

Abstract
The risk of insertional mutagenesis inherent to all integrating exogenous expression cassettes was the impetus for the development of various integration-defective lentiviral vector (IDLV) systems. These systems were successfully employed in a plethora of preclinical applications, underscoring their clinical potential. However, current production of IDLVs by transient plasmid transfection is not optimal for large-scale production of clinical grade vectors. Here, we describe the development of the first tetracycline-inducible stable IDLV packaging cell line comprising the D64E integrase mutant and the VSV-G envelope protein. A conditional self-inactivating (cSIN) vector and a novel polypurine tract (PPT)-deleted vector were incorporated into the newly developed stable packaging cell line by transduction and stable transfection, respectively. High-titer (~107 infectious units (IU)/ml) cSIN vectors were routinely generated. Furthermore, screening of single-cell clones stably transfected with PPT-deleted vector DNA resulted in the identification of highly efficient producer cell lines generating IDLV titers higher than 108 IU/mL, which upon concentration increased to 1010 IU/ml. IDLVs generated by stable producer lines efficiently transduce CNS tissues of rodents. Overall, the availability of high-titer IDLV lentivirus packaging cell line described here will significantly facilitate IDLV-based basic science research, as well as preclinical and clinical applications.
Introduction
The recent successes of integration-competent lentiviral vectors (ICLVs) targeting patients’ hematopoietic stem cells (HSC) in altering the pathologic course of fatal genetic diseases have opened a new avenue in treating incurable diseases.1–4 However, the prospect of broadening the usage of the above therapeutic methodology to include nonfatal diseases is limited by the risk of insertional mutagenesis inherent to all integrating vectors. Indeed, data accumulated from various preclinical studies have raised major concerns regarding the oncogenic potential of gamma-retroviral and lentiviral vectors.5–10 These concerns have materialized in several human clinical trials in which autologous bone marrow transplantation of gamma-retroviral vector-transduced HSC resulted in tumor development.11–13 Taking a similar therapeutic approach, Cavazzana-Calvo et al.4 have successfully employed ICLV’s to deliver a β-globin gene expression cassette to patients’ HSC as a means to treat β-thalassemia. However, reports from this trial describe clonal expansion of the myeloid lineage due to vector-mediated insertional mutagenesis, leading to dysregulation of the HMGA2 gene in progenitor cells.
Biosafety concerns regarding ICLV-associated insertional mutagenesis and the desire to harness the advantages of lentiviral vectors—including low immunogenicity, high capacity, and the ability to transduce nondividing cells—were the impetus for the development of integration-defective lentiviral vectors (IDLVs). Generally, IDLVs are packaged into vector particles carrying class I integrase mutants in which one amino acid in the DDE conserved catalytic triad (predominantly the D64 residue) is mutated.14–21 Several studies have demonstrated different levels of illegitimate integrase-independent integration (up to one integration event per 200 vector genomes).14,20,22–24 However, the development of the polypurine tract (PPT)-deleted IDLVs by Kantor et al. has significantly reduced the levels of illegitimate integration.24,25
IDLVs have efficiently transduced a plethora of cell lines and various primary cells including human embryo fibroblasts, human macrophages, HSC, primary neuronal cells, and embryonic stem cells.14,16,18–20,22,24,26–33 Different laboratories have demonstrated the ability of IDLVs to maintain long-term expression of reporter genes in non-dividing cells in various target organs, including the CNS, spinal cord, muscle, eye, and liver.14,16,18,20,27,31,34,35 Furthermore, Thrasher et al. and Suwanmanee et al. have successfully employed IDLVs in mouse models of gene replacement therapy for degenerative retinal disease and hemophilia B.35,36 The efficacy of IDLVs in cancer immunotherapy and as a means of inducing protective immune responses to human pathogens has been characterized in different experimental settings.37–46 Importantly, a growing number of manuscripts have described IDLVs carrying either zinc-finger nucleases, and the CRISPR-Cas 9 system as an effective means of gene editing for clinical and basic science applications.47–52
In the early stages of retroviral vector development, production of gamma-retroviral vectors was premised on stable packaging cell lines.53 The establishment of the highly transfectable 293T in the early 90’s allowed Pear et al.54 to produce high-titer retroviral vectors by transient transfection. The ability to pseudotype retroviral vectors with the VSV-G envelope further facilitated production of high-titer retroviral vector particles.55 Premised on the aforementioned advancements in retroviral vector production, Naldini et al.56 employed transient three-plasmid transfection to produce and concentrate VSV-G pseudotyped lentiviral vector to transduce murine neurons in vivo. Similar successes employing transient plasmid transfection in producing high-titer lentiviral vectors rendered this approach most popular. Notwithstanding the ease of producing high-titer lentiviral vectors by transient transfection, the need to scale up vector production, to reduce variability between vector preparations, and to reduce the risk of emerging replication competent retroviruses (RCRs) were the drive for the establishment of several stable ICLV packaging cell lines.57–62
Here, we describe the establishment of the first IDLV packaging cell line, which generates high titer, VSV-G pseudotyped conditional SIN and PPT-deleted IDLVs. The vectors efficiently transduce rodent CNS. The novel packaging cell line will facilitate the scaling up in production of safer IDLVs more suitable for clinical trials.
Results
The establishment of the IDLV inducible stable packaging cell line
Maximizing vector biosafety and titers are the main guidelines to be followed in establishing all lentiviral viral vector stable packaging cell lines. Encouraged by earlier studies of VSV-G pseudotyped lentiviral vectors demonstrating efficient transduction of brain and liver tissues in vivo and HSC ex vivo, we based the first IDLV packaging cell line on this highly effective and broadly used envelope protein. To minimize the likelihood of emerging RCRs, we split the process of incorporating the VSV-G envelope and the packaging expression cassettes into the genome of the future packaging cells to two independent stable transfection procedures (Figure 1). To address the potential hurdle associated with constitutive expression of the cytotoxic HIV-1 protease and the VSV-G envelope proteins,63–65 the aforementioned expression cassettes were placed under the control of tetracycline-inducible promoters. The IDLV packaging construct (pTK1574) was established by placing an HIV-1 packaging cassette expressing the HIV-1 Gag, Pol, Tat, Rev, and Vpu under the control of a tetracycline inducible promoter (Figure 2a). A D64E HIV-1 integrase mutant encoded by the above Pol gene renders the vector particles generated by the novel inducible packaging cassette integration defective. The efficiency of the novel packaging system at generating IDLVs in a tetracycline regulated fashion was evaluated by transiently transfecting it along with a conventional HIV-1 vector expressing the green fluorescent protein (GFP) under the control of a cytomegalovirus (CMV) promoter16 (pTK945, Figure 2a) and a VSV-G envelope expression cassette (pMDG) into SODk0 cells, either in the presence or absence of doxycycline (dox). The SODk0 cell line constitutively expresses the synthetic tetracycline regulated trans-activator tTA.60 Physical and infectious titers of vector particles in culture media at 72 hours post-transfection were determined by p24gag enzyme-linked immunosorbent assay (ELISA) and by scoring GFP expression following serial vector dilutions on 293T cells. In the absence of dox, the novel packaging cassette supports efficient production of physical (p24gag 142.8 ng/ml) and infectious vector particles (1.42 ± 0.11 × 107 IU/ml). Per contra, addition of Dox to culture media effectively inhibited vector production.
Figure 1.
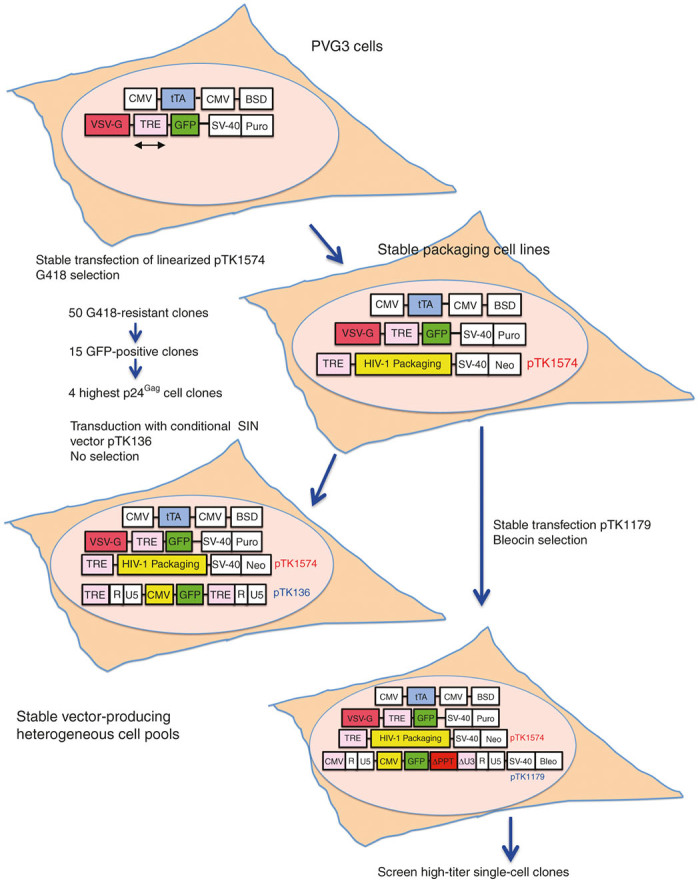
Schematic outline for generating tetracycline-regulated integration-defective lentiviral vector packaging system. The PVG3 cell line constitutively expresses the synthetic tetracycline-regulated trans activator tTA under the control of a cytomegalovirus promoter, as well as the VSV-G envelope protein and the green fluorescent protein (GFP) marker gene from a bidirectional tetracycline-regulated expression cassette (depicted by a bidirectional arrow). Packaging cell lines were generated by stably transfecting PVG3 cells with the tetracycline-regulated HIV-1 packaging construct pTK1574, followed by G418 selection. p24gag enzyme-linked immunosorbent assay was used to identify and screen for high levels of particle production in cell clones expressing minimal—yet detectable—levels of GFP (in the presence of doxycycline (Dox)). Producer cell lines were further established by either transduction or stable transfection.
Figure 2.

Generating tetracycline highly regulated integration-defective lentiviral vector (IDLV) packaging cell lines. (a) Depiction of the tetracycline inducible packaging vector pTK1574 containing the gag, pol (encoding the integrase-mutant D64E), vpu, rev, and tat genes under the control of a tetracycline-regulated promoter and the HIV-1 vector pTK945 carrying the GFP marker gene under the control of a cytomegalovirus promoter. (b) Tight regulation of the VSV-G envelope and the IDLV packaging cassettes in PVG3 cells and in the stable packaging cell lines 3–8 and 3–12. The aforementioned cell lines were cultured either in presence of 1 µg/ml Dox or in the absence of Dox and in the presence of 5 mmol/l sodium butyrate. Expression of HIV-1 Gag (p55 and p24), Pol (p66 and p51) and the VSV-G envelope proteins in the above cell lines in the presence and absence of Dox were determined by western blot analysis. (c) Regulation of GFP expression in PVG3 and the packaging cell lines 3–8 and 3–12. The aforementioned cell lines were cultured either in presence of 1 µg/ml Dox or in the absence of Dox and in the presence of 5 mmol/l sodium butyrate. Expression of GFP was determined by fluorescence microscopy. Note that very weak expression of GFP could be detected in the presence of Dox.
Encouraged by these results, we sought to stably transfect the inducible IDLV packaging cassette into the human embryo kidney (HEK) 293-based PVG3 cell line, which was established in an earlier study.58 The PVG3 cells constitutively express the synthetic tetracycline-regulated trans activator tTA66 under the control of a CMV promoter, as well as the VSV-G envelope protein and the GFP marker gene from a bi-directional tetracycline-regulated expression cassette (Figure 1). As shown in Figure 2b,c, VSV-G and GFP expression is tightly regulated in PVG3 cells. In the presence of Dox, PVG3 cells demonstrate a barely detectable VSV-G, and GFP expression. However, Dox withdrawal and addition of sodium butyrate (SB), a broad-spectrum histone deacetylase inhibitor resulted in robust VSV-G and GFP expression (Figure 2b,c).
As a first step in establishing the stable IDLV producing cell line, PVG3 cells were transfected with the inducible IDLV expression cassette (pTK1574, Figures 1 and 2) and a total of 50 G418-resistant cell clones were isolated and screened for minimal, yet detectable GFP expression in the presence of 1 µg/ml Dox. A total of 15 GFP expressing cell clones were identified, and p24gag ELISA was used to determine their ability to produce high levels of vector particles upon Dox withdrawal and addition of SB. Interestingly, conditioned media obtained from the above 15 cell clones at 48 hours postexposure to SB exhibited a wide range of p24gag concentrations, varying from undetectable levels up to 1,307.1 ng/ml. Next, we characterized the kinetics of vector particle production upon Dox withdrawal and addition of SB in the four cell clones exhibiting the highest p24gag levels (3–6, 3–8, 3–10, and 3–12). As shown in Table 1, only moderate levels of p24gag were detected in conditioned media of the four cell clones at 6 days post-Dox withdrawal (up to 817 ng/ml). However, all cell clones demonstrated a significant increase in vector particle production at 2 days post-SB addition, and maintained high concentrations of p24gag in conditioned media up to 4 days postaddition of SB. However, increased cell death and morphological changes were observed in the above cell clones at 4 days post-SB exposure, indicating that between 2 to 3 days post-SB addition is the optimal time frame for vector particles production.
Table 1. Stable production of vector particles by packaging cell lines.
| Clone name |
P24gag (ng/ml) |
||||||
|---|---|---|---|---|---|---|---|
| +Dox | -Dox day6 | +SB day1 | +SB day2 | +SB day3 | +SB day4 | ||
| 3–6 | Beforea | N/Ac | 335.78 | 415.40 | 991.79 | 1,286.36 | 1,550.02 |
| Afterb | N/A | 358.01 | 1,009.93 | 1,518.69 | |||
| 3–8 | Before | N/A | 817.59 | 552.53 | 1,061.29 | 1,493.37 | 1,470.97 |
| After | N/A | 820.37 | 1,279.10 | 1,769.85 | |||
| 3–10 | Before | N/A | 629.42 | 557.78 | 1,272.87 | 1,282.77 | 1,546.23 |
| After | N/A | 635.27 | 1,688.21 | 2,079.15 | |||
| 3–12 | Before | N/A | 461.47 | 910.57 | 1,369.99 | 1,361.68 | 1,436.45 |
| After | N/A | 628.28 | 1,279.05 | 1,444.14 | |||
Vector particle production by four packaging cell lines before (a) and after (b) four passages in culture (in the absence of G418) was initiated by Dox withdrawal from culture media. Sodium butyrate (SB, 5mM) was added to culture media after 6 days of induction. After 72 hours in conditioned media, vector particle concentration was determined by p24gag ELISA. N/A(c) indicates p24gag measurement not higher than background.
Aware of the possibility that a residual expression of the HIV-1 GagPol gene products in the presence of Dox may result in negative selection of vector particle producing cell clones, we sought to characterize the above cell clones’ stability. To this end, we cultured the four cell clones in the presence of Dox and in the absence of G418 for 4 weeks, after which production of vector particles was induced by Dox withdrawal and addition of SB. As shown in Table 1, all four cell clones exhibited kinetics and levels of vector particle production comparable to the levels detected prior to four weeks of cell culturing and expansion. These data indicate that the above cell clones are potential precursors of IDLV stable producer cell lines.
Establishing the first IDLV producer cell lines via transduction with conditional SIN vectors
Stable incorporation of vector genomes into packaging cells’ chromatin constitutes the last step in the process of establishing stable vector producer cell lines. This step can be mediated either by transducing or by transfecting stable packaging cell lines, either with vector particles or with vector plasmid DNAs, respectively. Introducing the vector cassettes by transduction is highly efficient and time saving, and was employed successfully in the past to generate high titer lentiviral and simple retroviral vector producer cell lines.60,67,68 Since self-inactivating (SIN) vectors cannot be mobilized from transduced target cells, this approach cannot be employed as a means to generate conventional SIN vector producing cell lines. This limitation was addressed by the development of the conditional-SIN (cSIN) vectors, in which a tetracycline-regulated promoter replaced the parental U3-enhancer/promoter sequences. Consequently, cSIN vectors are efficiently mobilized from tTA expressing packaging cell lines, yet retain their SIN features in all tTA-lacking target cells.62 We sought to employ this technology to establish the first IDLV stable producer cell lines and to further characterize the ability of the above four packaging cell lines (exhibiting the highest p24gag production) to generate high-titer IDLVs.
To this end, we transduced the four packaging cell lines with the cSIN vector pTK136 (ref. 62) (Figures 1 and 3), from which the GFP reporter gene is expressed under the control of a CMV promoter. Heterogeneous pools of vector-transduced cell lines were induced to generate vector particles by Dox withdrawal and addition of 5 mmol/l SB. Samples of conditioned media were collected between days 2–4 post-SB addition and IDLV titers were determined by scoring GFP positive cells following serial dilution of conditioned media on 293T cells. As shown in Figure 3b, between days 2 and 4 post-SB exposure, the four novel IDLV producer cell lines generated vector titers higher than 106 IU/ml. However, IDLV titers (4–5 × 106 IU/ml) generated by cell lines 3-8-136 and 3-12-136 were significantly higher than the titers (1–1.5 × 106 IU/ml) obtained from clones 3-6-136 and 3-10-136. Altogether, these results support the notion that cSIN vectors can rapidly and efficiently establish high titer IDLV producing cell lines.
Figure 3.
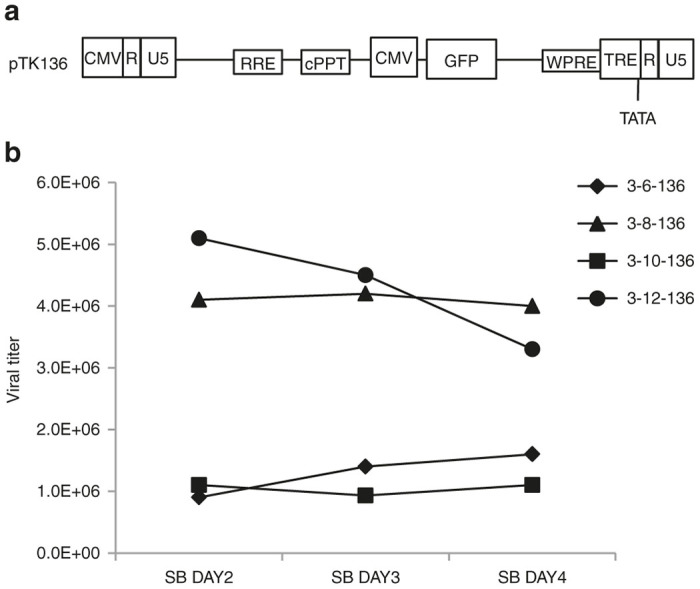
Generating tetracycline-regulated integration-defective lentiviral vector producer cell lines by transduction. (a) Depiction of the cSIN vector pTK136. Note that the 3’U3 region is replaced by tetracycline-regulated element. (b) Vector production by the aforementioned vector-producing cell lines (3-6-136, 3-8-136, 3-10-136, 3-12-136) was initiated by Dox withdrawal from culture media. Sodium butyrate (5 mmol/l) was added to culture media after 6 days of induction. Conditioned media were collected at 2–4 days after addition of sodium butyrate. Viral titers (IU/ml) were determined by scoring GFP expression by fluorescence microscopy following serial dilution of conditioned media on 293T cells.
Establishing the first IDLV PPT-deleted producer cell lines by stable transfection
Although time and labor consuming, stable transfection of plasmid vector DNA into stable packaging cells has been successfully employed to establish stable producer cell lines generating high titer SIN vectors.57–59,61 The fact that the efficiency of stable transfections is independent of the vector’s ability to successfully complete its life cycle renders this methodology highly suitable for generating stable producer cell lines of vectors unable to efficiently complete reverse transcription and integration. These include PPT-deleted vectors, which were developed as a means to further enhance vector biosafety by minimizing illegitimate, integrase-independent integration of IDLVs.24 PPT-deleted vectors cannot efficiently complete reverse-transcription and thus generate minimal levels of linear vector genomes. The majority of PPT-deleted vectors are in the form of 1-LTR circles. Consequently, both integrase-mediated integration of PPT-deleted ICLVs and illegitimate integration of PPT-deleted IDLVs are severely impaired.24 To combine the advantages conferred by employing a stable vector producing cell line with the enhanced biosafety properties of PPT-deleted vectors, we chose to stably transfect a PPT-deleted vector into stable packaging cell lines. Since both packaging cell lines (3–8 and 3–12) exhibited long-term Dox-dependent expression of GagPol and GFP/VSV-G, and produced high titer cSIN IDLVs (Figures 2 and 3; Table 1), we chose to base the establishment of the first stable PPT-deleted IDLV producer cell lines on the 3–8 and 3–12 packaging cells. To this end, plasmid DNA comprising both the PPT-deleted vector pTK1179 cassette (expressing the GFP marker gene under the control of a CMV promoter) and the Bleocin resistance gene encoding cassette (Figures 1 and 4a) was transfected into the stable packaging cell lines 3–8 and 3–12.
Figure 4.
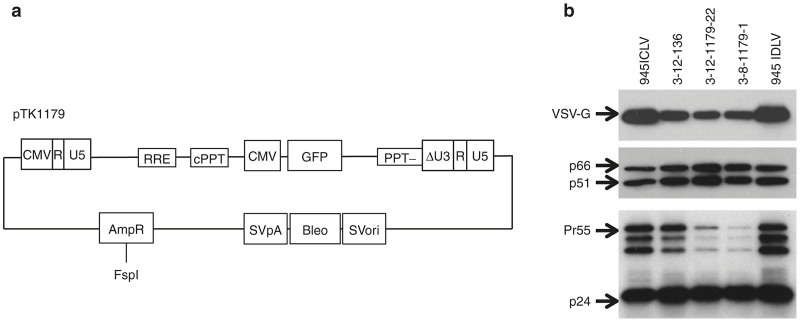
Generating stable producer cell lines by transfection. (a) Depiction of the PPT-deleted vector pTK1179. SVori: SV40 promoter and origin; Bleo: Zeocin resistance gene; SVpA: SV40 polyadenylation signal; AmpR: Ampicillin resistance gene: (b) Western blot analysis of HIV-1 Gag (p55 and p24), Pol (p66 and p51) and the VSV-G envelope protein in vector particles generated by either transient transfection (pTK945 integration-competent lentiviral vectors and integration-defective lentiviral vector (IDLV)) and IDLV generated by the stable vector producer cell lines. Equal amounts of concentrated virus (2 ng p24gag per lane for P24, 20 ng p24gag per lane for all other proteins) were loaded.
Two heterogeneous 3–8 and 3–12 cell pools and 30 single cell clones were isolated following Bleocin selection. Vector particle production from the above Bleocin resistant cell clones was induced by withdrawal of Dox and addition of 5 mmol/l SB. Concentration of p24gag and vector titers in conditioned media samples collected at 72 hours post-SB treatment was determined by ELISA and by scoring GFP expression following serial dilutions of conditioned media on 293T cells. As shown in Table 2, both 3–8 and 3–12 cell pools generated comparable levels of p24gag concentrations and high titers of the PPT-deleted IDLV particles (1.1 ± 0.4 × 107 to1.6 ± 0.5 × 107 IU/ml). As expected, the above isolated cell clones exhibited a wide range of vector titers, which varied between 1.7 ± 0.5 × 106 IU/ml (clone 3-12-1179-11) to 1.2 ± 0.3 × 108 IU/ml (clone 3-8-1179-1). Note that one cell clone (3-12-1179-14) failed to produce detectable levels of IDLVs (3-12-1179-14). The concentrations of p24gag in the above conditioned media samples varied between 621 ± 146 ng/ml (clone 3-12-1179-8) and 3,914 ± 1,275 ng/ml (clone 3-8-1179-2). Although cell lines producing low vector titers (<107 IU/ml) generated p24gag concentrations lower than 2,000 ng/ml, there was no direct correlation between p24gag concentrations and vector titers. To further characterize the above vector producing cells, vector copy number per diploid host genome (VCN) was determined by qPCR in the two cell pools and in 10 cell clones generating various vector titers. Interestingly, notwithstanding the fact that both cell pools produce comparable vector titers, the VCN in cell-pool 3-12-1179 was more than fourfold higher than the VCN in cell-pool 3-8-1179. Similarly, VCN in the various cell clones was not correlated with vector titers generated by these vector producing cell lines. To characterize the above cell clones’ stability, we cultured the four producer cell lines (8–136, 12–136, 8-1179-1, and 8-1179-22) in the presence of Dox and in the absence of G418 for 5 weeks, after which production of vector particles was induced by Dox withdrawal and addition of SB. As shown in Table 3, all four cell lines exhibited levels of vector particle and infectious unit production comparable to the levels detected prior to 5 weeks of cell culturing and expansion.
Table 2. Characterization of selected vTK1179 producer cell lines.
| Clone No. | IUa/ml | P24gag (ng/ml)b | VCNc |
|---|---|---|---|
| 3-8-1179 pool | 1.1 ± 0.4 × 107 | 1,824 ± 445 | 13.9 |
| 3-12-1179 pool | 1.6 ± 0.5 × 107 | 1,285 ± 443 | 58.9 |
| 3-8-1179-1 | 1.2 ± 0.3 × 108 | 2,801 ± 1,025 | 4.4 |
| 3-8-1179-2 | 2.8 ± 1.1 × 107 | 3,914 ± 1,275 | 2.7 |
| 3-12-1179-1 | 2.5 ± 0.2 × 107 | 2,024 ± 807 | 5.0 |
| 3-12-1179-2 | 2.6 ± 0.5 × 107 | 1,897 ± 699 | 7.6 |
| 3-12-1179-3 | 1.5 ± 0.3 × 107 | 1,296 ± 362 | |
| 3-12-1179-4 | 3.4 ± 1.1 × 107 | 1,255 ± 261 | |
| 3-12-1179-5 | 2.5 ± 0.9 × 106 | 1,698 ± 482 | 12.4 |
| 3-12-1179-6 | 2.6 ± 1.2 × 107 | 693 ± 116 | 2.3 |
| 3-12-1179-7 | 2.1 ± 1.0 × 107 | 1,853 ± 851 | |
| 3-12-1179-8 | 4.3 ± 1.1 × 106 | 621 ± 146 | |
| 3-12-1179-9 | 4.7 ± 1.1 × 107 | 885 ± 2 | |
| 3-12-1179-10 | 1.8 ± 0.9 × 107 | 1,490 ± 306 | |
| 3-12-1179-11 | 1.7 ± 0.5 × 106 | 852 ± 265 | |
| 3-12-1179-12 | 5.6 ± 1.9 × 106 | 1,980 ± 423 | 28.5 |
| 3-12-1179-13 | 6.8 ± 0.6 × 106 | 1,581 ± 534 | |
| 3-12-1179-14 | N/A | 629 ± 74 | 10.2 |
| 3-12-1179-15 | 2.6 ± 1.0 × 107 | 1,744 ± 522 | |
| 3-12-1179-17 | 3.7 ± 0.9 × 106 | 810 ± 157 | |
| 3-12-1179-18 | 9.0 ± 0.5 × 106 | 1,226 ± 168 | |
| 3-12-1179-19 | 5.2 ± 0.4 × 107 | 1,861 ± 270 | 10.2 |
| 3-12-1179-20 | 2.8 ± 0.9 × 107 | 1,174 ± 181 | |
| 3-12-1179-21 | 3.1 ± 0.4 × 107 | 1,153 ± 126 | |
| 3-12-1179-22 | 7.5 ± 0.2 × 107 | 2,999 ± 109 | 43.8 |
| 3-12-1179-23 | 3.8 ± 0.8 × 107 | 938 ± 144 | |
| 3-12-1179-24 | 2.8 ± 0.4 × 107 | 1,169 ± 210 | |
| 3-12-1179-25 | 1.9 ± 1.9 × 107 | 640 ± 129 | |
| 3-12-1179-26 | 2.2 ± 1.2 × 107 | 1,190 ± 570 | |
| 3-12-1179-27 | 3.3 ± 0.9 × 107 | 2,722 ± 1,366 | |
| 3-12-1179-28 | 4.0 ± 0.7 × 107 | 1,027 ± 13 | |
| 3-12-1179-29 | 2.8 ± 0.2 × 107 | 1,458 ± 293 |
Vector production was initiated by Dox withdrawal. Sodium butyrate (5 mmol/l) was added to culture media after 6 days of induction. After 72 hours in conditioned media, vector titers (IU/ml, a) were determined by scoring GFP-positive cells following serial dilution on 293T cells. P24gag level (b) was determined by ELISA. Numbers represent mean ± SD (n = 3). VCNs (viral copy numbers, c) were measured by qPCR.
Table 3. Stability of vector producer cell lines.
| 8–136 | 12–136 | 8-1179-1 | 12-1179-22 | ||
|---|---|---|---|---|---|
| P24gag ng/ml | Beforea | — | — | 2801 ± 1025 | 2999 ± 109 |
| Afterb | 2374 ± 172 | 1047 ± 70 | 1716 ± 56 | 2952 ± 163 | |
| Titer IU/ml | Before | 4.0 ± 0.2 × 106 | 6.6 ± 1.2 × 106 | 1.2 ± 0.3 × 108 | 7.5 ± 0.2 × 107 |
| After | 5.7 ± 0.5 × 106 | 5.2 ± 0.4 × 106 | 1.1 ± 0.2 × 108 | 7.8 ± 0.3 × 107 | |
Production of integration-defective lentiviral vector’s by four packaging cell lines before (a) and after (b) five passages in culture (in the absence of G418) was initiated by Dox withdrawal from culture media. Sodium butyrate (5 mmol/l) was added to culture media after 6 days of induction. After 72 hours in conditioned media, vector particle concentration was determined by p24gag ELISA. Vector titers (IU/ml) were determined by scoring GFP-positive cells following serial dilution on 293T cells. Numbers represent mean ± SD (n = 3).
Efficient transduction of rodent CNS with stable producing cell line-generated IDLVs
The establishment of a stable IDLV producing cell line was aimed at facilitating large-scale IDLV production for in vivo applications. Thus, we sought to characterize the efficacy of IDLVs generated by the above stable vector producing cell lines at transducing rat CNS. To this end, IDLVs generated by cell clones 3-12-136, 3-12-1179-22 and 3-8-1179-1 were concentrated and purified by ultracentrifugation as described by Kafri et al.60 Vector titers and concentration of p24gag were determined by scoring GFP expression following serial dilution on 293T cells and by p24gag ELISA, (3-12-1179-22: 2.70 × 1010 IU/ml, p24gag 1.24 × 106 ng/ml; 3-8-1179-1: 3.34 × 1010 IU/ml, p24gag 1.59 × 106 ng/ml; 3-12-136, 1.28 × 109 IU/ml, p24gag 6.68 × 105 ng/ml, respectively). Western blot analysis of concentrated vector particles using primary antibodies to HIV-1 p24gag and reverse-transcriptase proteins, as well as the VSV-G envelope proteins, demonstrated efficient vector particle formation and protein processing, which was comparable to ICLV and IDLV particles generated by transient transfection (Figure 4).
PPT-deleted IDLVs (pTK1179) generated by the novel stable producer cell line 3-8-1179-1 and conventional ICLV’s (pTK945) generated by transient transfection were injected into the striatum of 300–350 g male sprague dawley rats (total of 0.66 × 107 IU and 1.2 × 107 IU respectively). Transduction efficiency was determined at week 5 postinjection by confocal microscopy. As shown in Figure 5, in the rat striatum, ICLVs generated by transient transfection appeared to support moderately greater overall transduction in comparison to the cell line-generated PPT-deleted IDLVs (Figure 5a,b). These findings are in line with the higher number of injected ICLV IU and with an earlier study by Bayer et al.16 demonstrating reduced transgene expression from episomal lentiviral vectors. Also, we observed that IDLV’s generated by a novel producer cell line efficiently transduced murine striatum (data not shown). Interestingly, both ICLVs and IDLVs proved highly neurotrophic, such that almost all of the GFP-positive cells colocalized with the neuronal marker NeuN (Figure 5c,d). Furthermore, in the striatum virtually no GFP-positive cells exhibited colocalization with glial fibrillary acidic protein, a cellular marker of astrocytes (Figure 5e). However, a major difference between the two vectors was observed in the corpus callosum dorsal to the striatum. The nonintegrating vector exhibited little ability to transduce either oligodendrocytes or astrocytes in the corpus callosum, while the integrating vector efficiently transduced both oligodendrocytes and astrocytes in the corpus callosum dorsal to the striatum (Figure 5f).
Figure 5.
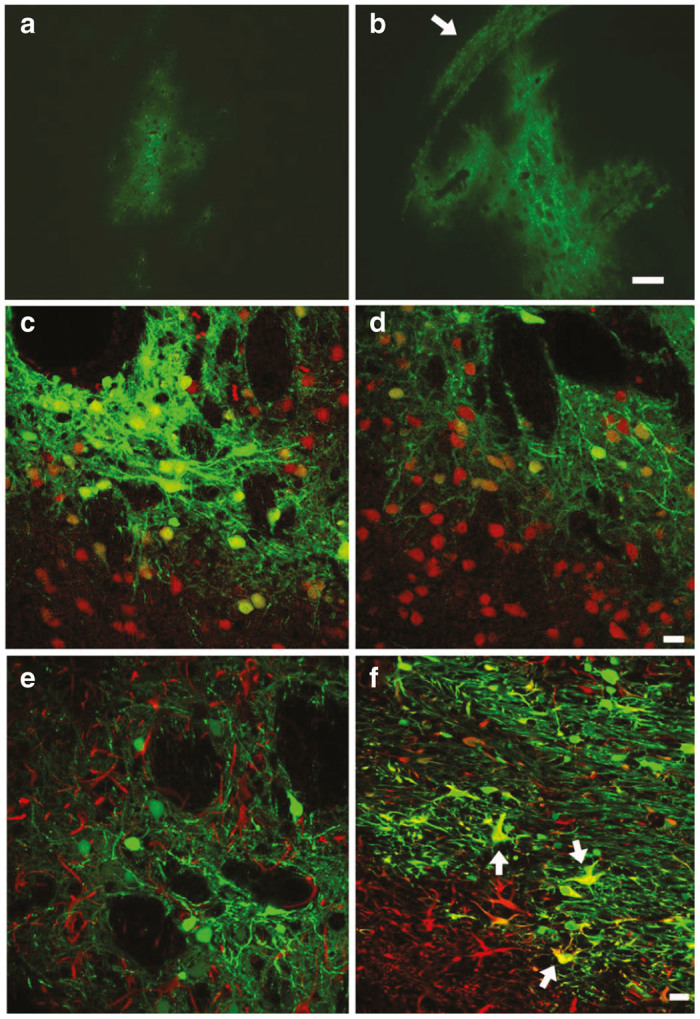
In vivo striatal transduction by integrating and nonintegrating lentiviral vectors. Five weeks after bilateral 1 µl vector infusions, both the nonintegrating (panel a) and the integrating (panel b) lentiviral vectors transduced neurons in the rat striatum. The vast majority of transduced cells were neurons that colocalized with NeuN (panel c, integration-competent lentiviral vectors (ICLV); panel d, integration-defective lentiviral vector (IDLV); NeuN, red). Further example of this neuronal tropism is illustrated in panel e where confocal microscopy showed that glial fibrillary acidic protein (GFAP)-positive cells did not colocalize with GFP for the ICLV (GFAP, red). In contrast, the ICLV, but not the IDLV, transduced both oligodendrocytes and astrocytes (GFAP, red) in the corpus callsosum dorsal to the striatum (panel f, arrows indicate GFP-GFAP-positive cells). Panels a,b magnification bar = 250 µm; panels c–f, magnification bars = 10 µm.
Discussion
First stable IDLV packaging
IDLVs offer efficient gene delivery of large genetic cargos into dividing and nondividing cells with minimal risk of insertional mutagenesis (inherent in ICLVs). Increasingly, research groups have been employing these vectors as a means to deliver marker genes and therapeutic genetic cargos to a plethora of cell lines and primary cells in vitro and to various target organs in vivo. The successful IDLV-based gene replacement therapy in mouse models of retinal degeneration35 and hemophilia B36 underscore their clinical potential. Furthermore, IDLVs have been used to transiently express genome-editing enzymes in dividing cells.49 Indeed, Yi et al.52 have recently conferred resistance to HIV-1 infection by mutating the HIV-1 coreceptor CCR5 gene with IDLV-delivered zinc-finger-nucleases.
To further facilitate the use of IDLV’s, we report here on the establishment of the first stable IDLV cell lines. Titers of VSV-G-pseudotyped IDLVs generated by the novel cell lines were higher than 108 and 1010 IU/ml prior to and after concentration, respectively, and are comparable to or higher than titers of most currently used IDLVs and ICLVs generated by transient transfection.
Using the tetracycline inducible system, we circumvented the difficulties associated with overexpressing the cytotoxic HIV-1 protease and the VSV-G envelope proteins. Importantly, the stable packaging cell lines generated in this study exhibited stability and maintained long-term, Dox-regulated expression of both the VSV-G envelope and HIV-1 GagPol gene products.
Our strategy for establishing the IDLV producer cell lines focused on minimizing the likelihood of emerging RCRs. Thus, all expression cassettes comprising the novel packaging system were introduced to the host genome separately using several independent rounds of stable transfection/selection procedures (Figure 1). Although time consuming, this approach minimizes the likelihood of recombination-mediated RCR formation. Recent studies described the use of gamma-retroviral vectors as a means to incorporate the tetracycline-regulated trans-activator (tTA), envelope protein expression cassettes and the HIV-1 packaging system to 293T as a means to establish stable ICLV packaging cell lines.59,61 Although efficient, this approach raises biosafety concerns associated with mobilization of retroviral vectors carrying various components of the HIV-1 vector producing system, as well as increased likelihood of recombination-mediated RCR formation. These biosafety concerns were underscored by a recent publication demonstrating low yet detectable levels of lentiviral vector packaging of gamma-retroviral genomes.69 The tight transcriptional regulation of the VSV-G envelope and the HIV-GagPol expression cassettes inhibits ongoing vector particle formation and consequently prevents superinfection of the stable packaging cell line with VSV-G-pseudotyped particles. This further reduces the likelihood of emerging RCRs.
In this study, we employed two different methodologies to introduce the vector cassette into the packaging cell line. In one approach, we used cSIN vectors generated by transient transfection to transduce the newly developed integrase-deficient packaging cell lines. This methodology is highly efficient and time saving. A pool of cSIN vector-transduced packaging cells generated IDLV titers higher than 5 × 106 and 1 × 109 IU/ml prior to and after concentration. We predict that screening and isolating highly efficient single cell clones could further increase vector titers. The cSIN-based approach is optimal for rapid establishment of stable producer cell lines, yet it is premised on the ability of vectors generated by transient transfection to efficiently integrate into target cell genomes. Thus, it cannot be employed to transfer integration-impaired vectors such as att sites- or PPT- deleted vectors24,29 to packaging cell lines. Stable transfection of vector DNA to packaging cells is the second methodology described in this study for generating stable IDLV producing cell lines. We employed this methodology (which is independent of the vector’s integration efficiency) to incorporate PPT-deleted vectors into packaging cell lines. Transfection of linearized (nonconcatemerized) vector plasmid DNA followed by Bleocin selection resulted in the establishment of two stable heterogeneous vector-producing cell pools. These exhibited vector titers higher than 107 IU/ml with VCN’s of 14 and 59. As expected, 8 out of 30 isolated vector producing cell clones exhibited titers 2–10-fold higher (>108 IU/ml) than the titers generated by the above cell pools. The lack of correlation between p24gag concentration and VCN to vector titers suggests that identification of highly efficient vector producing cell clones cannot be based on these two parameters. The establishment of the first PPT-deleted IDLV producer cell lines enhances the overall biosafety of the IDLV system as it combines the advantage of minimal illegitimate integration conferred by PPT-deleted vectors with the safety and efficiency gained by vector production by stable producer cell lines.
Overall, the establishment of stable PPT-deleted and conventional IDLV producing cell lines facilitates the usage and enhances the safety of the IDLV system, rendering it more suitable for clinical trials.
Materials and Methods
Plasmids
The lentiviral vector pTK136, pTK1179, pTK945, and the packaging cassettes ΔNRF (Int+), pTK939 (Int-) were generated, as described previously.24,62 The inducible packaging system pTK1574 was derived by cloning a SacII/PvuI fragment containing the tetracycline-regulated element and the human CMV minimal promoter into similar sites in pTK939. The resultant pTK1574 expresses the integrase-deficient (D64E) HIV packaging cassette (excluding the HIV-1 nef, vif, and vpr genes) under the control of a tetracycline regulated promoter.
Cells
The inducible IDLV packaging system is based on the PVG3 cell line,58 in which the synthetic trans-activator tTA is expressed under the control of a CMV promoter. In addition, PVG3 cells express the VSV-G envelope and the GFP marker gene from a bidirectional tetracycline-inducible promoter. All cell types were maintained in Dulbecco’s Modified Eagle Medium (DMEM)-High Glucose (Thermo Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS) (Atlantic Biologicals, Miami, FL), 100 U/ml penicillin, 100 µg/ml streptomycin, and 250 ng/ml amphotericin B (Corning Cellgro, Manassas, VA). The PVG3 cell line and all its derivatives were cultured on poly-lysine 0.001% (Sigma, St Louis, MO) coated plates in the presence of 1 µg/ml Dox.
Viral vector production, concentration, and titration
Lentiviral vector production via three plasmids transient transfection was performed as previously described.56,62 The following plasmid amounts were used: 15 µg pTK945, 10 µg ΔNRF, and 5 µg of the VSV-G envelope plasmid pMD.G. Vector producing cells were regularly maintained in 1 µg/ml Dox. To induce vector production, cells were washed with phosphate-buffered saline (PBS) for three times, and passaged onto poly-lysine precoated plates in Dox-free medium. At 6 days post-Dox withdrawal, culture media were supplemented with 5 mmol/l SB. Viral vectors containing media were collected at 60 hours after addition of SB.
For animal studies, viral vectors were concentrated by ultracentrifugation as previously described.60 The emergence of RCRs was ruled out by three independent safety assays (GFP rescue assay, Tat transfer assay, and Gag transfer assay), as previously described.60
Titers were determined by scoring GFP expression following serial dilutions on 293T cells. VCN was determined by multiplex PCR36 on the ABI7300 realtime PCR system. NotI794 primer/prober set (Left primer: 5′-taagaccaccgcacagca-3′; Right primer: 5′-cacttctccaattgtccctca-3′; Roche Universal Probe Library #25, 4686993001, Indianapolis, IN) was used for vectors detection, and paired with human GUSB primer/probe set (Roche 5190525001, Indianapolis, IN) as the reference gene.
HIV-1 p24 ELISA
Titers of physical vector particles were determined by p24gag ELISA using the National Institutes of Health p24 Antigen Capture Assay kit, as previously documented.18
Western blot
Cells and vector particles were lysed in RIPA buffer and characterized for specific protein content by standard western blot analysis using denaturing 10% SDS–PAGE gels. Blots were probed with a murine anti-VSV-G monoclonal antibody (Ab) (1:1,000; P5D4, Santa Cruz, Dallas, TX), murine anti-HIV-1 RT (1:200, 5B2B2; NIH AIDS Research & Reference Reagent Program, Germantown, MD), and murine anti-HIV-1 p24 Gag (1:5,000, 6457; NIH AIDS Research & Reference Reagent Program) followed by a polyclonal goat anti-murine secondary Ab labeled with horseradish peroxidase (1:10,000; Pierce, Grand Island, NY). Signals were detected by enhanced chemiluminescence (ECL) reagent (GE Health Amersham, Pittsburgh, PA).
Experimental animals
All of the animals were pathogen-free male Sprague–Dawley rats obtained from Charles Rivers. The animals were maintained in a 12-hour light–dark cycle and had free access to food and water. All care and procedures were in accordance with the Guide for the Care and Use of Laboratory Animals (DHHS Publication No. (NIH) 85-23), and all procedures received prior approval by the University of North Carolina Institutional Animal Care and Usage Committee.
For virus vector infusions, rats first were anesthetized with 50 mg/kg pentobarbital and placed into a stereotaxic frame. Subsequently, each rat received a bilateral, striatal infusion (1 µl/side) of either the integrating (N = 3) or the nonintegrating (N = 3) lentiviral vector using a 32-gauge stainless steel injector and a Sage infusion pump (interaural line (IAL) 10.0 mm, lateral 3.0 mm, vertical 5.5 mm, according to the atlas of Paxinos and Watson, 1985). The 1 µl infusion occurred over a period of 5 minutes, and the injector was left in place for 3 minutes postinfusion to allow diffusion.
Immunohistochemistry
Five weeks after the vector infusions, the rats received an overdose of pentobarbital (100 mg/kg pentobarbital, i.p.) and subsequently were perfused transcardially with ice-cold 0.1M sodium PBS (pH = 7.4), followed by 4% paraformaldehyde in 0.1M phosphate buffer (pH = 7.4). After overnight fixation in the paraformaldehyde-phosphate buffer, vibratome sections (40 µm thick) were taken and rinsed in PBS. Tissue sections were incubated in 10% normal goat serum and 0.1% Triton X-100 in PBS for 30 minutes. Next, sections were incubated with a primary antibody to NeuN (1:1,000, Chemicon, Pittsburgh, PA) or glial fibrillary acidic protein (1:4,000, DAKO A/S, Glostrup, Denmark) overnight in 3% normal goat serum, 0.2% Triton X-100 and PBS. Tissue sections were then rinsed in PBS, incubated in blocking serum (10% normal goat serum, 0.1% Triton X-100, PBS) for 1 hour and then incubated with a secondary fluorescent antibody (Alexa-fluor 594 goat anti-rabbit, or goat-anti-mouse, Molecular Probes) for 1 hour at 4 °C. Following three rinses in PBS, the sections were mounted on slides and coverslipped with fluorescent mounting media. The eGFP and Alexa-fluor 594 fluorescence were visualized on either an Olympus IX 70 fluorescence microscope or a Leica SP2 laser scanning confocal microscope. All cases of colocalization were determined using the confocal microscope and the presence of colocalization within a Z-stack.
Acknowledgments
This study was supported by the National Institutes of Health (NIH) grant 5R01DK058702-13 to T.K. and P.H. and by the University of North Carolina (UNC) Center for AIDS Research. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH: pD64E from Vinay K. Pathak; HIV-1 p24 monoclonal antibody (183-H12-5c), murine anti-HIV-1 RT (5B2B2), and murine anti-HIV-1 p24 Gag (6457). We wish to thank Leschek for her support. In memory of the Meds Yeghern/tseghasbanutiun. In honor of Chiune Sugihara, Feng-Shan Ho, and Pan Jun-Shun.
The technology of the PPT-deleted vector was licensed by UNC to commercial entities and provides royalties to UNC and T.K.
References
- Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard BC, Dickerson D, Beebe K, Gooch C, Fletcher J, Okbinoglu T. Comparison of HIV-derived lentiviral and MLV-based gammaretroviral vector integration sites in primate repopulating cells. Mol Ther. 2007;15:1356–1365. doi: 10.1038/sj.mt.6300159. [DOI] [PubMed] [Google Scholar]
- Cesana D, Sgualdino J, Rudilosso L, Merella S, Naldini L, Montini E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J Clin Invest. 2012;122:1667–1676. doi: 10.1172/JCI62189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove PW, Kepes S, Hanawa H, Obenauer JC, Pei D, Cheng C. Globin lentiviral vector insertions can perturb the expression of endogenous genes in beta-thalassemic hematopoietic cells. Mol Ther. 2008;16:525–533. doi: 10.1038/sj.mt.6300394. [DOI] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Keating K, Thorpe R. Comparison of toxicogenomic profiles of two murine strains treated with HIV-1-based vectors for gene therapy. Toxicol Appl Pharmacol. 2007;225:189–197. doi: 10.1016/j.taap.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Zhou S, Ma Z, Lu T, Janke L, Gray JT, Sorrentino BP. Mouse transplant models for evaluating the oncogenic risk of a self-inactivating XSCID lentiviral vector. PLoS One. 2013;8:e62333. doi: 10.1371/journal.pone.0062333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- Apolonia L, Waddington SN, Fernandes C, Ward NJ, Bouma G, Blundell MP. Stable gene transfer to muscle using non-integrating lentiviral vectors. Mol Ther. 2007;15:1947–1954. doi: 10.1038/sj.mt.6300281. [DOI] [PubMed] [Google Scholar]
- Banasik MB, McCray PB. Integrase-defective lentiviral vectors: progress and applications. Gene Ther. 2010;17:150–157. doi: 10.1038/gt.2009.135. [DOI] [PubMed] [Google Scholar]
- Bayer M, Kantor B, Cockrell A, Ma H, Zeithaml B, Li X. A large U3 deletion causes increased in vivo expression from a nonintegrating lentiviral vector. Mol Ther. 2008;16:1968–1976. doi: 10.1038/mt.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Bushman FD, Craigie R. Identification of discrete functional domains of HIV-1 integrase and their organization within an active multimeric complex. EMBO J. 1993;12:3269–3275. doi: 10.1002/j.1460-2075.1993.tb05996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor B, Ma H, Webster-Cyriaque J, Monahan PE, Kafri T. Epigenetic activation of unintegrated HIV-1 genomes by gut-associated short chain fatty acids and its implications for HIV infection. Proc Natl Acad Sci USA. 2009;106:18786–18791. doi: 10.1073/pnas.0905859106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit C, Schwartz O, Mammano F. The karyophilic properties of human immunodeficiency virus type 1 integrase are not required for nuclear import of proviral DNA. J Virol. 2000;74:7119–7126. doi: 10.1128/jvi.74.15.7119-7126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe S, Sarkis C, Barkats M, Mammeri H, Ladroue C, Petit C. Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proc Natl Acad Sci USA. 2006;103:17684–17689. doi: 10.1073/pnas.0606197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanisch K, Yáñez-Muñoz RJ. Integration-deficient lentiviral vectors: a slow coming of age. Mol Ther. 2009;17:1316–1332. doi: 10.1038/mt.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornu TI, Cathomen T. Targeted genome modifications using integrase-deficient lentiviral vectors. Mol Ther. 2007;15:2107–2113. doi: 10.1038/sj.mt.6300345. [DOI] [PubMed] [Google Scholar]
- Ellis S, Fong-Wong L, Iqball S, Thoree V, Mitrophanous KA, Binley K. Assessment of Integration-defective HIV-1 and EIAV Vectors In Vitro and In Vivo. Mol Ther Nucleic Acids. 2012;1:e60. doi: 10.1038/mtna.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor B, Bayer M, Ma H, Samulski J, Li C, McCown T. Notable reduction in illegitimate integration mediated by a PPT-deleted, nonintegrating lentiviral vector. Mol Ther. 2011;19:547–556. doi: 10.1038/mt.2010.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareen SU, Kelley-Clarke B, Nicolai CJ, Cassiano LA, Nelson LT, Slough MM. Design of a novel integration-deficient lentivector technology that incorporates genetic and posttranslational elements to target human dendritic cells. Mol Ther. 2014;22:575–587. doi: 10.1038/mt.2013.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick HE, Nowrouzi A, Fronza R, McDonald RA, Kane NM, Alba R. Integrase-deficient lentiviral vectors mediate efficient gene transfer to human vascular smooth muscle cells with minimal genotoxic risk. Hum Gene Ther. 2012;23:1247–1257. doi: 10.1089/hum.2012.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewen N, Leske DA, Chen Y, Teo WL, Saenz DT, Peretz M. Comparison of wild-type and class I integrase mutant-FIV vectors in retina demonstrates sustained expression of integrated transgenes in retinal pigment epithelium. J Gene Med. 2003;5:1009–1017. doi: 10.1002/jgm.447. [DOI] [PubMed] [Google Scholar]
- Michelini Z, Negri DR, Baroncelli S, Spada M, Leone P, Bona R. Development and use of SIV-based Integrase defective lentiviral vector for immunization. Vaccine. 2009;27:4622–4629. doi: 10.1016/j.vaccine.2009.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale SJ, Hollis RP, Pepper KA, Petersen D, Yu XJ, Yang C. Transient gene expression by nonintegrating lentiviral vectors. Mol Ther. 2006;13:1121–1132. doi: 10.1016/j.ymthe.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Pelascini LP, Janssen JM, Gonçalves MA. Histone deacetylase inhibition activates transgene expression from integration-defective lentiviral vectors in dividing and non-dividing cells. Hum Gene Ther. 2013;24:78–96. doi: 10.1089/hum.2012.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluffo H, Foster E, Ahmed SG, Lago N, Hutson TH, Moon L. Efficient gene expression from integration-deficient lentiviral vectors in the spinal cord. Gene Ther. 2013;20:645–657. doi: 10.1038/gt.2012.78. [DOI] [PubMed] [Google Scholar]
- Saenz DT, Barraza R, Loewen N, Teo W, Poeschla EM. Feline immunodeficiency virus-based lentiviral vectors. Cold Spring Harb Protoc. 2012;2012:71–76. doi: 10.1101/pdb.ip067579. [DOI] [PubMed] [Google Scholar]
- Vargas J, Gusella GL, Najfeld V, Klotman ME, Cara A. Novel integrase-defective lentiviral episomal vectors for gene transfer. Hum Gene Ther. 2004;15:361–372. doi: 10.1089/104303404322959515. [DOI] [PubMed] [Google Scholar]
- Mátrai J, Cantore A, Bartholomae CC, Annoni A, Wang W, Acosta-Sanchez A. Hepatocyte-targeted expression by integrase-defective lentiviral vectors induces antigen-specific tolerance in mice with low genotoxic risk. Hepatology. 2011;53:1696–1707. doi: 10.1002/hep.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez-Muñoz RJ, Balaggan KS, MacNeil A, Howe SJ, Schmidt M, Smith AJ. Effective gene therapy with nonintegrating lentiviral vectors. Nat Med. 2006;12:348–353. doi: 10.1038/nm1365. [DOI] [PubMed] [Google Scholar]
- Suwanmanee T, Hu G, Gui T, Bartholomae CC, Kutschera I, von Kalle C. Integration-deficient lentiviral vectors expressing codon-optimized R338L human FIX restore normal hemostasis in Hemophilia B mice. Mol Ther. 2014;22:567–574. doi: 10.1038/mt.2013.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger G, Goujon C, Darlix JL, Cimarelli A. SIVMAC Vpx improves the transduction of dendritic cells with nonintegrative HIV-1-derived vectors. Gene Ther. 2009;16:159–163. doi: 10.1038/gt.2008.128. [DOI] [PubMed] [Google Scholar]
- Coutant F, Frenkiel MP, Despres P, Charneau P. Protective antiviral immunity conferred by a nonintegrative lentiviral vector-based vaccine. PLoS One. 2008;3:e3973. doi: 10.1371/journal.pone.0003973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai B, Yang L, Yang H, Hu B, Baltimore D, Wang P. HIV-1 Gag-specific immunity induced by a lentivector-based vaccine directed to dendritic cells. Proc Natl Acad Sci USA. 2009;106:20382–20387. doi: 10.1073/pnas.0911742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Guan J, Wen B, Zhu N, Chen H, Song J. Induction of broadly neutralising HCV antibodies in mice by integration-deficient lentiviral vector-based pseudotyped particles. PLoS One. 2013;8:e62684. doi: 10.1371/journal.pone.0062684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Dai B, Wang P. Vaccines delivered by integration-deficient lentiviral vectors targeting dendritic cells induces strong antigen-specific immunity. Vaccine. 2010;28:6675–6683. doi: 10.1016/j.vaccine.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Yang H, Dai B, Tai A, Wang P. Nonintegrating lentiviral vectors can effectively deliver ovalbumin antigen for induction of antitumor immunity. Hum Gene Ther. 2009;20:1652–1664. doi: 10.1089/hum.2009.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwacz K, Mukherjee S, Apolonia L, Blundell MP, Bouma G, Escors D. Nonintegrating lentivector vaccines stimulate prolonged T-cell and antibody responses and are effective in tumor therapy. J Virol. 2009;83:3094–3103. doi: 10.1128/JVI.02519-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri DR, Bona R, Michelini Z, Leone P, Macchia I, Klotman ME. Transduction of human antigen-presenting cells with integrase-defective lentiviral vector enables functional expansion of primed antigen-specific CD8(+) T cells. Hum Gene Ther. 2010;21:1029–1035. doi: 10.1089/hum.2009.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri DR, Michelini Z, Baroncelli S, Spada M, Vendetti S, Bona R. Nonintegrating Lentiviral Vector-Based Vaccine Efficiently Induces Functional and Persistent CD8+ T Cell Responses in Mice. J Biomed Biotechnol. 2010;2010:534501. doi: 10.1155/2010/534501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri DR, Michelini Z, Baroncelli S, Spada M, Vendetti S, Buffa V. Successful immunization with a single injection of non-integrating lentiviral vector. Mol Ther. 2007;15:1716–1723. doi: 10.1038/sj.mt.6300241. [DOI] [PubMed] [Google Scholar]
- Coluccio A, Miselli F, Lombardo A, Marconi A, Malagoli Tagliazucchi G, Gonçalves MA. Targeted gene addition in human epithelial stem cells by zinc-finger nuclease-mediated homologous recombination. Mol Ther. 2013;21:1695–1704. doi: 10.1038/mt.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar AV, Hollis RP, Kuftinec G, Senadheera S, Chan R, Kohn DB. Integrase-defective lentiviral vectors as a delivery platform for targeted modification of adenosine deaminase locus. Mol Ther. 2013;21:1705–1717. doi: 10.1038/mt.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A, Cesana D, Genovese P, Di Stefano B, Provasi E, Colombo DF. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat Methods. 2011;8:861–869. doi: 10.1038/nmeth.1674. [DOI] [PubMed] [Google Scholar]
- Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18:807–815. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–5705. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi G, Choi JG, Bharaj P, Abraham S, Dang Y, Kafri T. CCR5 Gene Editing of Resting CD4(+) T Cells by Transient ZFN Expression From HIV Envelope Pseudotyped Nonintegrating Lentivirus Confers HIV-1 Resistance in Humanized Mice. Mol Ther Nucleic Acids. 2014;3:e198. doi: 10.1038/mtna.2014.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AD. Retrovirus packaging cells. Hum Gene Ther. 1990;1:5–14. doi: 10.1089/hum.1990.1.1-5. [DOI] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci USA. 1993;90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Bryson PD, Zhang C, Lee CL, Wang P. A tetracycline-regulated cell line produces high-titer lentiviral vectors that specifically target dendritic cells. J Vis Exp. 2013;76:50606. doi: 10.3791/50606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockrell AS, Ma H, Fu K, McCown TJ, Kafri T. A trans-lentiviral packaging cell line for high-titer conditional self-inactivating HIV-1 vectors. Mol Ther. 2006;14:276–284. doi: 10.1016/j.ymthe.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Takeuchi Y, Martin F, Cosset FL, Mitrophanous K, Collins M. Continuous high-titer HIV-1 vector production. Nat Biotechnol. 2003;21:569–572. doi: 10.1038/nbt815. [DOI] [PubMed] [Google Scholar]
- Kafri T, van Praag H, Ouyang L, Gage FH, Verma IM. A packaging cell line for lentivirus vectors. J Virol. 1999;73:576–584. doi: 10.1128/jvi.73.1.576-584.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Throm RE, Ouma AA, Zhou S, Chandrasekaran A, Lockey T, Greene M. Efficient construction of producer cell lines for a SIN lentiviral vector for SCID-X1 gene therapy by concatemeric array transfection. Blood. 2009;113:5104–5110. doi: 10.1182/blood-2008-11-191049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Ma H, McCown TJ, Verma IM, Kafri T. Generation of a stable cell line producing high-titer self-inactivating lentiviral vectors. Mol Ther. 2001;3:97–104. doi: 10.1006/mthe.2000.0238. [DOI] [PubMed] [Google Scholar]
- Kaplan AH, Swanstrom R. The HIV-1 gag precursor is processed via two pathways: implications for cytotoxicity. Biomed Biochim Acta. 1991;50:647–653. [PubMed] [Google Scholar]
- Planelles V, Bachelerie F, Jowett JB, Haislip A, Xie Y, Banooni P. Fate of the human immunodeficiency virus type 1 provirus in infected cells: a role for vpr. J Virol. 1995;69:5883–5889. doi: 10.1128/jvi.69.9.5883-5889.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogel ME, Wu LI, Emerman M. The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J Virol. 1995;69:882–888. doi: 10.1128/jvi.69.2.882-888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang LH, Gilboa E. Expression of genes introduced into cells by retroviral infection is more efficient than that of genes introduced into cells by DNA transfection. J Virol. 1984;50:417–424. doi: 10.1128/jvi.50.2.417-424.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persons DA, Mehaffey MG, Kaleko M, Nienhuis AW, Vanin EF. An improved method for generating retroviral producer clones for vectors lacking a selectable marker gene. Blood Cells Mol Dis. 1998;24:167–182. doi: 10.1006/bcmd.1998.0184. [DOI] [PubMed] [Google Scholar]
- Cockrell AS, van Praag H, Santistevan N, Ma H, Kafri T. The HIV-1 Rev/RRE system is required for HIV-1 5’ UTR cis elements to augment encapsidation of heterologous RNA into HIV-1 viral particles. Retrovirology. 2011;8:51. doi: 10.1186/1742-4690-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]