22, 1681-1686. 10.1093/humrep/dem070 [DOI] [PubMed] [Google Scholar]
- Zhang S., Hulver M. W., McMillan R. P., Cline M. A. and Gilbert E. R. (2014). The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. (Lond.) 11, 10 10.1186/1743-7075-11-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuelke K. A. and Brackett B. G. (1992). Effects of luteinizing hormone on glucose metabolism in cumulus-enclosed bovine oocytes matured in vitro. Endocrinology 131, 2690-2696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.