

Abstract
Salmonella arizonae (also called Salmonella subgroup IIIa) is a Gram-negative, non-spore-forming, motile, rod-shaped, facultatively anaerobic bacterium. S. arizonae strain RKS2983 was isolated from a human in California, USA. S. arizonae lies somewhere between Salmonella subgroups I (human pathogens) and V (also called S. bongori; usually non-pathogenic to humans) and so is an ideal model organism for studies of bacterial evolution from non-human pathogen to human pathogens. We hence sequenced the genome of RKS2983 for clues of genomic events that might have led to the divergence and speciation of Salmonella into distinct lineages with diverse host ranges and pathogenic features. The 4,574,836 bp complete genome contains 4,203 protein-coding genes, 82 tRNA genes and 7 rRNA operons. This genome contains several characteristics not reported to date in Salmonella subgroup I or V and may provide information about the genetic divergence of Salmonella pathogens.
Keywords: S. enterica subspecies arizonae RKS2983, Facultative anaerobe, Genomic evolution, Host-adapted, Salmonella pathogenicity islands
Introduction
Salmonella are Gram-negative facultative anaerobic bacteria of the family Enterobacteriaceae inhabiting the gastrointestinal tract of a wide variety of animals. There are currently over 2,600 serotypes (also called serovars) documented in the genus Salmonella . By chromosomal DNA hybridization experiments and MLEE, Salmonella currently are classified into two species,S. enterica and S. bongori (formerly subgroup V). The species S. enterica is further divided into six subspecies, including S. enterica subspecies enterica , S. enterica subspecies salamae , S. enterica subspecies arizonae , S. enterica subspecies diarizonae , S. enterica subspecies houtenae , and S. enterica subspecies indica , corresponding to the former subgroups I, II, IIIa, IIIb, IV and VI, respectively. Additionally, subgroup VII was described by Boyd et al. [1],[2]. Salmonella taxonomy is a dynamic field of research and many issues remain unsolved, especially regarding species definition [3]-[5]. To avoid confusions, therefore, we use the traditional Salmonella classification system and the terms subgroup and serotype rather than subspecies or serovar (see more detailed explanation in [5]). Most of Salmonella infections in warm-blooded animals are caused by Salmonella subgroup I serotypes, and non-subgroup I serotypes are typically associated with cold-blooded vertebrates and rarely colonize the intestines of warm-blooded animals.
Salmonella evolved from a common ancestor with Escherichia coli about 120–150 million years ago [6],[7]. During the evolutionary process, several key genomic events might have led bacteria to diverge, such as gene mutation and gene acquisition or loss [8]. Importantly, numerous lines of evidence have indicated that gene acquisition and loss are the major force driving the evolution of virulence in Salmonella [9]. In fact, it has been postulated that the evolution of Salmonella -specific virulence can be divided into three phases. The first phase is the split of Salmonella and E. coli by the Salmonella acquisition of Salmonella pathogenicity island 1, which is present in all lineages of Salmonella but absent from E. coli .SPI-1 encodes virulence factors that strengthen the infection of Salmonella serotypes by different mechanisms, including the invasiveness of the bacteria into intestinal epithelial cells [10], induction of neutrophil recruitment, and secretion of intestinal fluid [11]-[13]. The second phase is the divergence of Salmonella into S. bongori and S. enterica; this pathogenic lineage acquired SPI-2[14]-[17], which contains genes encoding a type III secretion system that is required for survival in macrophages [18]. The third phase is the adaptation of Salmonella subgroup I to warm-blooded animals, but the key genomic events involved remain unknown.
Genome sequencing efforts in Salmonella have mostly focused on Salmonella subgroup I serotypes, largely due to their pathogenicity in humans. In this study, we sequenced the genome of a strain from Salmonella subgroup IIIa (also known as Salmonella arizonae ), which lies somewhere between Salmonella subgroups I and V in evolution. Based on the important evolutionary position of Salmonella subgroup IIIa, we anticipated that its genomic comparisons with other Salmonella subgroups, especially subgroups I and V, may provide novel insights into the evolutionary transition of Salmonella adaptation from cold- to warm-blooded hosts.
Organism information
Classification and features
S. arizonae is classified to Class Gammaproteobacteria , Order Enterobacteriales , Family Enterobacteriaceae and Genus Salmonella (Table 1).S. arizonae was first described in 1939 by the name Salmonella dar es salaam and was categorized as Salmonella subgroup IIIa, later named S. arizonae [19]. S. arizonae is a rare cause of human infection and is naturally found in reptiles.
Table 1.
Classification and general features of S. arizonae RKS2983
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [34] | |
| Phylum Proteobacteria | TAS [35] | ||
| Class Gammaproteobacteria | TAS [35],[36] | ||
| Order "Enterobacteriales" | TAS [37]-[39] | ||
| Family Enterobacteriaceae | TAS [39],[40] | ||
| Genus Salmonella | TAS [40]-[41] | ||
| Species Salmonella enterica | TAS [41],[42] | ||
| Subspecies Salmonella enterica subsp. arizonae | TAS [42] | ||
| Strain RKS2983 | TAS [42] | ||
| Serovar 62:z36:- | TAS [42] | ||
| Gram stain | Negative | IDA | |
| Cell shape | Rod-shaped | IDA | |
| Motility | Motile | IDA | |
| Sporulation | Non-sporulating | IDA | |
| Temperature range | Mesophilic | IDA | |
| Optimum temperature | 35°C–37°C | IDA | |
| pH | 7.2–7.6 | IDA | |
| Carbon source | Glucose | IDA | |
| MIGS–6 | Habitat | Human | TAS [42] |
| MIGS-6.3 | Salinity | Medium | IDA |
| MIGS-22 | Oxygen requirement | Facultative anaerobes | IDA |
| MIGS-15 | Biotic relationship | Endophyte | IDA |
| MIGS-14 | Pathogenicity | Pathogenic | IDA |
| MIGS-4 | Geographic location | California, USA | TAS [42] |
| MIGS-5 | Sample collection time | 1985 | TAS [42] |
| MIGS-4.1 | Latitude | Not report | NAS |
| MIGS-4.2 | Longitude | Not report | NAS |
| MIGS-4.3 | Depth | Not report | NAS |
| MIGS-4.4 | Altitude | Not report | NAS |
a.) Evidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [43].
We obtained RKS2983 from the Salmonella Genetic Stock Center (SGSC) as one of the strains in the set of Salmonella Reference Collection C strain (SARC6) [2]; it was initially isolated from a human of California in 1985. It is, like other Salmonella bacteria, Gram-negative with diameters around 0.7 to 1.5 μm and lengths of 2 to 5 μm, facultatively anaerobic, non-spore-forming, and predominantly motile with peritrichous flagella. The bacteria were grown at 37°C in Luria broth with pH of 7.2-7.6. Detailed information on the strain can be found at SGSC [20].
Genome sequencing information
Genome project history
This complete genome project was deposited in the Genomes On-Line Database (GOLD) and the complete genome sequence of strain RKS2983 was deposited at DDBJ/EMBL/GenBank under the accession CP006693.1. Table 2 presents the project information and its association with MIGS version 2.0 [21].
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Illumina Paired-End library and SOLiD mate_pair library (2 x 50 bp) |
| MIGS-29 | Sequencing platforms | Illumina HiSeq 2000 and SOLiD 3.0 |
| MIGS-31.2 | Fold coverage | 100 × |
| MIGS-30 | Assemblers | SOAPdenovo v1.05 |
| MIGS-32 | Gene calling method | Glimmer software that used in the RAST pipeline |
| Genbank ID | CP006693.1 | |
| Genbank date of release | September 22, 2014 | |
| GOLD ID | GI686507741 | |
| BIOPROJECT | PRJNA215272 | |
| MIGS 13 | Source material identifier | CDC 409–85 |
| Project relevance | Evolution in bacteria |
Growth conditions and DNA isolation
S. arizonae RKS2983 was cultured to mid-logarithmic phase in 50 ml of Luria Broth on a gyratory shaker at 37°C. DNA was isolated from the cells using a CTAB bacterial genomic DNA isolation method [22].
Genome sequencing and assembly
The genome of S. arizonae RKS2983 was sequenced by use of two sequencing platforms, SOLiD 3.0 and Illumina HiSeq 2000. First, genomic DNA was sequenced with the Illumina sequencing platform by the paired-end strategy (2×100 bp) and the details of library construction and sequencing can be found at the Illumina web site [23]. The sequence data from Illumina HiSeq 2000 were assembled by SOAPdenovo v1.05 and the assembly contained 103 scaffolds with a genome size of 4.5 Mb. Then, the genomic DNA was sheared into 3 kb fragments by the Hydroshear instrument and was sequenced on a SOLiD sequencer by the mate-pair strategy (2 × 50 bp) according to the manual for the instrument (Applied Biosystems). The two sets of data from different methods were assembled by the velvet v1.2.09 software. The final assembly contained 20 scaffolds. Gaps between contigs were closed by PCR amplification using ABI3730 sequencer.
Genome annotation
Genes were predicted by Rapid Annotation using Subsystem Technology [24] with Glimmer 3 [25] followed by manual curation. The predicted coding sequences (CDSs) were translated and used to search the National Center for Biotechnology Information non-redundant database and Clusters of Orthologous Groups databases. These data sources were combined to assert a product description for each predicted protein. Then, we compared them with the annotated genes from four available Salmonella genomes, including S. typhi Ty2, S. typhimurium LT2 (AE006468) [26], S. arizonae RKS2980 (CP000880) [27] and S. bongori NCTC12419 (NC_015761) [17]. Non-coding genes and miscellaneous features were predicted using tRNAscanSE [28], RNAMMer [29], Rfam [30] and TMHMM [31].
Genome properties
The genome (Figure 1) consists of a chromosome of 4,574,836 bp (51.5% GC content) with 4,390 genes predicted, including 4,203 protein-coding genes, 22 rRNA genes, 82 tRNA genes and 98 pseudogenes. The properties and the statistics of the genome are summarized in Tables 3 and 4.
Figure 1.

Graphical circular map of the S. arizonae RKS2983 genome. From the outside to the center: genes on forward strand (color by COG categories), genes on reverse strand (color by COG categories), GC content, and GC skew. The map was generated with the CGviewer software.
Table 3.
Nucleotide content and gene count levels of the genome
| Attribute | Value | % of total a |
|---|---|---|
| Genome Size (bp) | 4,574,836 | |
| G + C content (bp) | 2,356,040 | 51.50 |
| Coding region (bp) | 3,924,843 | 85.79 |
| Total genesb | 4,390 | |
| rRNA genes | 22 | 0.50 |
| tRNA genes | 82 | 1.87 |
| Protein-coding genes | 4,203 | 95.70 |
| Pseudogenes | 98 | 2.23 |
| Frameshifted Genes | 78 | 1.78 |
| Genes assigned to COGs | 3,383 | 77.06 |
a.) The total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome.
Table 4.
Number of genes associated with the 25 general COG functional categories
| Code | Value | % of totala | Description |
|---|---|---|---|
| J | 168 | 3.83 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.02 | RNA processing and modification |
| K | 0 | 0.00 | Transcription |
| L | 216 | 4.92 | Replication, recombination and repair |
| B | 0 | 0.00 | Chromatin structure and dynamics |
| D | 32 | 0.73 | Cell cycle control, mitosis and meiosis |
| Y | 0 | 0.00 | Nuclear structure |
| V | 44 | 1.00 | Defense mechanisms |
| T | 106 | 2.41 | Signal transduction mechanisms |
| M | 223 | 5.08 | Cell wall/membrane biogenesis |
| N | 89 | 2.03 | Cell motility |
| Z | 0 | 0.00 | Cytoskeleton |
| W | 0 | 0.00 | Extracellular structures |
| U | 44 | 1.00 | Intracellular trafficking and secretion |
| O | 137 | 3.12 | Posttranslational modification, protein turnover, chaperones |
| C | 240 | 5.47 | Energy production and conversion |
| G | 307 | 6.99 | Carbohydrate transport and metabolism |
| E | 314 | 7.15 | Amino acid transport and metabolism |
| F | 76 | 1.73 | Nucleotide transport and metabolism |
| H | 142 | 3.23 | Coenzyme transport and metabolism |
| I | 88 | 2.00 | Lipid transport and metabolism |
| P | 182 | 4.15 | Inorganic ion transport and metabolism |
| Q | 53 | 1.21 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 311 | 7.08 | General function prediction only |
| S | 348 | 7.93 | Function unknown |
| - | 1007 | 22.94 | Not in COGs |
a.) The total is based on the total number of protein coding genes in the annotated genome.
Insights from the genome sequence
We first looked into the genetic relatedness of Salmonella and E. coli . For this, we concatenated the 945 genes common to the 25 sequenced strains analyzed in this study and conducted comparisons using BLAST with the parameters set at >70% DNA identity and >0.7 gene length ratio to categorize genes into common genes. The multiple sequence alignment program MAFFT program [32] was used to align the gene sequences of the Salmonella and E. coli strains. Phylogenetic trees were constructed with the aligned gene sequences using the Neighbor-Joining methods based on 1,000 randomly selected bootstrap replicates by MEGA 4.0 software [33]. The tree showed that S. bongori positioned between Salmonella subgroup I and E. coli , S. arizonae RKS2983 positioned between Salmonella subgroup I and S. bongori , and all Salmonella subgroup I strains were clustered together (Figure 2).
Figure 2.

Phylogenetic tree highlighting the position of S. arizonae RKS2983 (shown in bold) relative to strains of other Salmonella lineages. The corresponding GenBank accession numbers are displayed in parentheses. The tree was built based on the comparison of concatenated nucleotide sequences of 945 conserved genes in all strains. Individual orthologous sequences were aligned by the MAFFT program [32] and concatenated. The phylogenetic tree was constructed by using the MEGA 4.0 software [33] with Neighbor-Joining method. The bootstrap values are shown at branch points.
The core gene data of S. arizonae RKS2983, S. bongori NCTC 12419 and S. typhimurium LT2 (representing Salmonella subgroup I)were presented in Figure 3. There are 2823 genes common to all three genomes and 926 genes specific in RKS2983. SPI-2 is in the set of 516 genes common to RKS2983 and LT2 and is absent in S. bongori NCTC 12419. As many as 1017 genes are in LT2 but not in the other two genomes; we postulate that some of these genes may be associated with virulence to warm-blooded hosts.
Figure 3.
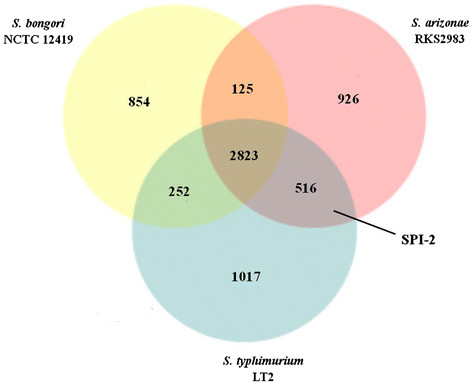
Venn diagram showing the core genes in S. arizonae RKS2983, S. bongori NCTC 12419 and S. typhimurium LT2. The core genes conducted using BLAST with the parameters set at “>70% DNA identity and >0.7 gene length ratio”.
We compared these genomes for presence or absence of Salmonella pathogenecity islands (SPIs) and found that S. arizonae RKS2983 shared some of the SPIs with S. bongori NCTC 12419 and others with S. typhimurium LT2 or S. typhi Ty2 (Table 5), providing opportunities of evolutionary studies about acquisition of SPIs during transition of Salmonella from cold- to warm-blooded animal pathogens.
Table 5.
Distribution of known SPIs in four representation genomes of Salmonella genus
| Genomic Island | S. bongori 12419 | S. arizonae RKS2983 | S. typhimurium LT2 | S. typhi Ty2 |
|---|---|---|---|---|
| SPI-1 | + | + | + | + |
| SPI-2 | - | + | + | + |
| SPI-3 | + | - | + | + |
| SPI-4 | + | + | + | + |
| SPI-5 | + | + | + | + |
| SPI-6 | - | - | + | + |
| SPI-7 | - | - | - | + |
| SPI-8 | - | - | - | + |
| SPI-9 | + | + | + | + |
| SPI-10 | - | - | - | + |
| SPI-11 | + | + | + | + |
| SPI-12 | - | - | + | + |
| SPI-13 | + | + | + | - |
| SPI-14 | - | + | + | - |
| SPI-15 | - | - | - | + |
| SPI-16 | - | - | + | + |
| SPI-17 | - | - | - | + |
| SPI-18 | - | - | - | + |
| SPI-19 | - | - | - | - |
| SPI-20 | + | + | - | - |
| SPI-21 | + | + | - | - |
| SPI-22 | - | - | - | - |
+ means SPI is present in the serotype.
- means SPI is absent in the serotype.
Conclusions
S. arizonae is phylogenetically positioed between S. bongori and Salmonella subgroup I and shares some pathogenicity-associated genes with S. bongori and some others with Salmonella subgroup I lineages. Therefore S. arizonae genome analyses may provide important clues to key genomic events that might have facilitated the evolution of warm-blooded animal pathogens from cold-blooded parasites.
Abbreviations
SARC: Salmonella reference collection C
CTAB: Cetyl trimethyl ammonium bromide
MLEE: Multilocus enzyme electrophoresis
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CXW carried out the genome sequence analysis and drafted the manuscript. SLZ and BL participated in genome sequence analysis. XYW participated in PCR amplification and sequencing of the PCR products by ABI3130 sequencer. RJ, JZ and GRL participated in the study design and provided reagents for the project. YGL and JZ provided the SOLiD and ABI Sequencing platform. SLL conceived the study and finalized the manuscript. All authors read and approved the final manuscript.
Contributor Information
Chun-Xiao Wang, Email: chunxiao_wang@foxmail.com.
Song-Ling Zhu, Email: zhu.maxim@gmail.com.
Xiao-Yu Wang, Email: wang55616918@163.com.
Ye Feng, Email: pandafengye@hotmail.com.
Bailiang Li, Email: libailianghmu@gmail.com.
Yong-Guo Li, Email: liyongguo@54dr.com.
Randal N Johnston, Email: rnjohnst@ucalgary.ca.
Gui-Rong Liu, Email: grliu@ucalgary.ca.
Jin Zhou, Email: jinzhouh85@163.com.
Shu-Lin Liu, Email: slliu@ucalgary.ca.
Acknowledgements
This work was supported by a Heilongjiang Innovation Endowment Award for graduate studies to CXW (YJSCX2012-235HLJ) and to XYW (YJSCX2012-198HLJ); National Natural Science Foundation of China (NSFC30970078 and NSFC81201248) and a grant of Natural Science Foundation of Heilongjiang Province of China to GRL; and grants of the National Natural Science Foundation of China (NSFC30970119, 81030029, 81271786, NSFC-NIH 81161120416) to SLL.
References
- Brenner FW, Villar RG, Angulo FJ, Tauxe R, Swaminathan B. Salmonella nomenclature. J Clin Microbiol. 2000;38(7):2465–2467. doi: 10.1128/jcm.38.7.2465-2467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd EF, Wang FS, Whittam TS, Selander RK. Molecular genetic relationships of the salmonellae. Appl Environ Microbiol. 1996;62(3):804–808. doi: 10.1128/aem.62.3.804-808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Wang CX, Zhu SL, Li Y, Deng X, Johnston RN. et al. Genetic boundaries to delineate the typhoid agent and other Salmonella serotypes into distinct natural lineages. Genomics. 2013;102(4):331–337. doi: 10.1016/j.ygeno.2013.07.014. [DOI] [PubMed] [Google Scholar]
- Tang L, Li Y, Deng X, Johnston RN, Liu GR, Liu SL. Defining natural species of bacteria: clear-cut genomic boundaries revealed by a turning point in nucleotide sequence divergence. BMC Genomics. 2013;14:489. doi: 10.1186/1471-2164-14-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Liu SL. The 3Cs provide a novel concept of bacterial species: messages from the genome as illustrated by Salmonella. Antonie Van Leeuwenhoek. 2012;101(1):67–72. doi: 10.1007/s10482-011-9680-0. [DOI] [PubMed] [Google Scholar]
- Baumler AJ, Tsolis RM, Ficht TA, Adams LG. Evolution of host adaptation in Salmonella entswerica. Infect Immun. 1998;66(10):4579–4587. doi: 10.1128/iai.66.10.4579-4587.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle RF, Feng DF, Tsang S, Cho G, Little E. Determining divergence times of the major kingdoms of living organisms with a protein clock. Science (New York, NY) 1996;271(5248):470–477. doi: 10.1126/science.271.5248.470. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci U S A. 1998;95(16):9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porwollik S, McClelland M. Lateral gene transfer in Salmonella. Microbes and infection / Institut Pasteur. 2003;5(11):977–989. doi: 10.1016/S1286-4579(03)00186-2. [DOI] [PubMed] [Google Scholar]
- Jones BD, Ghori N, Falkow S. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer’s patches. J Exp Med. 1994;180(1):15–23. doi: 10.1084/jem.180.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick BA, Miller SI, Carnes D, Madara JL. Transepithelial signaling to neutrophils by salmonellae: a novel virulence mechanism for gastroenteritis. Infect Immun. 1995;63(6):2302–2309. doi: 10.1128/iai.63.6.2302-2309.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Hensel M. Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev. 2004;17(1):14–56. doi: 10.1128/CMR.17.1.14-56.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Groisman EA. Distribution of pathogenicity islands in Salmonella spp. Infect Immun. 1996;64(12):5410–5412. doi: 10.1128/iai.64.12.5410-5412.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensel M, Shea JE, Baumler AJ, Gleeson C, Blattner F, Holden DW. Analysis of the boundaries of Salmonella pathogenicity island 2 and the corresponding chromosomal region of Escherichia coli K-12. J Bacteriol. 1997;179(4):1105–1111. doi: 10.1128/jb.179.4.1105-1111.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensel M, Shea JE, Waterman SR, Mundy R, Nikolaus T, Banks G. et al. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol Microbiol. 1998;30(1):163–174. doi: 10.1046/j.1365-2958.1998.01047.x. [DOI] [PubMed] [Google Scholar]
- Cirillo DM, Valdivia RH, Monack DM, Falkow S. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol Microbiol. 1998;30(1):175–188. doi: 10.1046/j.1365-2958.1998.01048.x. [DOI] [PubMed] [Google Scholar]
- Fookes M, Schroeder GN, Langridge GC, Blondel CJ, Mammina C, Connor TR, et al. Salmonella bongori provides insights into the evolution of the Salmonellae. PLoS Pathogens. 2011;7(8):e1002191. [DOI] [PMC free article] [PubMed]
- Ochman H, Soncini FC, Solomon F, Groisman EA. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc Natl Acad Sci U S A. 1996;93(15):7800–7804. doi: 10.1073/pnas.93.15.7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosa JH, Brenner DJ, Ewing WH, Falkow S. Molecular relationships among the Salmonelleae. J Bacteriol. 1973;115(1):307–315. doi: 10.1128/jb.115.1.307-315.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SGSC web site. [www.ucalgary.ca/~kesander]
- Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P. et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JDJ. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15. [Google Scholar]
- Illumina web site. [http://www.illumina.com/technology/sequencing_technology.ilmn]
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with glimmer. Bioinformatics (Oxford, England) 2007;23(6):673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L. et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413(6858):852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- Desai PT, Porwollik S, Long F, Cheng P, Wollam A, Bhonagiri-Palsikar V, Hallsworth-Pepin K, Clifton SW, Weinstock GM, McClelland M: Evolutionary Genomics of Salmonella enterica Subspecies. mBio.2013;4(2):e00198-13. [DOI] [PMC free article] [PubMed]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31(1):439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87(12):4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyL. In Bergey’s manual of systematic bacteriology, volume 2, part B. 2nd ed. New York: Springer; 2005:1.
- Garrity GM, Bell JA, Lilburn T. Class III. Gammaproteobacteria class.In Bergey’s manual of systematic bacteriology, volume 2, part B. 2nd ed. New York: Springer; 2005:1.
- Garrity GM, Holt JG. Taxonomic outline of the archaea and bacteria. In Bergey’s manual of systematic bacteriology, volume 1. 2. Springer, New York; 2001. [Google Scholar]
- Rahn O. New principles for the classification of bacteria. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg. 1937;96:273–286. [Google Scholar]
- Commission J. Conservation of the family name Enterobacteriaceae, of the name of the type genus, and designation of the type species Opinion No. 15. Int Bull Bacteriol Nomencl Taxon. 1958;8:74. [Google Scholar]
- Goullet P. Esterase electrophoretic pattern relatedness between Shigella species and Escherichia coli. J Gen Microbiol. 1980;117:493–500. doi: 10.1099/00221287-117-2-493. [DOI] [PubMed] [Google Scholar]
- Tindall BJ, Grimont PA, Garrity GM, Euzeby JP. Nomenclature and taxonomy of the genus Salmonella. Int J Syst Evol Microbiol. 2005;55(Pt 1):521–524. doi: 10.1099/ijs.0.63580-0. [DOI] [PubMed] [Google Scholar]
- Le Minor L, Popoff MY. Request for an opinion. Designation of Salmonella enterica sp. nov., nom. rev., as the type and only species of the genus Salmonella. Int J Syst Bacteriol. 1987;37:465–468. doi: 10.1099/00207713-37-4-465. [DOI] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM. et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]