

Abstract
Phosphofructokinase-1 (PFK1), the “gatekeeper” of glycolysis, catalyses the committed step of the glycolytic pathway by converting fructose 6-phosphate (F6P) to fructose 1,6-bisphosphate. Allosteric activation and inhibition of PFK1 by over 10 metabolites and in response to hormonal signaling fine-tune glycolytic flux to meet energy requirements1. Mutations inhibiting PFK1 activity cause glycogen storage disease type VII, also known as Tarui disease2, and mice deficient in muscle PFK1 have decreased fat stores3. Additionally, PFK1 is suggested to have important roles in metabolic reprograming in cancer4,5. Despite its critical role in glucose flux, the biologically relevant crystal structure of the mammalian PFK1 tetramer has not been determined. We report here the first structures of the mammalian PFK1 tetramer, for the human platelet isoform (PFKP), in complex with ATP-Mg2+ and ADP at 3.1 and 3.4 Å, respectively. The structures reveal substantial conformational changes in the enzyme upon nucleotide hydrolysis as well as a unique tetramer interface. Mutations of residues in this interface can affect tetramer formation, enzyme catalysis and regulation, indicating the functional importance of the tetramer. With altered glycolytic flux being a hallmark of cancers6, these new structures allow a molecular understanding of the functional consequences of somatic PFK1 mutations identified in human cancers. We characterized three of these mutations and show they have distinct effects on allosteric regulation of PFKP activity and lactate production. The PFKP structural blueprint for somatic mutations as well as the catalytic site can guide therapeutic targeting of PFK1 activity to control dysregulated glycolysis in disease.
Previous attempts to obtain the structure of mammalian tetrameric PFK1 used native protein or recombinant protein generated in yeast or bacteria. A limitation of using native PFK1 is that most mammalian tissues express all three isoforms – muscle (PFKM), liver (PFKL) and platelet (PFKP)7. Although there are structures of PFK from prokaryotes8–11 and eukaryotes12–14, including dimeric rabbit PFKM expressed in E. coli12, these structures provide limited information on the catalytic interface or the conformational changes with regulation of the tetrameic mammalian enzyme. To overcome current limitations with structural studies of human PFK1, we produced recombinant PFKP by using a baculovirus expression system. The recombinant enzyme, purified to homogeneity (Extended Data Fig. 1a), is tetrameric as shown by transmission electron microscopy (TEM; Fig. 1a). The activity and regulation of recombinant PFKP, including high cooperativity for F6P, a high affinity for ATP-Mg2+, and high sensitivity to ATP inhibition (Extended Data Fig. 1b, c), was similar to previously reported mouse PFKP expressed in yeast15.
Figure 1. Structure of ATP-bound tetrameric PFKP is in the active conformation.
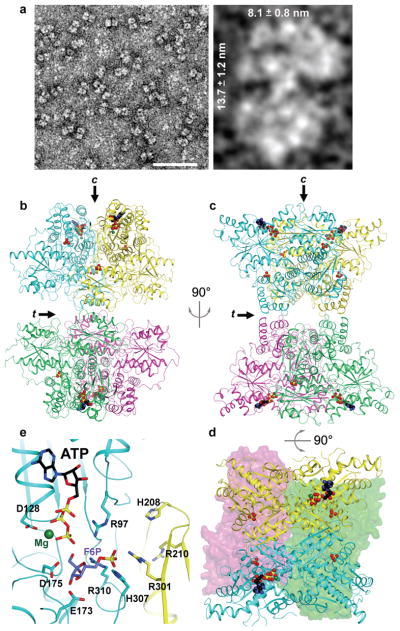
a, TEM images of PFKP. Left panel – bar represents 50 nm. Right panel – Indicated dimensions are the mean ± SD of 37 individual particles. b-c, Ribbon diagrams of PFKP displaying the relative orientation of PFKP tetramer subunits. Each subunit is individually coloured. Arrows labeled “c” and “t” indicate the catalytic and tetramer interfaces, respectively. View in (c) is rotated 90° from (b). d, View rotated 90° from (c) displaying the catalytic sites. The front subunits are shown in ribbon representation and the rear subunits depicted as surface models. ATP, black; Mg2+, dark green; phosphate, yellow. e, The binding mode of ATP-Mg2+ at the active site of PFKP. The binding mode of the F6P substrate in ScPFK12 is also shown.
We determined the crystal structure of the PFKP tetramer in complex with ATP-Mg2+ at 3.1 Å resolution (Fig. 1b–d, Extended Data Fig. 2). The atomic model has good agreement with the crystallographic data and the expected geometric parameters (Extended Data Table 1). The asymmetric unit contained two tetramers, and the eight protomers have essentially the same conformation (with rmsd of ~0.3 Å between any pair of them, Extended Data Fig. 3). The overall organizations of the two tetramers are slightly different, reflected in part by changes in the relative orientations of the two dimers (Extended Data Fig. 3).
Each PFKP tetramer measures 13.8 nm by 10.3 nm, similar in size and shape to what we calculated from TEM images (Figs. 1a, 1b). The tetramer is composed of a dimer of dimers, and the interface between the two dimers is relatively small, with a buried surface area of 700 Å2 for each subunit (arrow labeled “t” in Figs. 1b–c). The two subunits of the dimer are arranged in an antiparallel orientation, confirming previous predictions16, with a buried surface area of 1800 Å2 for each subunit. The active site is located at the interface between the two subunits (arrow labeled “c” in Figs. 1b,1e).
The structure of PFKP likely represents the active conformation of the enzyme. The crystal was prepared at pH 7, near physiological pH, and residues in the active site that are important for substrate binding and/or catalysis have similar conformations in PFKP as in other PFK structures (Fig. 1e). The F6P substrate, as observed in the S. cerevisiae PFK (ScPFK) structure12, can be readily accommodated in the PFKP active site for catalysis. The invariant substrate binding residues His208 and Arg210 from the second protomer of the dimer are located ~6 Å away from F6P, suggesting that a closure of this region of the active site may occur upon F6P binding and catalysis. PFKP contains only one ATP in each subunit bound to the active site, despite the presence of 10 mM ATP during crystallization and even though the allosteric adenine nucleotide-binding sites are functional (Extended Data Fig. 1b,c). We also observed the binding of two phosphate groups in each protomer at positions corresponding to the prokaryotic PFK effector sites (Extended Data Fig. 2c,d). The enzyme activity, regulation and stability of PFK1 is controlled by binding phosphate or sulfate ions17,18. PFKP displayed a loss of ATP inhibition in the presence of 10 mM sodium sulfate (Extended Data Fig. 2e) suggesting that phosphate-binding and inhibitory site ATP-binding are mutually exclusive in the tetrameric structure.
We also determined the crystal structure of PFKP in complex with ADP at 3.4 Å resolution, at pH 7.5. The relatively low resolution of this structure precludes a detailed structural comparison with that of the ATP-Mg2+ complex. However, it is clear there is a dramatic change in the relative positions of the two domains in each protomer (Fig. 2a), and especially the overall structures of the dimer and tetramer (Fig. 2b). A rotation of ~12° is observed between the subdomains of the ADP complex protomer relatively to the ATP complex, leading to an 8 Å shift in the substrate binding domain relative to the nucleotide binding domain. An effect of this conformational change is to open the catalytic site (Figs. 2c, 2d), which may play a role in the release of products. The conformational changes observed between the ATP and ADP complexes of PFKP are different from those seen for the R- and T-states of bacterial PFK (Extended Data Fig. 4)8,19.
Figure 2. PFKP undergoes a large conformational change upon ATP hydrolysis.

a, Structural overlay of ATP-bound (cyan) and ADP-bound (grey) PFKP subunits. b, Structural overlay of ATP-bound (coloured ribbon) and ADP-bound (grey surface) PFKP tetramers. Arrows labeled “c” and “t” represent the catalytic and tetramer interfaces, respectively. c–d, Conformation of the active site in the ATP-Mg2+-bound (c) and ADP-bound (d) structures.
We tested the importance of hydrophobic and electrostatic interactions at the tetramer interface for enzyme activity (Fig. 3a). The majority of residues at the interface are hydrophobic. Tyr645 and Phe649 from the two subunits form a π-stack of four aromatic side chains in the interface, with Phe649 in the middle (Fig. 3a). Despite the overall similarities in organization between the PFKP tetramer to that of ScPFK12, there are significant differences in the tetramer interface between the two enzymes (Extended Data Fig. 5). Phe649, which is evolutionarily conserved in metazoans but not in yeasts, is a leucine residue in ScPFK α subunit. However, this Leu residue has a completely different local environment in ScPFK compared to Phe649 in PFKP. We generated recombinant PFKP with Phe649 mutated to Leu (Extended Data Fig. 6a) to test whether Phe649 is required for tetramer formation. Previous studies showed that PFK1 assembles into tetramers in a concentration- and ligand-dependent manner, with allosteric activators favoring the formation of tetramers and allosteric inhibitors favoring the formation of dimers20–22. In a buffer containing ADP, ATP and F6P, TEM showed that wild-type (WT) PFKP particles had the dimensions and appearance of tetramers (Figs. 3b,c). In contrast, PFKP-F649L particles were the same width but half the length of WT, consistent with dimer formation along the catalytic interface (Fig. 3b,c; Extended Data Fig. 6b). We compared the PFKP-F649L particles to those induced by the inhibitor citrate, which was shown to cause PFKM to form dimers23. In a buffer containing 1 mM citrate, we saw two sizes of particles with WT PFKP; one with dimensions of tetramers and the other with dimensions of dimers along the catalytic interface (Fig. 3b,c; Extended Data Fig. 6c), further confirming dimer formation by PFKP-F649L. The catalytic activity of PFKP-F649L was reduced 98% (Extended Data Fig. 6d) compared with wild type enzyme, indicating that tetramer formation is necessary for PFK1 activity.
Figure 3. Interactions at the tetramer interface of PFKP regulate enzyme activity.

a, Interface of the PFKP tetramer, with one of two tetramer interfaces involving residues from subunit A (cyan) and subunit D (magenta). Two views of the hydrophobic interactions at the tetramer interface and predicted electrostatic interactions between Arg613 of one subunit with Glu657 of the adjacent subunit. Arrows indicate the position of the two-fold symmetry axis in the tetramer interface, relating the two subunits. b, TEM images of wild type (WT) and PFKP-F649L PFKP particles in buffer containing activator and substrates or wild type in buffer containing the inhibitor citrate. c, Scatter plot of length vs width for particles observed in TEM. WT with activator and substrate (black, n=53); F649L with activator and substrate (blue, n=77); WT with citrate, tetramers (green, n=76) and dimers (red, n=41). d, F6P dependence of PFKP wild type (black squares) and E657A (grey circles) at 0.25 mM ATP. Data are means ± SEM of 8 (wild type) and 5 (E657A) determinations from 2 independent protein preparations.
The structures suggest that an electrostatic interaction at the tetramer interface between Arg613 of one subunit and Glu657 of the adjacent subunit (Fig. 3a) may be important for enzyme function. This salt bridge was only observed in the ATP-bound structure but not in the ADP-bound PFKP structure or dimeric rabbit PFKM structures12, suggesting that it may contribute to maintaining an active form of the mammalian tetramer. PFKP-E657A had reduced affinity of ~4.5 mM for F6P, compared with ~0.8 mM in wild type, and an ~2-fold decrease in maximum activity (Fig. 3d, Extended Data Fig. 6a and Table 2). Our data indicate that hydrophobic interactions are essential for the formation of tetramers while electrostatic interactions are required for optimal enzyme activity.
The structure of PFKP provides a foundation for understanding the functional effects of somatic PFK1 mutations identified in cancers. Cancer cells rely on aerobic glycolysis to provide energy and cellular building blocks required to support rapid proliferation6. PFK1 activity is increased in cancer cell lines and primary tumour tisues24 and expression of PFKP is upregulated in breast25 and liver26 cancers. The effect of somatic mutations in PFK1 on metabolic adaptation has not been reported. We mapped the 44 reported somatic mutations in cancers27 that were not associated with SNPs28 onto the structure of PFKP (Fig. 4a, Extended Data Table 3). Analysis by Mutation Assessor29 predicted that 28 of these mutations would alter enzyme activity.
Figure 4. Somatic cancer mutations of PFKP alter enzymatic activity and allosteric regulation.

a–c, Location of indicated PFKP mutations in human cancers identified from the COSMIC database and mapped onto the catalytic interface of the PFKP subunit. Mutations chosen for further analysis are denoted with coloured boxes, including Arg48Cys (a, red) and location of Arg48 at PO42−-binding site (b), Asp564Asn (a, green) and ionic bond of Asp564 with Arg319 (b), and Asn426Ser (a, blue) and location of Ans425 at the catalytic interface (c). d–f, The effect of mutations on citrate inhibition (d), ATP activation and inhibition (e), and affinity for F6P (f). Data are means ± SEM of 7 (d), 5 (e), and 7 (f) determinations from 2 independent protein preparations. WT, black circles; Arg48Cys, red squares; Asn426Ser, blue triangles; Asn564N, green triangles. g, Lactic acid excretion (μmol lactate excreted per hour per μg of total cell lysate) from MTLn3 rat mammary adenocarcinoma cells expressing wild type and mutant PFKP-GFP. Data are means ± SEM of 4 experiments. * p < 0.05; ** p < 0.01.
We selected three identified somatic mutations for biochemical analysis. Arg48 interacts with a bound phosphate ion in the structure (Fig. 4b), and the R48C mutant had reduced citrate inhibition, shifting E50Citrate from 0.4 mM for wild type to greater than 4 mM (Fig. 4d) but did not markedly change effects of ATP and F6P (Fig. 4e–f, Extended Data Table 2). Analogous mutations in PFKM have been described in Tarui disease30,20. These data indicate that Arg48 is located in the citrate-binding site, which is occupied by the phosphate ion in the current structure30. A serine substitution for Asn426, located close to the catalytic interface, is predicted to disrupt interactions with the backbone carbonyls of Gln472, Gly473, and Gly474 and the main-chain amide of Ile476, which are involved in positioning a loop at the catalytic interface (Fig. 4c). The N426S mutant partially relieves ATP inhibition, shifting EC50ATP from ~1 mM to greater than 3 mM (Fig. 4e). Located across the catalytic interface from Asn426, Asp564 forms an electrostatic interaction with Arg319 (Fig. 4b). The D564N mutant had decreased maximum velocity and affinity for F6P (Fig. 4f, Extended Data Table 2). We also stably expressed PFKP wild type and mutants tagged with GFP in MTLn3 rat mammary adenocarcinoma cells (Extended Data 7b). In cell lysates, PFK1 activity was greater with expression of wild type, N426S, and D564N but not R48C compared with untransfected or GFP controls (Extended Data 7c). Lactic acid excretion was also greater with cells expressing wild type and N426S but significantly less with D564N compared with GFP controls (Fig. 4g). Inhibition of glycolytic flux by loss of function mutations, such as D564N, may confer a selective advantage for cancer cell growth and metastasis by redirecting carbon flow through the pentose phosphate pathway, similar to that observed by glycosylation-dependent PFK1 inhibition4. However, relief of inhibition by allosteric regulators had no effect on lactate production in the glutamine-free cell culture conditions we used. This finding could reflect the ability of PFK1 to dynamically alter metabolic states by integrating multiple signals. Additionally, the functional significance of selective PFKP mutations will depend on the mutational signature of the respective cancer in which they occur as well as the relative expression of other PFK1 isoforms.
In addition to cancer, aberrant glycolytic flux is increasingly recognized to contribute to a number of other diseases such as obesity, diabetes and Tarui disease. The biologically relevant tetrameric structures of the PFKP provide information on the catalytic interface and conformational changes upon ATP hydrolysis that contributes to a mechanistic understanding of the functional impact of disease-associated mutations. Additionally, these new structural insights will enable rational drug design for therapeutic development.
Methods
Cloning, expression, and purification of recombinant human PFKP
Homo sapiens PFKP cDNA (NM_002627.4) encoding the 784 amino acid isoform 1 was cloned into the pFastBac HTa vector and baculovirus was generated using the Bac-to-Bac Expression system (Invitrogen, Grand Island, NY) as per manufacturer protocols. 2×109 sf21 or Hi5 cells were used to express PFKP at a multiplicity of infection of 1 for 48 hours. Cell pellets were resuspended in lysis buffer (20 mM tris(hydroxymethyl)aminomethane (Tris-HCl; pH 7.5); 50 mM potassium phosphate; 1 mM 2-mercaptoethanol; 10% glycerol; 10 mM imidazole; cOmplete Protease Inhibitor Cocktail tablet (Roche)) and lysed with 15 passes of a dounce homogenizer. Cell debris was removed by centrifugation and the pellet discarded. The supernatant was incubated with Talon resin (Clontech, Mountain View, CA), washed with 20 bed volumes of lysis buffer, and eluted with a minimal volume of elution buffer (lysis buffer with 100 mM imidazole). Protein was concentrated using an Amicon Ultracel-30K Centrifugal Filter Unit (Milipore, Billerica, MA) and buffer exchanged into FPLC buffer (20 mM HEPES, pH 7.5, 100 mM KCl, 1 mM TCEP, 1 mM ATP, 1 mM MgCl2, and 5 % glycerol). PFKP was passed over a Superose 6 10/300 GL column (GE Healthcare, Piscataway, NJ) and the peak corresponding to the tetrameric fraction collected. Buffer was exchanged to crystallization buffer (20 mM HEPES, pH 7.5, 100 mM KC, 1 mM TCEP, 10 mM MgCl2, and 5 % glycerol) containing either 10 mM ADP or 10 mM ATP using an Amicon Ultracel-30K Centrifugal Filter Unit and recombinant PFKP concentrated to >5 mg/ml. Protein was stored at 4°C. Recombinant PFKP was tested for activity and allosteric regulation prior to crystallization.
PFK1 activity assays
Activity assays for PFK1 were preformed using an auxiliary enzyme assay31. Kinetic studies were performed in 200 μl reaction containing 50 mM HEPES pH 7.4, 100 mM KCl, 10 mM MgCl2, 0.15 mM NADH, 0.675 units/ml aldolase, 5 units/ml triosephosphate isomerase, and 2 units/ml glycerol phosphate dehydrogenase. ATP and fructose 6-phosphate were used as indicated. Auxiliary enzymes were desalted using an Amicon Ultracel-10K Centrifugal Filter Unit prior to use. The concentration of PFKP was normalized and samples diluted as a 10X stock in 10% glycerol, 20mM Tris-HCl (pH 7.5) and 1mM DTT immediately prior to the assay. The temperature was equilibrated to 25°C for 10 minutes prior to initiating the reaction with the addition of PFKP. The absorbance at 340 nm was measured using a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA). Kinetic parameters were generated by linear regression analysis of the Hill equation using Prism (GraphPad Software, La Jolla, CA) and are the average of a minimum of 3 measurements from 2 independent preparations of protein (R2 >0.95 for all analyses). An unpaired t-test with equal variance was used to compare the activity of wild type and F649L PFKP. One unit (U) of activity is defined as the amount of enzyme that catalyzes the formation of 1 μmol of fructose 1,6-bisphosphate per minute at 25°C. Data on the effect of sulfate on PFK1 activity were obtained in the presence of 10 mM sodium sulfate or 10 mM sodium chloride as a control.
Transmission Electron Microscopy
20 μl of 25 μg/ml PFKP was applied to glow-discharged carbon-coated grids and stained with 2% (w/v) uranyl acetate. Grids were examined and photographed with a JEOL 100CX II (JEOL, Tokyo, Japan). For estimation of size of PFKP dimers and tetramers, the length and width of individual particles from TEM images were measured using FIJI ImageJ software32. The average length and width ± SD are reported. For experiments analyzing the shape and size of PFKP for crystallography studies, the protein was diluted in TEM buffer (20 mM HEPES, pH 7.5, 100 mM KCl, 1 mM DTT, 1 mM ATP, 1 mM MgCl2, and 5 % glycerol). For experiments analyzing the oligomeric state of the enzyme in the presence of activators, WT and F649L PFKP were diluted in TEM buffer containing 3mM ADP, 3mM ATP and 8mM F6P. For experiments analyzing the oligomeric state of the enzyme in the presence of inhibitors, PFKP was diluted in TEM buffer containing 1mM citrate.
Crystallization and structure determination
PFKP was crystallized in two different complexes of ADP and ATP-Mg2+ by a microbatch method at 18°C. For the ADP complex, 2 μl of protein solution containing PFKP (6.35 mg/ml) was mixed with 1 μl of the precipitant solution consisting of 200 mM potassium sodium tartrate tetrahydrate, pH 7.4, and 20% (w/v) PEG 3350. The same protein buffer was used for growing the crystals of the PFKP in complex with ATP and Mg2+. The crystals were obtained using microbatch method and the precipitant solution comprising 200 mM potassium thiocyanate, pH 7, and 20% (w/v) PEG 3350. All crystals were cryoprotected by addition of 20% (v/v) ethylene glycol in the respective mother liquor and flash-frozen in liquid nitrogen for data collection at 100 K.
Crystals of the PFKP complexes both belong to space group P21. However, the crystallographic asymmetric unit of the ADP-bound form contains 4 subunits of PFKP that are assembled as one tetramer, whereas that of ATP-Mg2+ form contains two tetramers. A single-wavelength native data set to resolution 3.1 Å was collected at the X4C beamline of the National Synchrotron Light Source (NSLS). The diffraction images were processed with the HKL package33. The structure of PFK from rabbit skeletal muscle (PDB id: 3O8L)12 was used to determine the ATP-Mg2+ structure of PFKP using the molecular replacement method, with the program MolRep34. Only a monomeric model of PFK from rabbit skeletal muscle resulted in a solution, which led to structure determination of the ATP-bound PFKP structure. The remaining of the PFKP model was built manually with the program XtalView35. The structure refinement was performed with CNS36. A similar methodology was used for data collection and processing of the ADP-bound structure of PFKP, the crystal of which diffracted to 3.4 Å at the X4C beamline of NSLS. The ADP-bound structure was subsequently determined using a monomeric model of the ATP-bound complex of PFKP, with the program MolRep34 followed by structure refinement by CNS36. The data processing and refinement statistics are summarized in Extended Data Table 1. The Ramachandran plots suggest that 85.4% and 74.9% of residues in ATP-bound complex and ADP-bound complex of PFKP are in most favored regions, and there is no residue in disallowed regions, respectively. The trajectory between the ATP-Mg2+-bound and the ADP-bound structures was generated using UCSF Chimera40. The structures were aligned with Matchmaker tool and the trajectory calculated with the Morph Conformation tool.
Selection of cancer mutations and generation of point mutants
Somatic mutations identified in human cancers were selected from the COSMIC database27 and known SNPs were disregarded28. The mutations were modeled onto the structure of PFKP and selected for further analysis. Point mutants at the tetramer interface, F649L and E657A, and cancer mutants, R48C N426S, and D564N, were generated by using a commercially available site-directed mutagenesis kit (QuikChange Lightning, Aligent, Santa Clara, CA). DNA primers were designed using the online primer design tool (http://www.genomics.agilent.com/primerDesignProgram.jsp) and purchased from Elim Biopharmaceuticals (Hayword, CA).
Analysis of cells expressing PFKP
A mammalian PFKP expression construct was generated by PCR amplification and the cDNA inserted into the multiple cloning site of pEGFP-N1 using the restriction enzymes XhoI and BamHI. Cancer mutations were generated by site-directed mutagenesis as described above. Constructs were expressed by transfecting MTLn3 rat mammary adenocarcinoma cells37 using FugeneHD (Promega, Madison WI) transfection reagent. One day post-transfection cells were replated into 100 mm dishes and 800 μg/ml G418 was added to select for transfected cells. After one week of selection, FACS was used to sort cells expressing GFP. For metabolic assays, cells were seeded into a 6-well plate at a density of 3 × 105 cell per well. One day after replating, cells were washed twice in serum- and glutamine-free media and cells were incubated for 2 hours in 1 ml of the same media. 50 μl of the media was collected in triplicate and the amount of lactic acid in the media measured using an enzyme-linked assay38. 100 μl of Reagent A (300 mM hydrazine; 200 mM glycine, pH 9.5; 20 mM β-nicotinamide adenine dinucleotide) and 50 μl of Reagent B (200 U/ml L-lactate dehydrogenase from rabbit muscle (Sigma Aldrich, St. Louis MO) were added to each well and incubated for one hour at room temperature. The absorbance at 340 nm was measured and the amount of lactate was determined from a standard curve. Cells were lysed in buffer (10mM potassium phosphate, pH 7.5; 0.1% Triton X-100; cOmplete Protease Inhibitor Cocktail tablet (Roche)), cellular debris removed by centrifugation, and the protein concentration determined by the Bradford method. Lactic acid levels in the media were normalized to protein concentration. PFK1 activity assays were preformed on the lysate as previously described39. Enzyme-linked PFK1 activity assays were performed on 10 μg of total cell lysate as described above with the exception that 10 mM ammonium sulphate was added to the assay mixture and the auxiliary enzymes were not desalted. Levels of PFKP expression were determined by immunoblotting using rabbit anti-GFP (Invitrogen A-11122, 1DB-001-0000868907) and mouse anti-actin clone C4 (ED Millipore MAB1501, 1DB-001-0000850281) antibodies. Two-sided paired t-tests were used to determine statistical significance.
Extended Data
Extended Data Figure 1. Activity of purified recombinant PFKP.
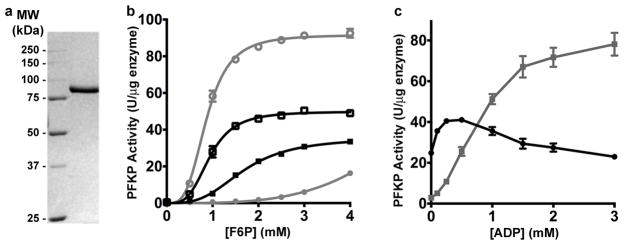
a, Coomassie-stained SDS-PAGE of purified PFKP. The molecular weight (MW) of protein standards is shown in kilodalton (kDa). b, Allosteric regulation of PFKP by ATP and ADP; F6P saturation curve PFKP in the presence of 0.25 mM ATP (filled black squares), 3 mM ATP (filled grey circles), 0.25 mM ATP and 0.25 mM ADP (open black squares), and 3 mM ATP and 3 mM ADP (open grey circles). c, Effect of ADP on kinetic behaviour of PFKP in the presence of 0.25 mM (black squares) or 3 mM (grey circles) ATP. Data in (b) and (c) are means ± SEM of 10 (b) or 5 (c) determinations from 2 separate protein preparations.
Extended Data Figure 2. Structure, nucleotide binding, and phosphate ion binding of PFKP.

a, The structure of PFKP protomer can be divided into two halves: the N-terminal (cyan) and the C-terminal (yellow) subdomains. The N-terminus of each subdomain begins with a nucleotide-binding domain (NBD) followed by a smaller substrate-binding domain (SBD). Each NBD closely resembles a canonical Rossmann fold composed of 7-stranded β-sheet surrounded by 6 α helices. Each SBD consists of 4-stranded β-sheet surrounded by 5 α helices. Two phosphate ions (stick drawings) are bound in pockets equivalent to the effector binding sites of the E. coli PFK. b, Final 2Fo–Fc electron density at 3.1 Å resolution for ATP-Mg2+, contoured at 1σ. A strong electron density is observed near β- and γ-phosphate of the nucleotide, which was unambiguously modeled as Mg2+ ion. An extended but weaker electron density is also observed near the γ-phosphate of the nucleotide, which is surrounded by three backbone carbonyls of strictly conserved Ser32, Gly34, and Gly172. This electron density was modeled as a second metal ion, although it may belong to a water molecule. c,d Structure of the two inorganic phosphate-binding sites in PFKP. e, Plot of concentration of ATP vs relative enzymatic activity of PFKP in the presence (black triangles) and absence (grey circles) of 10 mM sodium sulfate. Activity is expressed relative to maximal activity at this pH and fructose 6-phosphate concentration. Data are means ± SEM of 3 determinations.
Extended Data Figure 3. Structural comparison of the two PFKP tetramers in the ATP-Mg2+ complex.

a, Overlay of the structures of the eight PFKP subunits. Only two loops show substantial differences, indicated with the red arrows. b. Overlay of the two PFKP tetramers. A noticeable difference is the twisting of the second dimer in the two tetramers, indicated with the red arrow.
Extended Data Figure 4. Structural comparison of ATP- and ADP-bound of PFKP with R- and T-state of E. coli PFK.

a. Structural overlay of ATP-bound (coloured) and ADP-bound (grey) of PFKP. For the sake of comparison with structures of E. coli PFK, the N-terminal and C-terminal subdomains of PFKP are coloured cyan and blue for subunit A; yellow and orange for subunit B. b. The view in panel a is slabbed so as to highlight the difference between the two structures. c. Structural overlay of R-state (coloured; PDB code: 4PFK)11 and T-state (grey; PDB code: 6PFK)23 of E. coli PFK. d. The view in panel c is slabbed.
Extended Data Figure 5. A unique tetramer interface in PFKP.

a, Alignment of residues from PFKP surrounding Phe649 (arrow) with human PFKM and PFKL and Saccharomyces cerevisiae PFK1 alpha and beta subunits. b. Structure of PFKP tetramer. c. Structure of ScPFK tetramer15, viewed roughly in the same orientation as PFKP. The tetramer interface is highlighted in the red box. d. Structure of rabbit PFKM15 e. Stereo drawing of the overlay of the tetramer interface of PFKP (in color) and ScPFK.
Extended Data Figure 6. Purification and TEM analysis of PFKP tetramer mutants.
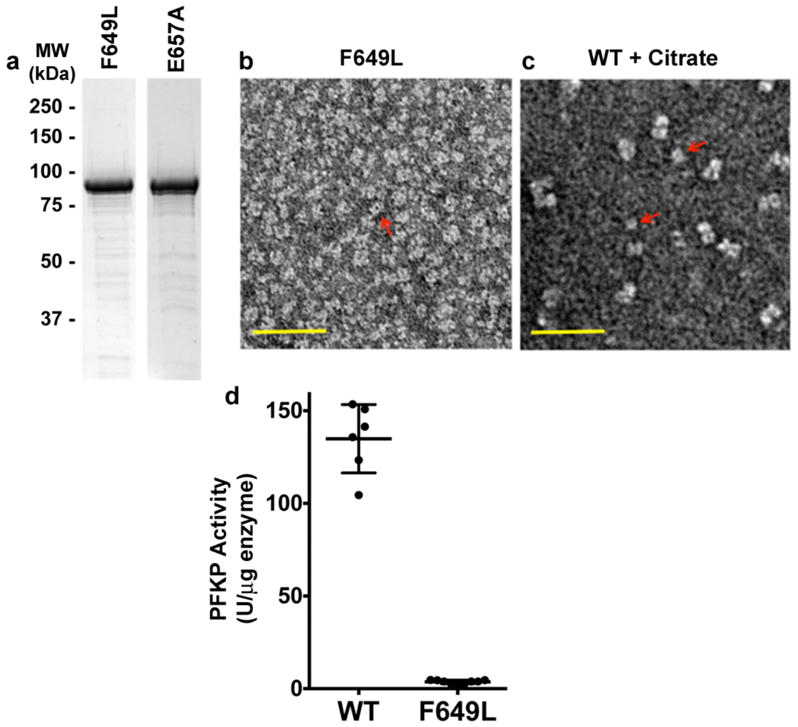
a, Coomassie-stained SDS-PAGE of PFKP F649L and E657A. b–c, TEM images of PFKP F649L (b) in buffer with activator and substrates (3 mM ADP, 3 mM ATP and 8 mM F6P) and wild type PFKP (c) in buffer containing inhibitor (1mM citrate). Red arrows indicate dimers. Scale bar, 50 nm. d, Activity of wild type PFKP and PFKP-F649L in buffer containing 3 mM ADP, 3 mM ATP and 8 mM F6P. Data are means ± SEM of 6 (wild type) and 9 (F649L) determinations from 2 independent protein preparations (p < 0.001).
Extended Data Figure 7. Purification of PFKP cancer mutants and their activity in cells.

a, Coomassie-stained SDS-PAGE of purified recombinant PFKP mutants R48C, N426S and D564N. b, Immunoblot of GFP and actin from total cell lysates of MTLn3 rat mammary adenocarcinoma cells expressing PFKP-GFP. Blots are representative of 3 experiments from individual preparations of cells. c, PFK1 activity (μmol F1,6bP produced per minute per ng of total cell lysate) was measured in 5 independent preparations of cells. A two-sided paired t-test was used to determine significance. ** P<0.01; ***P<0.001.
Extended Data Table 1.
Data collection and refinement statistics
One crystal was used for data collection for each structure.
| PFKP (ATP-Mg2+ complex) | PFKP (ADP complex) | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 137.2, 159.3, 170.5 | 79.3, 168.4, 133.3 |
| α, β, γ (°) | 90, 104.2, 90 | 90, 103.8, 90 |
| Resolution (Å) | 50-3.1 (3.2-3.1) * | 45.4-3.4 (3.5-3.4) * |
| Rmerge | 11.7 (68.4) | 15.6 (53.4) |
| I/σI | 13.9 (1.8) | 4.3 (1.2) |
| Completeness (%) | 93.0 (84.1) | 84.8 (74.3) |
| Redundancy | 6.4 (5.3) | 2.1 (1.9) |
| Refinement | ||
| Resolution (Å) | 50-3.1 (3.3-3.1) | 45.4-3.4 (3.6-3.4) |
| No. reflections | 109,577 (10,980) | 34,657 (3,486) |
| Rwork/Rfree | 22.8/25.8 | 24.2/29.4 |
| No. atoms | 47,148 | 23,644 |
| Protein | 46,792 | 23,500 |
| Ligand/ion | 316 | 144 |
| Water | 40 | 0 |
| B-factors | ||
| Protein | 62.8 | 74.5 |
| Ligand/ion | 45.6 | 66.1 |
| Water | 22.3 | |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.009 | 0.011 |
| Bond angles (°) | 1.3 | 1.2 |
Highest resolution shell is shown in parenthesis.
Extended Data Table 2.
Saturation kinetics on wild type and mutant PFKP.Kinetic properties of wild type and mutant protein were determined by modeling the sigmoidal part of the curve (Vmin to Vmax) to the Hill equations. Assays were performed at pH 7.4 with 0.25mM ATP. For F6P affinity, assays were performed at pH 7.4 with 0.25 mM ATP. ATP and citrate inhibition assays were performed at pH 7.4 with 2 mM F6P (wild type, R48C, and N426S) or 4 mM F6P (D564N). Citrate inhibition assays were performed with 0.25 mM ATP. ND, not determined
| Parameter | Wild type | E657A | R48C | N426S | D564N |
|---|---|---|---|---|---|
| Maximum Velocity | 59.27 | 32.29 | 58.19 | 67.41 | 30.60 |
| S0.5F6P (mM) | 0.83 | 4.51 | 0.84 | 0.82 | 2.04 |
| nHF6P (mM) | 3.41 | 4.13 | 2.94 | 3.64 | 3.18 |
| EC50ATP (mM) | 0.96 | ND | 1.19 | >3 | 0.68 |
| EC50Citrate (mM) | 0.40 | ND | >4 | 0.31 | 1.40 |
Extended Data Table 3.
Somatic mutations of PFKP in cancer.Residues highlighted in grey were chosen for further characterization.
| Missence Mutation* | Mutation ID* | Ligand Interactions** | Predicted Impact on Activity** | Tissue/Cancer Type* |
|---|---|---|---|---|
| S32R | 1603385 | ADP ATP | High | Liver Carcinoma |
| R48C | 1347553 | PGA ADP | High | Large Intestine Carcinoma |
| R48H | 917671 | PGA ADP | Medium | Endometrium Carcinoma |
| M49L | 1675020 | Medium | Lung Carcinoma | |
| I51V | 538534 | Medium | Lung Carcinoma | |
| D128G | 1492251 | F1,6bP ADP ATP | High | Kidney Carcinoma |
| G129W | 1187879 | ADP ATP | High | Lung Carcinoma |
| L131I | 330744 | ATP | High | Lung Carcinoma |
| Q153K | 1236264 | Neutral | Autonomic ganglia Neuroblastoma | |
| A158V | 917677 | Low | Endometrium Carcinoma | |
| D175Y | 1347563 | ATP F6P F1,6bP | High | Large Intestine Carcinoma |
| R219Q | 1603390 | ATP ADP F6P F1,6bP | High | Liver Carcinoma |
| E245Q | 684504 | ATP ADP | Low | Lung Carcinoma |
| R262Q | 1347567 | PGA ADP | Low | Large Intestine Carcinoma |
| E286K | 1474583 | Medium | Breast Carcinoma | |
| V293I | 255484 | Medium | Primitive neuroectodermal tumour - medulloblastoma | |
| R301H | 1347571 | F6P | High | Large Intestine Carcinoma |
| V308M | 917679 | Medium | Endometrium Carcinoma | |
| E328V | 1603392 | Medium | Liver Carcinoma | |
| A332T | 291604 | Medium | Large Intestine Carcinoma | |
| P407S | 233090 | Neutral | Skin Malignant melanoma | |
| A414D | 1347575 | High | Large Intestine Carcinoma | |
| N426S | 917681 | High | Endometrium Carcinoma | |
| A445T | 1220252 | Medium | Large Intestine Carcinoma | |
| W463C | 370998 | PGA ADP | High | Lung Carcinoma |
| G467A | 332540 | PGA ADP | Neutral | Lung Carcinoma |
| T470I | 1702000 | Neutral | Skin Malignant melanoma | |
| A492T | 1675022 | F1,6bP | High | Lymphoid neoplasm |
| A537S | 26929 | Low | Lung Carcinoma | |
| D564N | 241118 | Medium | Large Intestine Carcinoma | |
| R575Q | 1220254 | Low | Large Intestine Carcinoma | |
| A603T | 1492249 | Medium | Kidney Carcinoma | |
| K627E | 1347584 | PGA ADP | Low | Large Intestine Carcinoma |
| K627N | 72151 | PGA ADP | Medium | Ovary Carcinoma |
| D648N | 1560809 | Low | Large Intestine Carcinoma | |
| N667Y | 1347586 | Medium | Large Intestine Carcinoma | |
| P680A | 917689 | Low | Endometrium Carcinoma | |
| I689M | 1128065 | Neutral | Prostate Carcinoma | |
| E703D | 684499 | Medium | Lung Carcinoma | |
| K709I | 255302 | Low | Primitive neuroectodermal tumour - medulloblastoma | |
| T713A | 1239809 | Low | Oesophagus Carcinoma | |
| E734G | 98079 | Low | Upper aerodigestive tract Carcinoma | |
| M758I | 117577 | Medium | Ovary Carcinoma | |
| L761P | 538526 | Medium | Lung Carcinoma |
Missense mutations identified, Mutation ID number, and the Tissue/Cancer type each mutation was identified from COSMIC database31.
Ligand interactions and predicted impact on activity obtained from Mutation Assessor33.
Mutations with ‘High’ or ‘Medium” impact are predicted to alter enzyme activity.
Supplementary Material
Acknowledgments
We thank S Oakes for access to FPLC instrumentation and R Fletterick and K White for helpful discussions; Angela Lauricella and George DeTitta at the Hauptman-Woodward Institute for the crystallization screening. This work was supported by NIH R01 GM047413 to DLB, and a CIHR postdoctoral fellowship and a Pilot/Feasibility grant from the UCSF Liver Center (P30 DK026743) to BAW, and a grant from the Protein Structure Initiative of the National Institutes of Health U54-GM094597 to LT.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions
BAW and DLB conceived initial studies with recombinant PFKP. BAW expressed and purified recombinant PFKP, performed thermostability screens to identify buffer conditions for protein stability. Crystal screening hits were extensively optimized by FES and FF. JS performed X-ray diffraction data collection and processing. FF determined and refined the structures. BAW generated and biochemically characterized recombinant wild type and mutant PFKP, and generated and analyzed cells with heterologous PFKP expression. BAW, FF, LT and DLB contributed to writing the manuscript.
Structures of ATP-Mg2+- and ADP-bound PFKP have been deposited in PDB under accession numbers 4XYJ and 4XYK, respectively.
The authors declare no competing financial interests.
References
- 1.Schöneberg T, Kloos M, Brüser A, Kirchberger J, Sträter N. Structure and allosteric regulation of eukaryotic 6-phosphofructokinases. Biol Chem. 2013;394:977–993. doi: 10.1515/hsz-2013-0130. [DOI] [PubMed] [Google Scholar]
- 2.Tarui S, et al. Phosphofructokinase deficiency in skeletal muscle. A new type of glycogenosis. Biochem Biophys Res Commun. 1965;19:517–523. doi: 10.1016/0006-291x(65)90156-7. [DOI] [PubMed] [Google Scholar]
- 3.Getty-Kaushik L, et al. Mice Deficient in Phosphofructokinase-M Have Greatly Decreased Fat Stores. Obesity (Silver Spring) 2009 doi: 10.1038/oby.2009.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yi W, et al. Phosphofructokinase 1 Glycosylation Regulates Cell Growth and Metabolism. Science. 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moreno-Sánchez R, et al. Phosphofructokinase type 1 kinetics, isoform expression and gene polymorphisms in cancer cells. J Cell Biochem. 2012;113:1692–1703. doi: 10.1002/jcb.24039. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Dunaway GA. A review of animal phosphofructokinase isozymes with an emphasis on their physiological role. Mol Cell Biochem. 1983;52:75–91. doi: 10.1007/BF00230589. [DOI] [PubMed] [Google Scholar]
- 8.Evans PR, Farrants GW, Hudson PJ. Phosphofructokinase: structure and control. Philos Trans R Soc Lond, B, Biol Sci. 1981;293:53–62. doi: 10.1098/rstb.1981.0059. [DOI] [PubMed] [Google Scholar]
- 9.Rypniewski WR, Evans PR. Crystal structure of unliganded phosphofructokinase from Escherichia coli. Journal of Molecular Biology. 1989;207:805–821. doi: 10.1016/0022-2836(89)90246-5. [DOI] [PubMed] [Google Scholar]
- 10.Paricharttanakul NM, et al. Kinetic and structural characterization of phosphofructokinase from Lactobacillus bulgaricus. Biochemistry. 2005;44:15280–15286. doi: 10.1021/bi051283g. [DOI] [PubMed] [Google Scholar]
- 11.Mosser R, Reddy MCM, Bruning JB, Sacchettini JC, Reinhart GD. Redefining the role of the quaternary shift in Bacillus stearothermophilus phosphofructokinase. Biochemistry. 2013;52:5421–5429. doi: 10.1021/bi4002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banaszak K, et al. The Crystal Structures of Eukaryotic Phosphofructokinases from Baker’s Yeast and Rabbit Skeletal Muscle. Journal of Molecular Biology. 2011;407:284–297. doi: 10.1016/j.jmb.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 13.Sträter N, et al. Molecular architecture and structural basis of allosteric regulation of eukaryotic phosphofructokinases. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2011;25:89–98. doi: 10.1096/fj.10-163865. [DOI] [PubMed] [Google Scholar]
- 14.Mcnae IW, et al. The crystal structure of ATP-bound phosphofructokinase from Trypanosoma brucei reveals conformational transitions different from those of other phosphofructokinases. Journal of Molecular Biology. 2009;385:1519–1533. doi: 10.1016/j.jmb.2008.11.047. [DOI] [PubMed] [Google Scholar]
- 15.Sánchez-Martínez C, Estévez AM, Aragon JJ. Phosphofructokinase C isozyme from ascites tumor cells: cloning, expression, and properties. Biochem Biophys Res Commun. 2000;271:635–640. doi: 10.1006/bbrc.2000.2681. [DOI] [PubMed] [Google Scholar]
- 16.Ferreras C, Hernández ED, Martínez-Costa OH, Aragón JJ. Subunit Interactions and Composition of the Fructose 6-Phosphate Catalytic Site and the Fructose 2,6-Bisphosphate Allosteric Site of Mammalian Phosphofructokinase. 2009;284:9124–9131. doi: 10.1074/jbc.M807737200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rizzo SC, Eckel RE. Control of glycolysis in human erythrocytes by inorganic phosphate and sulfate. Am J Physiol. 1966;211:429–436. doi: 10.1152/ajplegacy.1966.211.2.429. [DOI] [PubMed] [Google Scholar]
- 18.Akkerman JW, Gorter G, Sixma JJ, Staal GE. Human platelet 6-phosphofructokinase. Purification, kinetic parameters and the influence of sulphate ions on enzyme activity. Biochim Biophys Acta. 1974;370:102–112. doi: 10.1016/0005-2744(74)90036-9. [DOI] [PubMed] [Google Scholar]
- 19.Schirmer T, Evans PR. Structural basis of the allosteric behaviour of phosphofructokinase. Nature. 1990;343:140–145. doi: 10.1038/343140a0. [DOI] [PubMed] [Google Scholar]
- 20.Hesterberg LK, Lee JC. Self-association of rabbit muscle phosphofructokinase: effects of ligands. Biochemistry. 1982;21:216–222. doi: 10.1021/bi00531a003. [DOI] [PubMed] [Google Scholar]
- 21.Leite T, Da Silva D, Coelho R, Zancan P, Sola-Penna M. Lactate favours the dissociation of skeletal muscle 6-phosphofructo-1-kinase tetramers down-regulating the enzyme and muscle glycolysis. Biochem J. 2007;408:123. doi: 10.1042/BJ20070687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zancan P, Marinho-Carvalho MM, Faber-Barata J, Dellias JMM, Sola-Penna M. ATP and fructose-2,6-bisphosphate regulate skeletal muscle 6-phosphofructo-1-kinase by altering its quaternary structure. IUBMB Life. 2008;60:526–533. doi: 10.1002/iub.58. [DOI] [PubMed] [Google Scholar]
- 23.Telford JN, Lad PM, Hammes GG. Electron microscope study of native and crosslinked rabbit muscle phosphofructokinase. Proc Natl Acad Sci USA. 1975;72:3054–3056. doi: 10.1073/pnas.72.8.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol. 2009;86:174–179. doi: 10.1016/j.yexmp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Moon JS, et al. Krüppel-like factor 4 (KLF4) activates the transcription of the gene for the platelet isoform of phosphofructokinase (PFKP) in breast cancer. Journal of Biological Chemistry. 2011;286:23808–23816. doi: 10.1074/jbc.M111.236737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park YY, et al. Tat-activating regulatory DNA-binding protein regulates glycolysis in hepatocellular carcinoma by regulating the platelet isoform of phosphofructokinase through microRNA 520. Hepatology. 2013;58:182–191. doi: 10.1002/hep.26310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Research. 2010;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.1000 Genomes Project Consortium et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Research. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Rivera D, Ru W, Gunasekera D, Kemp RG. Identification of allosteric sites in rabbit phosphofructo-1-kinase. Biochemistry. 1999;38:16407–16412. doi: 10.1021/bi991761l. [DOI] [PubMed] [Google Scholar]
- 31.Brüser A, Kirchberger J, Kloos M, Sträter N, Schöneberg T. Functional linkage of adenine nucleotide binding sites in mammalian muscle 6-phosphofructokinase. Journal of Biological Chemistry. 2012;287:17546–17553. doi: 10.1074/jbc.M112.347153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 35.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. Journal of Structural Biology. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 36.Brünger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 37.Segall JE, et al. EGF stimulates lamellipod extension in metastatic mammary adenocarcinoma cells by an actin-dependent mechanism. Clin Exp Metastasis. 1996;14:61–72. doi: 10.1007/BF00157687. [DOI] [PubMed] [Google Scholar]
- 38.Lundholm L, Mohme-Lundholm E, Vamos N. Lactic acid assay with L(+)lactic acid dehydrogenase from rabbit muscle. Acta Physiol Scand. 1963;58:243–249. doi: 10.1111/j.1748-1716.1963.tb02645.x. [DOI] [PubMed] [Google Scholar]
- 39.Abrantes JL, et al. Herpes simplex type 1 activates glycolysis through engagement of the enzyme 6-phosphofructo-1-kinase (PFK-1) BBA - Molecular Basis of Disease. 2012:1–9. doi: 10.1016/j.bbadis.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 40.Pettersen EF, et al. UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.