

ABSTRACT
The protein kinase Akt (for which there are three isoforms) is a key intracellular mediator of many biological processes, yet knowledge of Akt signaling dynamics is limited. Here, we have constructed a fluorescent reporter molecule in a lentiviral delivery system to assess Akt kinase activity at the single cell level. The reporter, a fusion between a modified FoxO1 transcription factor and clover, a green fluorescent protein, rapidly translocates from the nucleus to the cytoplasm in response to Akt stimulation. Because of its long half-life and the intensity of clover fluorescence, the sensor provides a robust readout that can be tracked for days under a range of biological conditions. Using this reporter, we find that stimulation of Akt activity by IGF-I is encoded into stable and reproducible analog responses at the population level, but that single cell signaling outcomes are variable. This reporter, which provides a simple and dynamic measure of Akt activity, should be compatible with many cell types and experimental platforms, and thus opens the door to new insights into how Akt regulates its biological responses.
KEY WORDS: Live-cell imaging, Akt, IGF-I, Signaling pathways, Signaling dynamics
Summary: Description of a simple and dynamic measure of Akt signaling activity in living cells that will provide new insights into how Akt regulates its biological responses.
INTRODUCTION
Cells respond to their environment through the actions of intracellular signaling pathways. An environmental agent, such as a peptide hormone or growth factor, typically binds to the extracellular surface of its trans-membrane receptor. Through changes in conformational energy, ligand binding triggers enzymatic activity that activates multiple signaling networks. Despite many advances in biochemistry that have identified and characterized components of these networks in intimate detail, our knowledge of how growth-factor-initiated inputs are encoded into signaling outputs remains limited. Studying the response of individual cells within a population has been particularly challenging because most experimental methods lack sufficient sensitivity or exhibit low temporal resolution. Moreover, signaling pathways do not function in isolation but are interconnected and non-linear, and contain a variety of feedback and feed-forward modifiers that complicate analysis (Albeck et al., 2013; Purvis and Lahav, 2013; Tay et al., 2010; Zhou et al., 2015).
Live-cell imaging using sensitive, specific and quantifiable biosensors resolves several of the limitations inherent in biochemical assays. By enabling analysis of many individual cells within a population, this approach can result in major improvements in both the amount and quality of the data, often generating new insights into the complexities of pathway regulation (Batchelor et al., 2011; Lahav et al., 2004; Purvis and Lahav, 2013; Zhou et al., 2015). Several different types of imaging sensors have been developed to address a variety of biological questions. Fluorescence resonance energy transfer (FRET)-based reporters were among the first to be used to assess signaling activity in single cells, and have been employed to study G-protein-coupled receptors (Clister et al., 2015) and other signaling molecules, including protein kinases (Albeck et al., 2013; Gao and Zhang, 2008; Komatsu et al., 2011; Kunkel et al., 2005; Miura et al., 2014; Yoshizaki et al., 2007; Zhou et al., 2015). These studies have greatly advanced our understanding of the temporal and spatial regulation of pathway activity, and have revealed that many individual signaling responses are often hidden within population averages.
More recently, a series of fluorescent reporters have been devised that undergo movement between subcellular compartments in response to changes in specific signaling molecule activity. Translocation reporters of this type have been developed for CDK2, JNK, Erk (ERK1 and ERK2, also known as MAPK3 and MAPK1) and the p38 mitogen-activated protein kinase (MAPK) families (Regot et al., 2014; Spencer et al., 2013), for the kinases that are upstream of the transcription factors NFAT1 and NFAT4 (also known as NFATC2 and NFATC3) (Yissachar et al., 2013), p53 (Batchelor et al., 2011; Purvis et al., 2012), and for subunits of NFκβ (Nelson et al., 2004; Tay et al., 2010). Results using these reporters have shown that signaling pathways encode stimuli into a variety of different output patterns. Some pathways produce transient outputs despite continuous stimulation, others yield constant responses, whereas in others the pattern varies depending upon the type of input. Signaling pathways also differ in the level of response to a stimulus. Some exhibit graded (or analog) outputs (Toettcher et al., 2013), whereas others show all-or-none (or digital) responses (Tay et al., 2010).
The enzymatic activity of Akt protein kinases is stimulated through activation of class Ia phosphoinositide 3-kinases (PI3Ks) by hormones and growth factors (Manning and Cantley, 2007). Once activated, Akt can directly phosphorylate many substrates within several subcellular compartments (Hay, 2011; Manning and Cantley, 2007; Toker, 2012). These substrate proteins include mediators of immediate changes in cell shape, movement and intermediary metabolism, and components of longer-term effects on gene expression, cell viability, division or differentiation (Hay, 2011; Manning and Cantley, 2007; Toker, 2012). A variety of FRET-based reporters have been developed to track Akt by live-cell imaging (Gao and Zhang, 2008; Komatsu et al., 2011; Kunkel et al., 2005; Miura et al., 2014; Yoshizaki et al., 2007). Collectively, they have yielded data demonstrating rapid induction of enzymatic function in response to signaling by different growth factors, but have provided little information about how Akt activity is encoded into signaling outputs or about the dynamics of responses within a cell population. As with other FRET biosensor systems, they have required substantial investment in high-end imaging equipment and extensive expertise, and have not been widely adopted. Alternative approaches have been presented, including a bioluminescent sensor employing a split-luciferase reporter (Zhang et al., 2007), but they offer few advantages over FRET-based reporter molecules.
Here, we describe a robust fluorescent translocation sensor for measuring Akt activity. Our reporter protein is based on FoxO1, an Akt substrate that transits between the nucleus and cytoplasm (Brunet et al., 1999; Rena et al., 1999, 2002; Van Der Heide et al., 2004; Woods et al., 2001; Zhang et al., 2002). With this sensor, we are able to quantify the dynamics of Akt activity over time, and to show that insulin-like growth factor I (IGF-I)-mediated Akt signaling is encoded into stable and reproducible analog responses at the population level, but that in individual cells Akt signaling outputs are variable and mostly stochastic. The tools and approaches defined in this paper open the door to characterizing how the Akt pathway functions under a range of biological conditions in different cell types.
RESULTS
Developing a reporter to track Akt activity in living cells
We have engineered a fluorescent fusion protein to assess Akt activity at the single cell level. We devised our reporter using as a base FoxO1, a well-characterized Akt kinase substrate (Hay, 2011). FoxO1 contains three Akt phosphorylation sites that modulate the functions of nuclear localization sequence (NLS) and nuclear export sequence (NES) motifs (Fig. 1A). NLS activity is inhibited by Akt phosphorylation, whereas NES activity is enhanced, shifting the equilibrium of subcellular localization from the nucleus to the cytoplasm (Brunet et al., 1999; Rena et al., 1999, 2002; Zhang et al., 2002) (Fig. 1B). We constructed the reporter by fusing the green fluorescent protein clover (Lam et al., 2012), to the C-terminus of FoxO1. We also engineered three amino acid substitutions into the Forkhead domain of FoxO1 to inhibit its DNA-binding activity (Tang et al., 1999), and to prevent effects from phosphorylation by the protein kinase Mst1 (also known as STK4) (Lehtinen et al., 2006) (Fig. 1A). After lentiviral delivery into mouse 10T1/2 fibroblasts and C2 myoblasts, stable selection, and cell sorting, we were able to visualize rapid and robust reporter transit from the nucleus to the cytoplasm in response to the growth factor IGF-I (Fig. 1B,C).
Fig. 1.
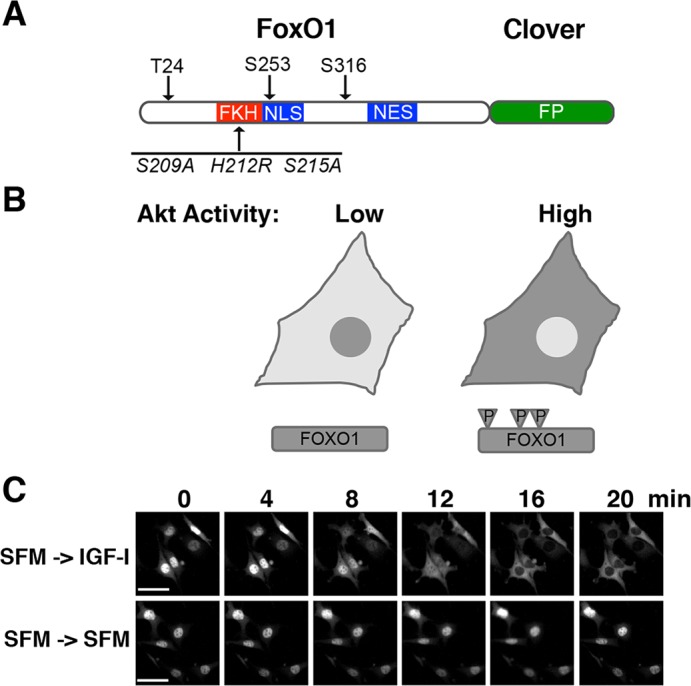
Development of a sensor for Akt activity. (A) Schematic of FoxO1–clover reporter protein showing locations of the three Akt phosphorylation sites (T24, S253 and S316) and three amino acid substitutions engineered into the Forkhead DNA binding domain (FKH) (S209A, H212R, and S215A). Also indicated are locations of the NLS and NES of FoxO1; FP, fluorescent protein. (B) Diagram of the expected location of the FoxO1–clover reporter in cells with low Akt activity, where FoxO1 is not phosphorylated (P) and is predominantly nuclear, or high activity, where FoxO1 is highly phosphorylated and is primarily cytoplasmic. (C) Time-lapse images of a representative experiment showing changes in the subcellular location of the FoxO1–clover reporter in 10T1/2 cells exposed to R3-IGF-I (250 pM) for the times indicated versus continual incubation in SFM. Scale bars: 50 µm.
Assessing growth factor specificity and responsiveness of the FoxO1–clover reporter
To study the response of the reporter to growth factors that stimulate Akt activity, we treated serum-starved 10T1/2 cells with 10% fetal bovine serum (FBS) or with individual growth factors in serum-free medium (SFM), and tracked the subcellular localization of FoxO1–clover. Cells incubated with FBS, PDGF-BB, or the IGF-I analog R3-IGF-I, for 60 min showed rapid and sustained translocation of the reporter from the nucleus to the cytoplasm in parallel with stimulation of Akt phosphorylation (Fig. 2; t1/2 of nuclear export of 6.1±1.3 min (mean±s.d.); n=50). In contrast, cells maintained in SFM or treated with BMP-2 for 60 min had a predominantly nuclear localization of FoxO1–clover, and exhibited minimal Akt phosphorylation. BMP-2 treatment stimulated phosphorylation of Smad5, one of its key intracellular signaling proteins (Katagiri and Tsukamoto, 2013; Wang et al., 2014), indicating that BMP-2 did activate its cognate receptor in 10T1/2 cells (Fig. 2). Longer-term treatment with FBS, PDGF-BB or IGF-I for up to 6 h led to sustained cytoplasmic accumulation of the reporter, whereas incubation with SFM or BMP-2 led to the maintenance of its nuclear localization (supplementary material Fig. S1).
Fig. 2.
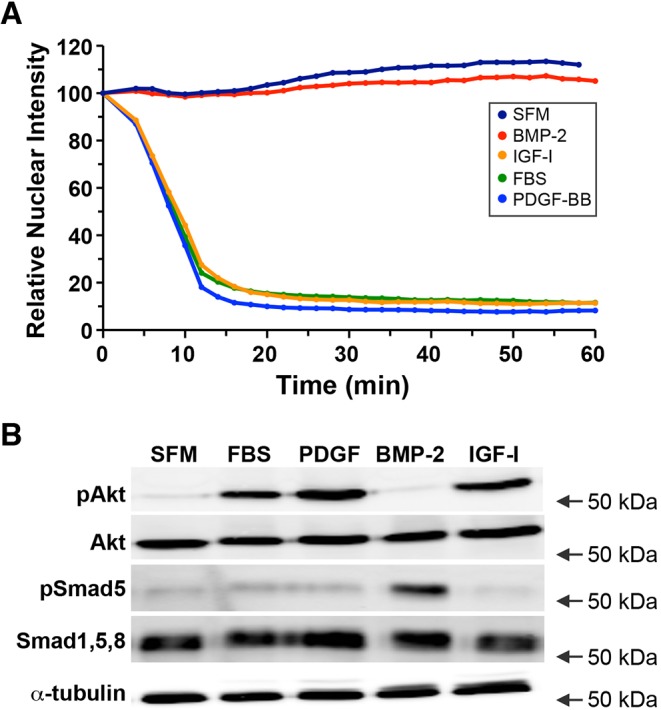
Reporter dynamics after exposure of 10T1/2 cells to different growth factors. (A) Time course of the relative nuclear intensity of the FoxO1–clover reporter in cells incubated in SFM and then exposed to SFM, BMP-2 (15 nM), R3-IGF-I (1 nM), 10% FBS or PDGF-BB (206 pM) for 60 min. Population means are presented (n=50 cells per incubation). The nuclear intensity of the reporter in each cell was normalized to its value at the start of imaging during incubation in SFM. (B) Expression of phosphorylated Akt (pAkt, at Thr308), total Akt, phosphorylated Smad5 (pSmad5), total Smad (Smad1,5,8) and α-tubulin by immunoblotting using whole-cell protein lysates from the same population analyzed in A after exposure to SFM or the indicated growth factors for 60 min. The position of the 50 kDa molecular mass marker is indicated to the right of each immunoblot.
It is possible that structural factors, such as changes in nuclear shape or volume, influence the apparent nuclear localization of the FoxO1–clover reporter protein, and thus might contribute to measurement errors, as could technical issues with our cell tracking process. To assess potential measurement errors, we re-analyzed the tracked images of five individual cells up to ten times during a 60-min incubation in SFM. Under these experimental conditions, we found that the intensity of nuclear fluorescence varied on average by ±3% from the mean value (supplementary material Fig. S2). As this value is smaller than the mean variability observed in cells incubated in serum-containing medium (see Fig. 3A below), the results suggest that our experimental system provides a sensitive readout of biological factors that act on the subcellular location of FoxO1.
Fig. 3.

Sustained Akt activity in 10T1/2 cells incubated in 10% serum. (A) Results of live tracking of 16 individual cells incubated in 10% FBS for 12 h, starting after mitosis and followed by incubation for 120 min in SFM (arrows). The relative nuclear intensity of the FoxO1–clover reporter protein recorded on the graph has been normalized to the mean value at 90 min after addition of SFM. (B) Time-lapse images from the experiment in A showing consistent cytoplasmic localization of the FoxO1–clover reporter in 10T1/2 cells during minutes 660–750 of a 750-min incubation in 10% FBS, and nuclear localization in the same cells after incubation in SFM (minutes 780, 810 and 840). Scale bars: 50 µm. (C) Heat map showing the consistently low nuclear intensity of the reporter protein in each of 65 individual cells analyzed for a complete cell cycle in 10% FBS. Cells have been aligned computationally beginning with the time since mitosis.
Establishing the half-life of the FoxO1–clover reporter
To accurately quantify signaling dynamics it is important to show that the reporter protein is stable over the duration of the experiments. Incubation of cells with or without growth factors for 6 h demonstrated that levels of FoxO1–clover were fairly constant (supplementary material Fig. S1). To formally assess the stability of the reporter molecule, we treated cells with the protein translation inhibitor cycloheximide. Under the conditions of our analyses, we found that FoxO1–clover had a half-life of >24 h, consistent with published data for FoxO1 (Sandoval et al., 2013). The half-life of CDK4 was <2 h in the same experiments, also consistent with published observations (Schwanhäusser et al., 2011), demonstrating the effectiveness of cycloheximide in blocking protein synthesis (supplementary material Fig. S3A). Thus, FoxO1–clover is a stable fusion protein, indicating that it will be a useful reagent for quantifying signaling responses over long experimental time courses.
Dynamic localization of the FoxO1–clover reporter protein in cycling cells
To test the behavior of the reporter protein over long time-course experiments, we tracked 10T1/2 fibroblasts during a 12-h incubation in medium with 10% FBS. The medium was then replaced with SFM, and cells were imaged for a further 120 min. We found that in the presence of 10% FBS the reporter was retained in the cytoplasm and exhibited only minor oscillations in intensity over the 12-h period (4% average absolute deviation from the mean) (Fig. 3A). Moreover, replacement of serum with SFM caused a rapid rise in nuclear fluorescence that was maintained for the 120-min incubation period (Fig. 3A,B; supplementary material Movie 1). Thus, levels of FoxO1–clover are relatively constant over long-term imaging studies, and the reporter remains dynamically responsive to changes in growth factor signaling.
During the 12-h incubation in serum-containing medium we noted that many cells underwent mitosis. We thus examined reporter localization during a full cell cycle, which averaged 23.8 h in 65 tracked cells incubated in medium with 10% FBS. When these individual fibroblasts were aligned based on the time since mitosis, we observed sustained cytoplasmic localization of the reporter molecule (Fig. 3C). Taken together, the results in Fig. 3 demonstrate that in proliferating 10T1/2 cells, Akt signaling activity remains steady in 10% FBS over long signaling periods and throughout the cell cycle.
Cells respond in an analog manner to IGF-I
We next assessed the effects of exposure to different concentrations of IGF-I on the rate and extent of cytoplasmic accumulation of the FoxO1–clover reporter protein. In SFM, the reporter was predominantly nuclear in 10T1/2 cells (Fig. 4A; supplementary material Movie 2). Addition of IGF-I caused a rapid and dose-dependent reduction in the nuclear levels of the reporter, with half-maximal translocation to the cytoplasm being reached by 6–10 min after the onset of incubation, and maximal values being attained within 14–16 min (Fig. 4A, supplementary material Movies 3 and 4). Similar results were seen in C2 myoblasts, but with an increase in sensitivity to IGF-I (Fig. 4C, compare with Fig. 4A), and a slower rate of cytoplasmic accumulation at the two lowest growth factor concentrations (Fig. 4C). Given that R3-IGF-I binds minimally to IGF-binding proteins, which typically inhibit acute IGF actions (Bach et al., 2005; Baxter, 2014), IGF binding proteins are probably not responsible for the variable responsiveness seen between these two cell types.
Fig. 4.
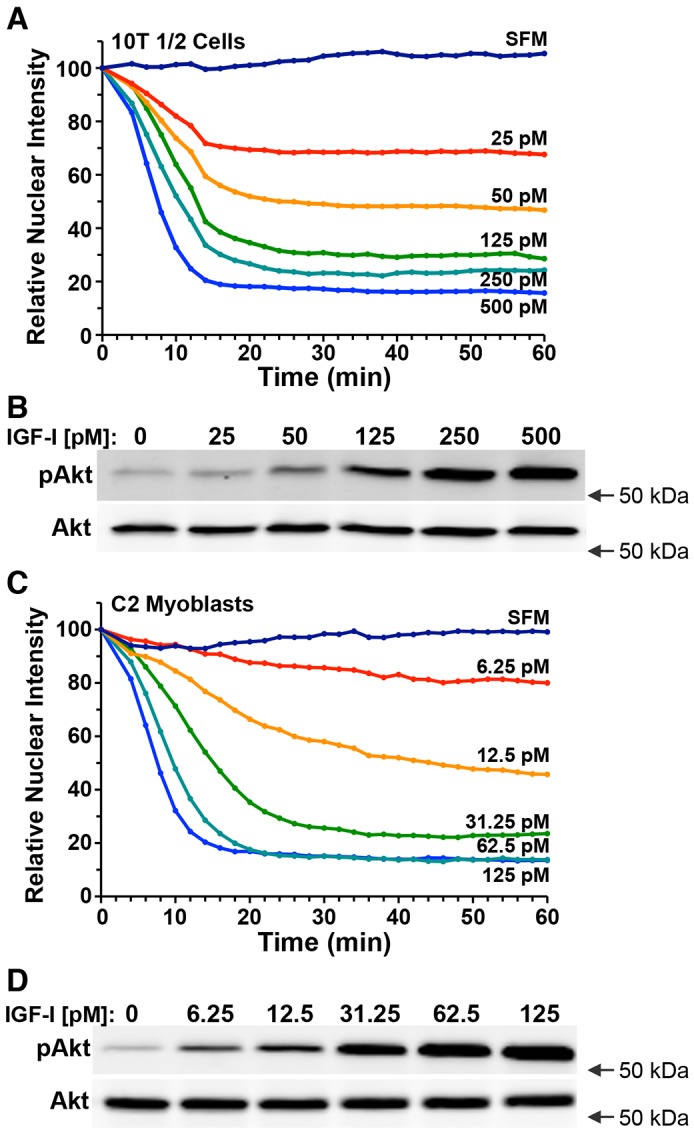
Graded responses of the FoxO1-clover reporter to different concentrations of IGF-I. (A) Time course of relative nuclear intensity of the FoxO1–clover reporter in 10T1/2 cells incubated in SFM and then exposed, starting at time 0, to different concentrations of R3-IGF-I as indicated for 60 min. Population means are presented (n=50 cells per incubation). (B) Expression of phosphorylated Akt (pAkt, at Thr308) and total Akt at 60 min after exposure to R3-IGF-I by immunoblotting using whole-cell protein lysates from the same population analyzed in A. (C) Time course of relative nuclear intensity of the FoxO1–clover reporter in C2 myoblasts incubated in SFM and then exposed to different concentrations of R3-IGF-I as indicated for 60 min. Population means are presented (n=50 cells per incubation). (D) Expression of pAkt and total Akt by immunoblotting at 60 min after incubation with R3-IGF-I using whole-cell protein lysates from the same population analyzed in C. Cells were imaged every 2 min in A and C, and the nuclear intensity of the reporter in each cell was normalized to its value at the start of imaging during incubation in SFM. Arrows in B and D represent the location of the 50 kDa molecular mass marker.
To confirm that reporter localization was tracking Akt activity, Akt phosphorylation was measured by immunoblotting whole-cell protein lysates from the same cells studied in Fig. 4A,C. In both 10T1/2 cells and C2 myoblasts, IGF-I caused a dose-dependent increase in the extent of Akt phosphorylation (Fig. 4B,D). Thus, there was a direct correspondence between the cytoplasmic localization of the FoxO1-clover reporter and the amount of Akt phosphorylation in response to treatment with IGF-I.
It is possible that expression of FoxO1–clover could disrupt Akt signaling by acting in a dominant-negative manner. To test this idea, we compared the effects of IGF-I on Akt activity between parental 10T1/2 cells and a line that stably expresses the reporter. We found similar dose-dependent increases in phosphorylation of the Akt substrate PRAS40 in both cell lines (supplementary material Fig. S3B). These data suggest that Akt signaling is not perturbed by expression of the reporter.
The time-course studies and immunoblotting results in Fig. 4 represent population averages, and thus do not provide insight into the behavior of individual cells exposed to different concentrations of IGF-I. We therefore studied single cell data. We found that individual responses to IGF-I were highly variable at lower growth factor concentrations for both 10T1/2 cells (50 pM) and C2 myoblasts (12.5 pM) (Fig. 5A,C). At higher levels of growth factor exposure [(500 pM) for 10T1/2 cells, (125 pM) for C2 cells], initial rates of export of FoxO1–clover from the nucleus were more consistent than at low IGF-I concentrations, but there was still substantial heterogeneity in the amount of reporter accumulating in the cytoplasm (Fig. 5B,D). A more in-depth visualization of these observations is depicted in Fig. 5E,F, which illustrate by frequency plots the range of signaling responses in both 10T1/2 and C2 cells during incubation with different IGF-I concentrations for 60 min. Taken together, the results in Fig. 5 show that effects of a given dose of IGF-I on individual cells are quite variable, even within populations that appear to respond consistently.
Fig. 5.

Heterogeneous responses of individual cells to IGF-I. (A,B) Time course results for each of 25 10T1/2 cells incubated with R3-IGF-I for 60 min (A, 50 pM; B, 500 pM). (C,D) Time course results for each of 25 C2 myoblasts incubated with R3-IGF-I for 60 min (C, 12.5 pM; D, 125 pM). For A–D, cells were imaged every 2 min. (E) Histograms of individual 10T1/2 cells exposed to SFM or to different concentrations of R3-IGF-I for 60 min showing the frequency of the final relative nuclear localization values (∼200 cells per each treatment). (F) Histograms of individual C2 myoblasts exposed to SFM or to different concentrations of R3-IGF-I for 60 min showing the frequency of the final relative nuclear localization values (∼200 cells per each treatment).
Reproducible population outcomes of the FoxO1–clover reporter but heterogeneous individual responses to repeated IGF-I exposures
To assess the effects of sequential exposures to IGF-I on the behavior of the FoxO1–clover reporter, cells were incubated with growth factor for 75 min, followed first by a washout period of 100 min in SFM, and then by a second incubation with IGF-I. We found that 10T1/2 cells exhibited qualitatively similar population responses to each IGF-I treatment (Fig. 6A, green tracing). Moreover, the second response to IGF-I closely matched results in cells exposed to growth factor only during the second time period (Fig. 6A, compare green and yellow tracings). As controls, cells incubated in IGF-I for the entire 250 min experiment maintained cytoplasmic expression of the reporter protein (Fig. 6A, lower blue tracing), and cells exposed to IGF-I just during the first period exhibited sustained nuclear localization after washout (Fig. 6A, red tracing). Thus, sequential population responses to IGF-I are similar to each other, and the magnitude of the second response is not influenced or conditioned by the first.
Fig. 6.
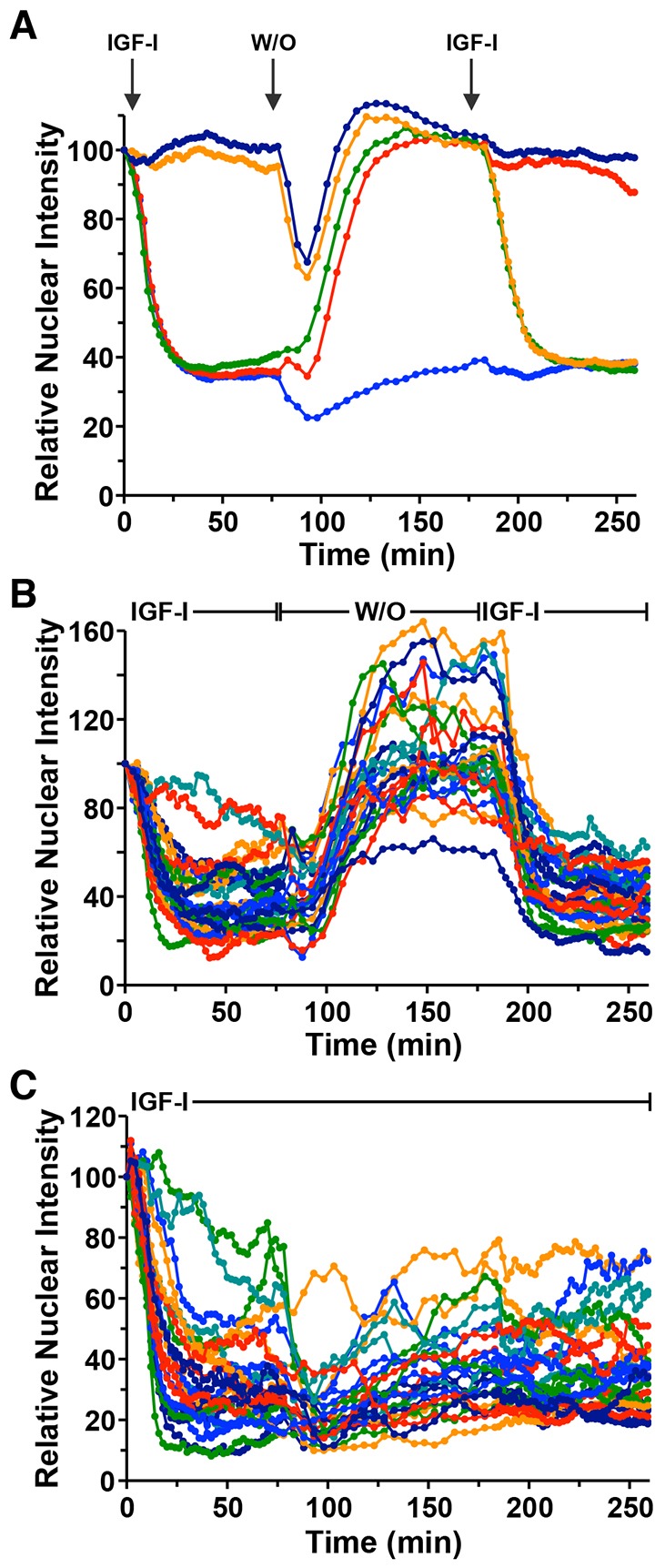
Repeated exposure to IGF-I yields similar population responses, but reveals heterogeneous effects on individual cells. (A) Time course of relative nuclear intensity of the FoxO1–clover reporter in 10T1/2 cells incubated with SFM (upper darker blue tracing), with R3-IGF-I [50 pM] (lower mid-blue tracing), sequentially with two exposures to R3-IGF-I interspersed with SFM (green tracing), with R3-IGF-I followed by SFM (red tracing), or with SFM followed by R3-IGF-I (orange tracing). Times of R3-IGF-I addition and washout (W/O) are indicated at the top. Population means are presented (n=50 cells per incubation). (B) Time course results for each of 25 individual cells incubated sequentially with R3-IGF-I interspersed with SFM as indicated. (C) Time course results for each of 25 individual cells incubated with R3-IGF-I. For A–C, cells were imaged every 2 min during each treatment period and every 5 min during the washout interval. The nuclear intensity of the reporter in each cell was normalized to its value at the start of imaging during incubation in SFM.
To assess potential signaling heterogeneity, we analyzed the behavior of 25 individual cells during either sequential or sustained treatment with IGF-I. The results revealed variable responses to the initial exposure to IGF-I (Fig. 6B, first incubation with IGF-I), and also to removal of growth factor from the medium. The data also indicated that individual cells responded variably to a second incubation with IGF-I, and to sustained treatment with IGF-I (Fig. 6C). To directly quantify these results, we plotted the relative nuclear localization of the reporter after the first incubation with IGF-I against the value at the end of the second treatment. We observed only a limited correlation for both sequential and sustained treatments (sequential R2=0.25; sustained R2=0.24), although the values were greater than seen if cells were randomly paired (supplementary material Fig. S4). These analyses reveal that individual cell responses to submaximal activation are variable across the population and partly stochastic.
Measuring the kinetics of subcellular localization with the FoxO1–clover reporter
We next focused on the kinetics of sub-cellular movement of the reporter protein in response to growth factor activity. Changes in phosphorylation at the Akt target sites in FoxO1 modulate both NLS and NES activity (Brunet et al., 1999; Rena et al., 1999, 2002; Zhang et al., 2002). This potentially creates four components that determine the rate of translocation of the FoxO1–clover reporter between subcellular compartments: rates of nuclear import when the protein is either phosphorylated or un-phosphorylated, and rates of nuclear export when it is either phosphorylated or un-phosphorylated. We attempted to study these components. To examine the rate of nuclear import of the un-phosphorylated reporter molecule, we incubated cells in SFM with leptomycin B, an inhibitor of nuclear export (Wolff et al., 1997). This resulted in a rapid increase in nuclear accumulation (rate constant of 0.126 min−1, Fig. 7A, upper blue tracing). By contrast, addition of leptomycin to cells pre-incubated with IGF-I led to a fourfold slower rate of nuclear localization (rate constant of 0.030 min−1, Fig. 7A, upper blue tracing; supplementary material Movie 5). Blocking IGF-stimulated Akt activity with PI-103, a dual PI3K and mammalian target of rapamycin complex 2 (mTorc2) inhibitor (Fan et al., 2006), more than doubled the rate of nuclear accumulation of the reporter in the presence of IGF-I plus leptomycin (rate constant of 0.069 min−1, Fig. 7A, red tracing; supplementary material Movie 6). The change in the rate of nuclear import seen with PI-103 under these conditions suggests that the FoxO1–clover reporter undergoes rapid de-phosphorylation upon inhibition of PI3K- and Akt-mediated signaling.
Fig. 7.
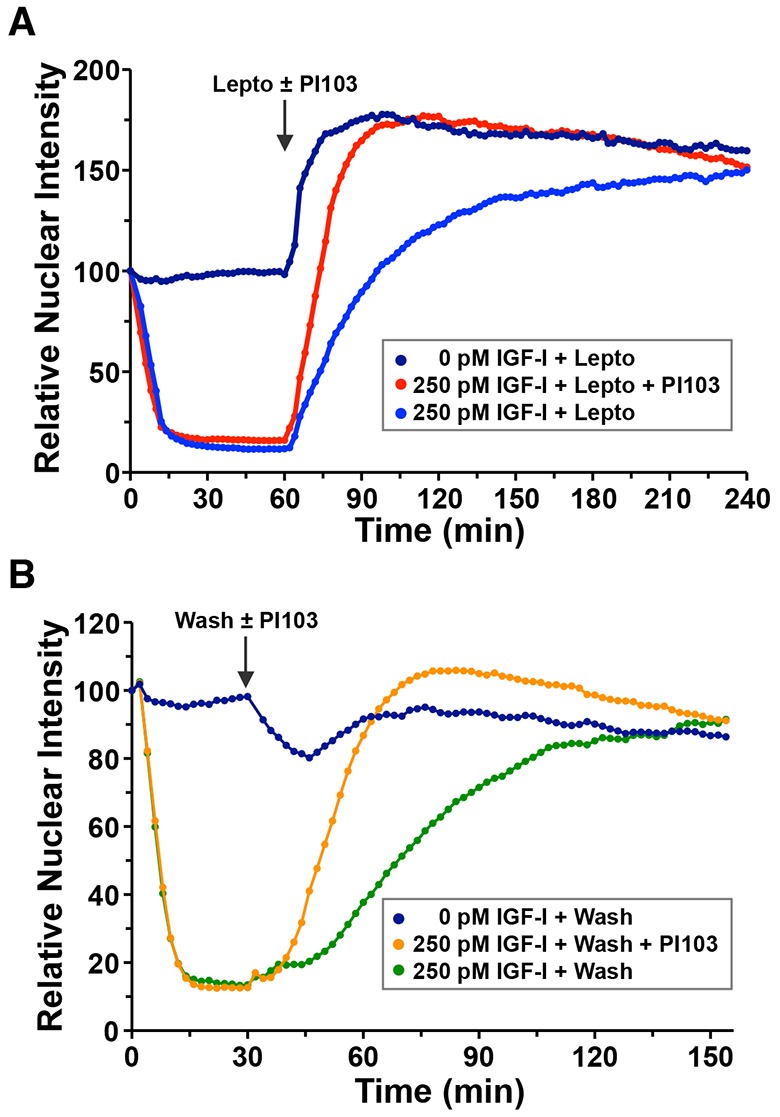
The FoxO1–clover reporter actively shuttles between the nucleus and cytoplasm. (A) Time course of the relative nuclear intensity of the FoxO1–clover reporter in 10T1/2 cells incubated with SFM or R3-IGF-I (250 pM) as indicated for 60 min, followed by addition of leptomycin B (100 nM, Lepto) alone or with PI-103 (500 nM) for 180 min. Population means are presented (n=50 cells per incubation). The arrow indicates the time of addition of Lepto or PI-103. (B) Time course of relative nuclear intensity of the FoxO1–clover reporter in 10T1/2 cells incubated with SFM or R3-IGF-I (250 pM) at time 0 followed by a wash and addition of SFM or PI-103 at 30 min. Cells were imaged every 2 min in both A and B.
To gain more insight into the kinetics of pathway activation and inactivation, we treated cells with IGF-I, washed out growth-factor-containing medium with SFM, and added SFM with or without PI-103. As expected, IGF-I caused FoxO1-clover to rapidly accumulate in the cytoplasm (Fig. 7B). The reporter then returned to the nucleus in the presence of SFM, and the rate of nuclear import increased by ∼threefold in the presence of PI-103 (0.015 min−1 in SFM versus 0.044 min−1 in SFM plus PI-103, Fig 7B, compare green and orange tracings), although this was nearly three times slower than in cells incubated in SFM plus leptomycin B (0.126 min−1, Fig. 7A, dark blue tracing). Taken together, these data indicate that the FoxO1–clover reporter moves continuously between the nuclear and cytoplasmic compartments, and that the rate constants of nuclear export are dependent on Akt kinase activity. In addition, the results show that IGF-I–PI3K–Akt signaling remains active after growth factor removal from the medium, as the half-time of nuclear accumulation was >30 min after IGF-I was washed out (Fig 7B, green tracing).
Quantifying fractional subcellular localization
Incubation of cells with leptomycin B also showed that nuclear import of the FoxO1–clover reporter could be increased significantly beyond the level seen in SFM, raising the possibility that a basal level of Akt signaling was present even in cells that were not stimulated by serum or IGF-I. To address this question, cells were incubated in SFM, followed by addition of PI-103. PI-103 caused only a small increase (∼10%) in the concentration of reporter in the nucleus compared with cells in SFM alone (Fig. 8A, compare red and green tracings). Subsequent addition of leptomycin caused a >50% rise in the nuclear intensity of the FoxO1–clover reporter (Fig. 8A, blue tracing). We conclude that in cells incubated in SFM, there is little basal Akt activity.
Fig. 8.
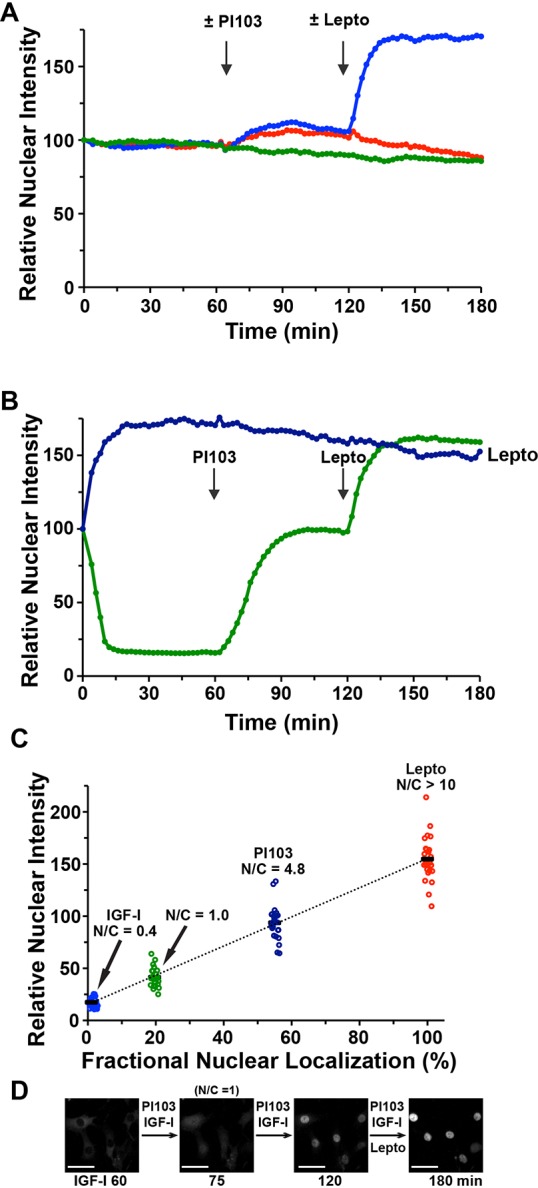
Quantifying the subcellular localization of the FoxO1–clover reporter. (A) Time course of relative nuclear intensity of the FoxO1–clover reporter in 10T1/2 cells incubated in SFM for 60 min and then exposed sequentially to PI-103 (500 nM; red and blue tracings) and leptomycin B (Lepto, 100 nM; blue tracing), as indicated by the vertical arrows. The green tracing represents cells incubated in SFM for the entire 180 min experimental period. (B) Time course of relative nuclear intensity of the FoxO1–clover reporter in 10T1/2 cells incubated with R3-IGF-I (250 pM) for 60 min, followed by PI-103 (500 nM) and leptomycin B (100 nM) for 60 min each (green tracing). The blue tracing represents results of cells incubated with leptomycin B for 180 min. Vertical arrows indicate time of additions. Cells were imaged every 2 min in A and B; population results were derived from analysis of 50 cells for each tracing. (C) Graphical view of quantitative data from individual cells (n=25) plotted from the experiments depicted in the green tracing in B. The mean ratio of nuclear to cytoplasmic fluorescence (N/C) is listed above each cluster of individual cells. See Materials and Methods for additional details. (D) Time-lapse images of a field of cells from the experiment plotted in B. Scale bars: 50 µm.
Having established that exposure of cells to higher concentrations of IGF-I could promote extensive nuclear exclusion of the FoxO1–clover reporter protein, and conversely finding that leptomycin could maximize nuclear localization, we attempted to use a series of manipulations to determine the actual fraction of reporter protein in the nucleus under different conditions. We thus measured nuclear and cytoplasmic fluorescence values for FoxO1–clover at different time points during a series of sequential treatments: after serum starvation (time 0), at 60 min after incubation with IGF-I (250 pM), at 60 min after subsequent addition of PI-103, and at 60 min after addition of leptomycin (summary population data appear in Fig. 8B and representative images in Fig. 8D). To place our observations in context with published studies using live-cell imaging (Regot et al., 2014; Tay et al., 2010), at each time point we also measured the ratio of nuclear to cytoplasmic fluorescence (denoted N/C), including when cytoplasmic and nuclear fluorescence intensities were identical (N/C=1). Although this varied among different cells, it typically occurred by ∼15 min after addition of PI-103 (Fig. 8C,D; supplementary material Movie 7). To calculate the fraction of the FoxO1–clover reporter in each subcellular compartment, we assigned the value at 60 min of leptomycin treatment as 100% nuclear localized, and the level at 60 min of exposure to IGF-I (250 pM) as 100% cytoplasmic. With leptomycin, we recorded no cytoplasmic fluorescence, but with IGF-I a small amount of nuclear fluorescence was detected, which was likely derived from the cytoplasm above and/or below the nucleus in the cells analyzed. By fitting the values of cells incubated with PI-103 and when N/C=1 between the two boundary conditions, we determined that ∼56% of the reporter was in the nucleus after PI-103 treatment and that ∼19% was in the nucleus when the nuclear and cytoplasmic fluorescence intensities were equivalent (Fig. 8C,D). These results reveal the importance of the method used for quantification, and suggest a source of variability when comparing cell types with proportionally different nuclear and cytoplasmic volumes.
DISCUSSION
Despite substantial progress in elucidating the biochemistry of many different signaling pathways, there remains a limited understanding of how components of these networks function in real time in cells and tissues (Purvis and Lahav, 2013). Here, we developed and tested a robust sensor that measures the activity of Akt protein kinases in individual cells with high sensitivity and specificity. The sensor protein, which is composed of a fusion between a modified FoxO1 transcription factor and the green fluorescent protein clover, was stably expressed and could be tracked for days under a variety of situations. This work provides new insights into how the Akt pathway functions, and further demonstrates the power of live-cell imaging to reveal and quantify dynamic cellular behaviors at the signal cell level.
Development of a translocation reporter for Akt activity
Most current methods to measure Akt signaling activity can be categorized as endpoint assays. These tend to be labor-intensive, and at best provide population averages. Some of these limitations have been overcome with the development of FRET reporters that measure kinase activity in real time by live-cell imaging (Gao and Zhang, 2008; Komatsu et al., 2011; Kunkel et al., 2005; Miura et al., 2014; Zhang et al., 2007), although these assays require investment in high-end equipment and substantial expertise. Recently, new biological sensors have been developed that translocate between the nucleus and the cytoplasm in response to a stimulus. These molecules typically maintain high signal-to-noise ratios, provide robust readouts in response to changes in signaling activity and are generally easier to use than FRET reporters (Hao et al., 2013; Regot et al., 2014; Spencer et al., 2013). Here, we have developed and characterized a translocation reporter for Akt kinase activity based on the transcription factor FoxO1. The reporter protein contains three Akt phosphorylation sites that are each crucial for mediating changes in subcellular localization (Brunet et al., 1999; Rena et al., 1999, 2002; Zhang et al., 2002). The presence of several phosphorylation sites, rather than one, is highly valuable in a kinase activity sensor, as they can extend the dynamic range of responses, and can potentially dampen signaling noise. In addition, having multiple phosphorylation sites helps insure specificity of the reporter, especially when other kinases might have partially overlapping substrate phosphorylation motifs.
Akt-mediated signaling is sustained
Many signaling pathways that have been examined at the single cell level, including the NFκβ and Erk pathways, are activated transiently in response to most signaling stimuli (Albeck et al., 2013; Regot et al., 2014; Tay et al., 2010). In our experiments, we find that several growth factors persistently activate Akt signaling, as judged by the continual cytoplasmic localization of the FoxO1–clover reporter (Figs 2–5). Sustained Akt activity under these conditions suggests that activation does not lead to significant negative feedback that otherwise would diminish responses over time. This presents an interesting contrast between the Akt and Erk signaling pathways, as both can be both stimulated by the same growth factors, but with the exception of carcinogenic mutations upstream of Erk, Akt remains persistently active whereas Erk becomes rapidly inhibited (De Luca et al., 2012; Sever and Brugge, 2015). It thus will be of interest to determine by live-cell imaging how Erk and Akt signaling behave in the same cells in response to growth factors, and whether and how their kinetics might vary.
Growth factor stimulation of Akt activity is encoded into analog signaling responses
IGF-I-induced activation of Akt leads to graded levels of signaling responses. In the two cell lines tested, we observed well-defined dose–response curves up to maximal values, with higher sensitivity in C2 myoblasts than in 10T1/2 fibroblasts (Fig. 4). In comparison to the overall population, individual cell responses to IGF-I were variable. Some cells exhibited rapid and extensive translocation of the FoxO1–clover fusion protein from the nucleus to the cytoplasm, whereas in others the reporter responded minimally (Figs 5 and 6). Heterogeneous responses to growth factor exposure were broader at lower doses of IGF-I than at higher values, but were present at both treatment levels (Figs 5 and 6). Thus, our data show that IGF-I-mediated signaling is encoded into analog outputs, and is highly variable at the level of individual cells.
Based on the variability observed in single cells to initial treatment with IGF-I, we compared responses to sequential stimuli separated by a wash out period (Fig. 6). Results showed a tight correlation in the population between the first and second signaling responses, but much less so at the individual cell level (Fig. 6A,B; supplementary material Fig. S4; R2=0.25 for sequential IGF-I treatment for single cells). These data indicate the existence of factors that vary among different cells within a population and influence signaling outcomes. One group of such factors might be IGF signaling components, including IGF-I receptor, IRS-1 or IRS-2 adaptors, PI3K subunits, PTEN, PDK1 or PDK2 (which regulate mTorc2), PHLPP or any of the three Akt proteins (Baserga, 2013; Hay, 2011; Manning and Cantley, 2007; Toker, 2012; Yee, 2012). It is conceivable that levels of these proteins change dynamically over time, and collectively that these alterations exert apparently stochastic effects on the extent of signaling responses across a population. Although speculative, this idea could be tested through targeted modifications in expression levels of one or more of these molecules.
Rapid activation and inactivation of Akt signaling
Our results show that at maximal doses of IGF-I, Akt signaling is activated within minutes. We detected measurable cytoplasmic levels of the FoxO1–clover reporter within 2 min of growth factor treatment, and observed nearly all of the reporter molecules in the cytoplasm within 15 min after growth factor exposure (Fig. 2A; Fig. 3A,C). These values are comparable to measurements obtained with some, but not all, Akt FRET reporters (Gao and Zhang, 2008; Komatsu et al., 2011; Kunkel et al., 2005; Miura et al., 2014; Zhang et al., 2007). We also detected comparably rapid nuclear translocation of the reporter after addition of the PI-103 kinase inhibitor to the medium of cells incubated with IGF-I (Fig. 7). Taken together, these results suggest that the FoxO1–clover reporter represents a faithful and dynamic readout of competing kinase and phosphatase activities. Our data also show that at the population level IGF-I-mediated signaling to Akt has a fairly long lifetime (t1/2 of ∼30 min after growth factor withdrawal, Fig. 7B).
Dynamic equilibrium of the FoxO1–clover reporter between nucleus and cytoplasm
Although the FoxO1–clover protein is primarily nuclear in SFM, addition of the nuclear export inhibitor, leptomycin B, led to a ∼50% increase in the amount of reporter in the nucleus (t1/2 of ∼5 min, Fig. 7A; Fig. 8A). These results indicate that, like FoxO1, the reporter protein shuttles continuously between subcellular compartments. Akt signaling changes this dynamic equilibrium in favor of the cytoplasm, as revealed when its activity is disrupted by PI-103 being added to cells treated with IGF-I (Fig. 8B), resulting in rapid nuclear accumulation of the reporter. Quantification of the amount of reporter protein in the cytoplasm and nucleus under different conditions also showed that nuclear to cytoplasmic ratios can be misleading. Although incubation of cells in SFM led to nuclear fluorescence being fourfold brighter than cytoplasmic fluorescence, the quantity of reporter in each compartment was roughly equivalent (Fig. 8C). This result reflects the fact that in fibroblasts, cytoplasmic volume is ∼four times greater than nuclear volume (Swanson et al., 1991). Given that the ratio of nuclear to cytoplasmic volume varies among cell types and even among the same cells in culture, a nuclear to cytoplasmic fluorescence ratio might incorrectly estimate the amount of reporter protein in either compartment.
Limitations of a translocation reporter
Despite the advantages of a translocation reporter there are also several assumptions inherent in its use. In our studies, we measured nuclear fluorescence intensity, but this value might vary independent of Akt activity if the size or shape of the nucleus or cytoplasm were to change during the time course of an experiment. This type of structural alteration might occur when cells elongate or migrate. Readouts from a translocation reporter also might vary with alterations in phosphatase activity, or in the activity of nuclear import or export machinery, although the latter seems to be relatively stable based on results using translocation reporters with mutated phosphorylation sites (Regot et al., 2014).
Future directions
In developing a translocation reporter for Akt activity, we sought to maintain the native substrate properties of the molecule as much as possible. As a consequence, this reporter provides a potential standard for comparison against future sensor variations. We envision the possibility of having multiple Akt reporter molecules based on other Akt substrates such as Bad or GSK3β (Manning and Cantley, 2007) that could respond in distinct ways to changes in Akt activity. A range of fluorescent reporters with specific properties also could provide a means of unraveling the unique functions of each of the three Akts present in mammalian cells (Manning and Cantley, 2007), and could better address broader questions about Akt kinase specificity, competition between substrates and network connectivity.
MATERIALS AND METHODS
Reagents
Fetal bovine serum (FBS) and newborn calf serum were obtained from Hyclone (Logan, UT). Okadaic acid was from Alexis Biochemicals (San Diego, CA); protease inhibitor and NBT/BCIP tablets were purchased from Roche Applied Sciences (Indianapolis, IN). Dulbecco's modified Eagle's medium (DMEM), FluoroBrite, phosphate-buffered saline (PBS), and trypsin-EDTA solution were from Gibco-Life Technologies (Carlsbad, CA). Puromycin was purchased from Enzo Life Sciences (Farmingdale, NY), polybrene was from Sigma-Aldrich (St Louis, MO), and leptomycin B was from Cell Signaling (Beverly, MA; 200 µM solution in ethanol). Cycloheximide was purchased from US Biochemical (Cleveland, OH). PI-103 was from Tocris (Bristol, UK), and was solubilized in DMSO. Cells for imaging were grown on Greiner Bio-One tissue culture plates (Monroe, NC). Restriction enzymes, buffers, ligases and polymerases were purchased from Roche Applied Sciences (Indianapolis, IN) and BD Biosciences-Clontech (Palo Alto, CA). AquaBlock EIA/WIB solution was from East Coast Biologicals (North Berwick, ME). R3-IGF-I was purchased from GroPep (Adelaide, Australia), recombinant human PDGF-BB was from Invitrogen (Carlsbad, CA), and recombinant human BMP-2 was purchased from R&D Systems (Mineapolis, MN). Growth factors were solubilized in 10 mM HCl with 1 mg/ml bovine serum albumin, stored in aliquots at −80°C, and diluted into FluoroBrite imaging medium immediately prior to use. Primary antibodies were purchased from the following suppliers: Cell Signaling, anti-phospho-PRAS40 (catalog number 2997), anti-PRAS40 (catalog number 2691), anti-GFP (catalog number 2955), anti-Akt (catalog number 4691), and anti-phospho-Akt (phosphorylated at Thr308; catalog number 2965); Santa Cruz Biotechnology (Santa Cruz, CA), anti-Cdk4 (sc-260) and anti-Smad (catalog number H-465); Abcam (Cambridge, UK), anti-phospho-Smad5 (phosphorylated at Ser463 and Ser465; catalog number 76296); and Sigma-Aldrich, anti-α-tubulin. Secondary antibodies included goat anti-rabbit-IgG and anti-mouse-IgG conjugated to Alexa Fluor 680 (Invitrogen), and IR800-conjugated goat anti-rabbit IgG, Rockland (Gilbertsville, PA). Other chemicals and reagents were purchased from commercial suppliers.
Production of recombinant lentiviruses
To construct a recombinant lentivirus encoding the FoxO1–clover fusion protein, a cDNA for full-length mouse FoxO1 was generated by PCR, using the cDNA insert from pdsRED-Mono-N1-FoxO1 as a template (plasmid number 34678, Addgene, Cambridge, MA). The 3′ end of the FoxO1 coding region was ligated in-frame to the 5′ end of the green fluorescent protein clover (Lam et al., 2012). The following three amino acid substitutions were introduced into the DNA of the Forkhead domain of FoxO1, using splice-overlap-extension PCR: S209A, H212R and S215A. All DNA modifications were confirmed by sequencing at the OHSU DNA Core Facility. Recombinant lentiviruses were prepared by co-transfecting a transfer vector containing the FoxO1–clover cDNA with third-generation packaging plasmids (plasmid numbers 12251, 12253, 12259, Addgene) into Hek293FT cells (Gibco-Life Technologies) as described previously (Tiscornia et al., 2006). Virus was purified and concentrated by centrifugation of cell culture supernatant at 19,000 g at 4°C for 2 h (Mukherjee et al., 2010).
Lentiviral infection and selection
C3H10T1/2 mouse embryonic fibroblasts (ATCC number CCL226) were incubated in DMEM supplemented with 10% FBS. Mouse C2 myoblasts (Yaffe and Saxel, 1977) were grown in DMEM supplemented with 10% FBS and 10% newborn calf serum. Cells were transduced at 50% of the confluent density with concentrated virus in the presence of 6 µg/ml polybrene, as described previously (Mukherjee et al., 2010). Cells were then selected by incubation with puromycin (2 µg/ml) for 1 week. Surviving cells were sorted by fluorescence intensity using a Becton-Dickinson Influx cell sorter at the OHSU Flow Cytometry Core Facility. Reporter expression was stable for at least 10 passages in each sorted cell population.
Cell imaging
All imaging studies were performed with FluoroBrite medium containing the following supplements: L-glutamine [final concentration: (2 mM)], selenium (0.005 mg/l), ethanolamine (1.9 mg/l), bovine serum albumin (400 mg/l) and transferrin (0.5 mg/l). Growth factors, leptomycin B and PI-103 were diluted into supplemented FluoroBrite just before use. Live-cell imaging was conducted using an EVOS FL Auto microscope with a built-in stage top incubator maintained at 37°C and in 95% air, 5% CO2. Images were collected at 100× magnification using a 10× fluorite objective with a numerical aperture (NA) of 0.3. Images were acquired at intervals of 2 to 10 min, using a GFP LED light cube (excitation peak, 472/22 nm; emission peak, 510/42 nm). All of the collected data were transformed prior to quantification, using ImageJ plug-ins (NIH, Bethesda, MD), as follows. To eliminate background fluorescence, each image underwent a polynomial fit, and the fit was subtracted from each image (Polynomial Fit module). To account for movement between frames, image sequences were registered using the rigid registration function in the Stack Reg plug-in. To reduce fluorescence heterogeneity in the nucleus, each image was subjected to a 2-pixel Gaussian blur using the Gaussian blur module. For image quantification, we employed the mTrackJ plug-in module (Meijering et al., 2012), and monitored individual cells by selecting a specific location in the nucleus. In addition, to account for differences across the population and between experiments, in each experiment the nuclear fluorescence intensity in individual cells was normalized to the intensity after incubation in SFM for 90 min. Cells that divided, migrated out of the image frame or overlapped with another cell were excluded from analysis. Detailed protocols for individual experiments can be found below.
Imaging protocols
Long-term imaging under cellular growth conditions
10T1/2 cells were imaged every 10-min for ∼12 h in supplemented FluoroBrite medium plus 10% FBS. Cells were then washed twice with DMEM and incubated for 120 min in SFM plus FluoroBrite. For tracking signaling responses throughout the cell cycle, 10T1/2 cells were imaged every 10 min for 48 h in supplemented FluoroBrite medium plus 10% FBS.
Reporter protein half-life
10T1/2 cells were incubated in 10% FBS with cycloheximide (100 µg/ml) and whole-cell lysates were collected 0, 2, 4, 8 and 24 h later.
Responses to different growth factors
10T1/2 cells were incubated in supplemented FluoroBrite plus IGF-I (1 nM), PDGF-BB (206 pM), BMP-2 (15 nM), 10% FBS or SFM. Cells were imaged every 2 min for 60 min, and every 5 min for 360 min. At the end of the 60-min imaging period, whole-cell lysates were collected.
Responses to different concentrations of IGF-I
10T1/2 cells and C2 myoblasts were incubated in SFM for 90 min. IGF-I was added in supplemented FluoroBrite (0 to 500 pM), and cells were imaged every 2 min for 60 min.
Responses to sequential IGF-I
10T1/2 cells were incubated in SFM for 90 min. IGF-I (50 pM) was added in supplemented FluoroBrite, and cells were imaged every 2 min for 75 min. Cells were then washed twice in DMEM and incubated in FluroBrite-containing medium for 100 min, with imaging every 5 min. This was followed by a second 75-min treatment with IGF-I with a similar imaging protocol. In addition, cells were incubated continuously with IGF-I (50 pM) for 250 min under the same conditions as for sequential treatment.
Effects of leptomycin and PI3K inhibition
For the results shown in Figs 7 and 8, 10T1/2 cells were incubated in SFM for 90 min. IGF-I (0 to 500 pM) was added in supplemented FluoroBrite, and cells were imaged every 2 min. In another series of experiments, after 60 min of IGF-I treatment, leptomycin B (100 nM), PI-103 (500 nM), or both drugs were added to medium, and imaging was continued for another 120 min. In other studies, after 30 min of incubation with IGF-I (250 pM), cells were washed twice with SFM, and then incubated in FluoroBrite with or without PI-103 (500 nM). For these latter experiments, the kinetics of nuclear import and export were calculated by fitting the mean response from 50 cells to a single exponential equation using GraphPad Prism (San Diego, CA).
Imaging data analysis
To assess signaling variability over time in cells incubated in 10% FBS, measurements of nuclear intensity of the FoxO1–clover reporter were summed from each of 50 cells for 4 h (total of 24 data points per cell) using information from Fig. 3A, and the mean value was determined for each cell. The absolute deviation from the mean was then calculated at each time point, and across all time points. To assess measurement error, the nuclear intensity of the FoxO1–clover reporter was determined in each of five cells for a total of ten times by analyzing the same video recordings (see supplementary material Fig. S1). These results were summed and the average absolute deviation was calculated. To determine the fraction of the FoxO1–clover reporter in the nucleus in Fig. 8C, fluorescence intensities were measured in the nuclear and cytoplasmic compartments of 10T1/2 cells treated with IGF-I (250 pM), PI-103 (500 nM) and leptomycin (100 nM) at five different time points: (1) in SFM; (2) after 60 min of IGF-I; (3) when nuclear fluorescence in the nucleus and peri-nuclear cytoplasm were equal (this time point varied, but usually occurred ∼15 min after the addition of PI-103); (4) 60 min after addition of PI-103; and (5) 60 min after addition of leptomycin. For subsequent quantification, the nuclear fluorescence intensity at 60 min after IGF-I treatment was assigned a value of 0% nuclear localization, and the intensity at 60 min after leptomycin was assigned 100%. From these two values, we constructed a linear equation to determine the percentage of nuclear localization based on nuclear fluorescence intensity, and used this information to calculate the percentage of nuclear localization after incubation of cells with PI-103, and when nuclear and cytoplasmic fluorescence intensities were equal (see Fig. 8C).
Protein extraction and immunoblotting
Whole-cell protein lysates were prepared as described previously (Mukherjee and Rotwein, 2008). Protein aliquots (15 µg/lane) were resolved by SDS-PAGE (10–12% separating gel), followed by transfer onto Immobilon-FL membranes, and blocking with a 50% solution of AquaBlock. Membranes were incubated sequentially with primary and secondary antibodies (Mukherjee and Rotwein, 2008). Primary antibodies were incubated for 12–16 h at a 1:1000 dilution, except for α-tubulin (1:10,000), and secondary antibodies for 90 min at 1:5000. Images were captured using the LiCoR Odyssey and version 3.0 analysis software (Lincoln, NE).
Supplementary Material
Acknowledgements
We appreciate the technical assistance of Courtney Roach.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
S.M.G. and P.R. conceived of experiments; S.M.G. performed experiments; S.M.G. and P.R. interpreted results and wrote the manuscript.
Funding
These studies were supported by National Institutes of Health (NIH) [grant number R01 grant DK042748 to P.R.]; and by an NIH Training Grant [grant number T32 CA106195 to S.M.G.]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.168773/-/DC1
References
- Albeck J. G., Mills G. B. and Brugge J. S. (2013). Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell 49, 249-261. 10.1016/j.molcel.2012.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach L. A., Headey S. J. and Norton R. S. (2005). IGF-binding proteins – the pieces are falling into place. Trends Endocrinol. Metab. 16, 228-234. 10.1016/j.tem.2005.05.005 [DOI] [PubMed] [Google Scholar]
- Baserga R. (2013). The decline and fall of the IGF-I receptor. J. Cell Physiol. 228, 675-679. 10.1002/jcp.24217 [DOI] [PubMed] [Google Scholar]
- Batchelor E., Loewer A., Mock C. and Lahav G. (2011). Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol. 7, 488 10.1038/msb.2011.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter R. C. (2014). IGF binding proteins in cancer: mechanistic and clinical insights. Nat. Rev. Cancer 14, 329-341. 10.1038/nrc3720 [DOI] [PubMed] [Google Scholar]
- Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J. and Greenberg M. E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857-868. 10.1016/S0092-8674(00)80595-4 [DOI] [PubMed] [Google Scholar]
- Clister T., Mehta S. and Zhang J. (2015). Single-cell analysis of G-protein signal transduction. J. Biol. Chem. 290, 6681-6688. 10.1074/jbc.R114.616391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A., Maiello M. R., D'Alessio A., Pergameno M. and Normanno N. (2012). The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 16 Suppl 2, S17-S27. 10.1517/14728222.2011.639361 [DOI] [PubMed] [Google Scholar]
- Fan Q.-W., Knight Z. A., Goldenberg D. D., Yu W., Mostov K. E., Stokoe D., Shokat K. M. and Weiss W. A. (2006). A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 9, 341-349. 10.1016/j.ccr.2006.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X. and Zhang J. (2008). Spatiotemporal analysis of differential Akt regulation in plasma membrane microdomains. Mol. Biol. Cell 19, 4366-4373. 10.1091/mbc.E08-05-0449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao N., Budnik B. A., Gunawardena J. and O'Shea E. K. (2013). Tunable signal processing through modular control of transcription factor translocation. Science 339, 460-464. 10.1126/science.1227299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N. (2011). Interplay between FOXO, TOR, and Akt. Biochim. Biophys. Acta 1813, 1965-1970. 10.1016/j.bbamcr.2011.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri T. and Tsukamoto S. (2013). The unique activity of bone morphogenetic proteins in bone: a critical role of the Smad signaling pathway. Biol. Chem. 394, 703-714. 10.1515/hsz-2012-0310 [DOI] [PubMed] [Google Scholar]
- Komatsu N., Aoki K., Yamada M., Yukinaga H., Fujita Y., Kamioka Y. and Matsuda M. (2011). Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 22, 4647-4656. 10.1091/mbc.E11-01-0072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel M. T., Ni Q., Tsien R. Y., Zhang J. and Newton A. C. (2005). Spatio-temporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J. Biol. Chem. 280, 5581-5587. 10.1074/jbc.M411534200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav G., Rosenfeld N., Sigal A., Geva-Zatorsky N., Levine A. J., Elowitz M. B. and Alon U. (2004). Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 36, 147-150. 10.1038/ng1293 [DOI] [PubMed] [Google Scholar]
- Lam A. J., St-Pierre F., Gong Y., Marshall J. D., Cranfill P. J., Baird M. A., McKeown M. R., Wiedenmann J., Davidson M. W., Schnitzer M. J. et al. (2012). Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 9, 1005-1012. 10.1038/nmeth.2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen M. K., Yuan Z., Boag P. R., Yang Y., Villén J., Becker E. B. E., DiBacco S., de la Iglesia N., Gygi S., Blackwell T. K. et al. (2006). A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125, 987-1001. 10.1016/j.cell.2006.03.046 [DOI] [PubMed] [Google Scholar]
- Manning B. D. and Cantley L. C. (2007). AKT/PKB signaling: navigating downstream. Cell 129, 1261-1274. 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E., Dzyubachyk O. and Smal I. (2012). Methods for cell and particle tracking. Methods Enzymol. 504, 183-200. 10.1016/b978-0-12-391857-4.00009-4 [DOI] [PubMed] [Google Scholar]
- Miura H., Matsuda M. and Aoki K. (2014). Development of a FRET biosensor with high specificity for Akt. Cell Struct. Funct. 39, 9-20. 10.1247/csf.13018 [DOI] [PubMed] [Google Scholar]
- Mukherjee A. and Rotwein P. (2008). Insulin-like growth factor-binding protein-5 inhibits osteoblast differentiation and skeletal growth by blocking insulin-like growth factor actions. Mol. Endocrinol. 22, 1238-1250. 10.1210/me.2008-0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A., Wilson E. M. and Rotwein P. (2010). Selective signaling by Akt2 promotes bone morphogenetic protein 2-mediated osteoblast differentiation. Mol. Cell. Biol. 30, 1018-1027. 10.1128/MCB.01401-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson D. E., Ihekwaba A. E. C., Elliott M., Johnson J. R., Gibney C. A., Foreman B. E., Nelson G., See V., Horton C. A., Spiller D. G. et al. (2004). Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 306, 704-708. 10.1126/science.1099962 [DOI] [PubMed] [Google Scholar]
- Purvis J. E. and Lahav G. (2013). Encoding and decoding cellular information through signaling dynamics. Cell 152, 945-956. 10.1016/j.cell.2013.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis J. E., Karhohs K. W., Mock C., Batchelor E., Loewer A. and Lahav G. (2012). p53 dynamics control cell fate. Science 336, 1440-1444. 10.1126/science.1218351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regot S., Hughey J. J., Bajar B. T., Carrasco S. and Covert M. W. (2014). High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724-1734. 10.1016/j.cell.2014.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rena G., Guo S., Cichy S. C., Unterman T. G. and Cohen P. (1999). Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 274, 17179-17183. 10.1074/jbc.274.24.17179 [DOI] [PubMed] [Google Scholar]
- Rena G., Woods Y. L., Prescott A. R., Peggie M., Unterman T. G., Williams M. R. and Cohen P. (2002). Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J. 21, 2263-2271. 10.1093/emboj/21.9.2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval P. C., Slentz D. H., Pisitkun T., Saeed F., Hoffert J. D. and Knepper M. A. (2013). Proteome-wide measurement of protein half-lives and translation rates in vasopressin-sensitive collecting duct cells. J. Am. Soc. Nephrol. 24, 1793-1805. 10.1681/ASN.2013030279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W. and Selbach M. (2011). Global quantification of mammalian gene expression control. Nature 473, 337-342. 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- Sever R. and Brugge J. S. (2015). Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 5, a006098 10.1101/cshperspect.a006098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer S. L., Cappell S. D., Tsai F.-C., Overton K. W., Wang C. L. and Meyer T. (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369-383. 10.1016/j.cell.2013.08.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J. A., Lee M. and Knapp P. E. (1991). Cellular dimensions affecting the nucleocytoplasmic volume ratio. J. Cell Biol. 115, 941-948. 10.1083/jcb.115.4.941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang E. D., Nunez G., Barr F. G. and Guan K.-L. (1999). Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274, 16741-16746. 10.1074/jbc.274.24.16741 [DOI] [PubMed] [Google Scholar]
- Tay S., Hughey J. J., Lee T. K., Lipniacki T., Quake S. R. and Covert M. W. (2010). Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature 466, 267-271. 10.1038/nature09145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G., Singer O. and Verma I. M. (2006). Production and purification of lentiviral vectors. Nat. Protoc. 1, 241-245. 10.1038/nprot.2006.37 [DOI] [PubMed] [Google Scholar]
- Toettcher J. E., Weiner O. D. and Lim W. A. (2013). Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell 155, 1422-1434. 10.1016/j.cell.2013.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker A. (2012). Achieving specificity in Akt signaling in cancer. Adv. Biol. Regul. 52, 78-87. 10.1016/j.advenzreg.2011.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Heide L. P., Hoekman M. F. and Smidt M. P. (2004). The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 380, 297-309. 10.1042/BJ20040167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. N., Green J., Wang Z., Deng Y., Qiao M., Peabody M., Zhang Q., Ye J., Yan Z., Denduluri S. et al. (2014). Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 1, 87-105. 10.1016/j.gendis.2014.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff B., Sanglier J.-J. and Wang Y. (1997). Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 4, 139-147. 10.1016/S1074-5521(97)90257-X [DOI] [PubMed] [Google Scholar]
- Woods Y. L., Rena G., Morrice N., Barthel A., Becker W., Guo S., Unterman T. G. and Cohen P. (2001). The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J. 355, 597-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D. and Saxel O. (1977). Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270, 725-727. 10.1038/270725a0 [DOI] [PubMed] [Google Scholar]
- Yee D. (2012). Insulin-like growth factor receptor inhibitors: baby or the bathwater? J. Natl. Cancer Inst. 104, 975-981. 10.1093/jnci/djs258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yissachar N., Sharar Fischler T., Cohen A. A., Reich-Zeliger S., Russ D., Shifrut E., Porat Z. and Friedman N. (2013). Dynamic response diversity of NFAT isoforms in individual living cells. Mol. Cell 49, 322-330. 10.1016/j.molcel.2012.11.003 [DOI] [PubMed] [Google Scholar]
- Yoshizaki H., Mochizuki N., Gotoh Y. and Matsuda M. (2007). Akt-PDK1 complex mediates epidermal growth factor-induced membrane protrusion through Ral activation. Mol. Biol. Cell 18, 119-128. 10.1091/mbc.E06-05-0467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Gan L., Pan H., Guo S., He X., Olson S. T., Mesecar A., Adam S. and Unterman T. G. (2002). Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J. Biol. Chem. 277, 45276-45284. 10.1074/jbc.M208063200 [DOI] [PubMed] [Google Scholar]
- Zhang L., Lee K. C., Bhojani M. S., Khan A. P., Shilman A., Holland E. C., Ross B. D. and Rehemtulla A. (2007). Molecular imaging of Akt kinase activity. Nat. Med. 13, 1114-1119. 10.1038/nm1608 [DOI] [PubMed] [Google Scholar]
- Zhou X., Clister T. L., Lowry P. R., Seldin M. M., Wong G. W. and Zhang J. (2015). Dynamic visualization of mTORC1 activity in living cells. Cell Rep. 10, 1767-1777. 10.1016/j.celrep.2015.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.