

Abstract
Rotavirus (RV) predominantly replicates in intestinal epithelial cells (IEC), and “danger signals” released by these cells may modulate viral immunity. We have recently shown that human model IEC (Caco-2 cells) infected with rhesus-RV release a non-inflammatory group of immunomodulators that includes heat shock proteins (HSPs) and TGF-β1. Here we show that both proteins are released in part in association with membrane vesicles (MV) obtained from filtrated Caco-2 supernatants concentrated by ultracentrifugation. These MV express markers of exosomes (CD63 and others), but not of the endoplasmic reticulum (ER) or nuclei. Larger quantities of proteins associated with MV were released by RV-infected cells than by non-infected cells. VP6 co-immunoprecipitated with CD63 present in these MV, and VP6 co-localized with CD63 in RV-infected cells, suggesting that this viral protein is associated with the MV, and that this association occurs intracellularly. CD63 present in MV preparations from stool samples from 36 children with gastroenteritis due or not due to RV were analyzed. VP6 co-immunoprecipitated with CD63 in 3/8 stool samples from RV-infected children, suggesting that these MV are released by RV-infected cells in vivo. Moreover, fractions that contained MV from RV-infected cells induced death and inhibited proliferation of CD4+ T cells to a greater extent than fractions from non-infected cells. These effects were in part due to TGF-β, because they were reversed by treatment of the T cells with the TGF-β-receptor inhibitor ALK5i. MV from RV-infected and non-infected cells were heterogeneous, with morphologies and typical flotation densities described for exosomes (between 1.10 and 1.18 g/mL), and denser vesicles (>1.24 g/mL). Both types of MV from RV-infected cells were more efficient at inhibiting T-cell function than were those from non-infected cells. We propose that RV infection of IEC releases MV that modulate viral immunity.
Introduction
Rotavirus (RV) is the single most important etiological agent causing severe gastroenteritis (GE) in young children (1). A better understanding of the immune response against RV is necessary to improve current vaccines and to develop new RV vaccines (1). Immunity to reinfection seems to be primarily mediated by intestinal IgA, and is not completely protective in people, since multiple infections can occur during a lifetime (1,2). Moreover, we have shown a relatively poor response of circulating RV-specific CD4+ and CD8+ T cells in acutely-infected adults and children (3–5). Since RV preferentially replicates in small intestine enterocytes, the tolerogenic gut environment could be responsible for regulating the immune response against RV.
Exosomes are a type of membrane vesicle (MV) with immunomodulatory properties (6). Exosomes released by intestinal epithelial cells (IEC) have been relatively well characterized (7–10), and have been proposed to modulate the immune response to dietary antigens. However, this function is not completely clear, since exosomes from IEC that enhance (8) or suppress immunity (11) have been described. Specific immune responses of both types may be modulated by exosomes due to their capacity to transfer immunogenic peptides (8,12,13). Moreover, the interaction of exosomes with dendritic cells has been shown to be important in their capacity to enhance immune responses (12). In contrast, their tolerogenic potential could be related to their capacity to induce death (14), or to inhibit proliferation of T cells (15), as has been observed in certain cancer cell models.
Recently we found that RV-infected functionally polarized Caco-2 cells (used as a model of IEC) produce a set of immunomodulators known to induce a non-inflammatory immune response, and we hypothesized that the relatively low RV-specific T-cell response observed in humans could be related to this finding (16). The RV-infected Caco-2 cells released the constitutive (HSC70) and the inducible (HSP70) 70-kDa heat shock proteins (HSPs) and TGF-β1. These molecules have been found associated with exosomes (6,15,17–21). Two previous articles have reported an incomplete characterization of exosomes secreted by Caco-2 cells (22,23).
Exosomes are not the only MV known to have an immunomodulatory function. Microparticles, typically identified in plasma and produced as a result of membrane shedding, and apoptotic vesicles, which are released after cell death, as well as other vesicles, have also been shown to possess this function (6,24). Apoptotic vesicles from IEC have been proposed to mediate transfer of self-antigens to APC and to favor tolerance (25). Also, apoptotic vesicles from pathogen-infected IEC can be captured and processed by APC to stimulate pathogen-specific T cells (26).
Because of the heterogeneity of the MV, there are different protocols to isolate them and markers to identify them (6). For the isolation of exosomes from IEC, protocols that use differential centrifugation to discard debris and bigger MV, followed by ultracentrifugation to concentrate the exosomes, have been described (7,11). Here we decided to use a protocol that uses 0.22-μm filtration instead of differential centrifugation (27). Although this protocol also removes large membrane fragments, it may allow us to concentrate both exosomes and small apoptotic bodies. Due to the endosomal origin of exosomes, characterization of these vesicles includes the detection of markers from this compartment, like CD63 (8). Although acetylcholinesterase (AChE) activity is commonly used as an exosome marker (19,28), it can be present in the plasma membrane, and is thus found in other types of MV (29).
In this study, we investigated whether HSPs and TGF-β1 released during RV infection by Caco-2 cells were associated with MV, and explored the inmunomodulatory capacity of these MV.
Materials and Methods
Subjects and sample collection
Thirty-six children admitted with GE to the pediatric emergency service of the San Ignacio Hospital in Bogotá, Colombia, and healthy adult volunteers were enrolled in this study. Children's parents/guardians and adult volunteers signed informed consent forms approved by the Ethics Committee of the Medical School of the Pontificia Universidad Javeriana. Blood samples were drawn from children to obtain plasma, stool samples were collected, and all samples were kept at −70°C until use (Supplementary Table 1; see online supplementary material at http://www.liebertonline.com). Antigen in stool and plasma samples and specific IgA in plasma samples were measured by ELISA (3). RV P typing was done by RT-PCR and nested PCR as previously described (30).
Cells and cell culture
Caco-2 cells (a gift from C. Sapin, INSERM U 538, Université Pierre et Marie Curie, Paris, France) were cultured in DMEM (Invitrogen-Gibco, Grand Island, NY), supplemented with 20% heat-inactivated FCS (Invitrogen-Gibco), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, 10 mM HEPES, and 0.1 mM non-essential amino acids (Invitrogen-Gibco), and used between passages 68 and 77. The cells used were free of Mycoplasma (Mycoplasma detection kit for conventional PCR, VenorGeM; Sigma-Aldrich, St. Louis, MO), and the experiments were performed in the presence of 0.5 μg/mL ciprofloxacin to maintain them in this state. The cells were seeded at a density of 10,000 cells/cm2 in tissue culture flasks, and were cultured for 15 d before infection. For some experiments the cells were seeded onto 6-well Transwell plates (Costar tissue-culture-treated polycarbonate membranes with 3.0-μm pores and a 24-mm insert; Corning Inc., Corning, NY), and cultured for 21 d, as previously described (16).
RV infection
Rhesus RV (RRV) was grown and titered on MA104 cells (31). RRV was used as an unpurified supernatant of infected MA104 cells, and the supernatant of mock-infected MA104 cells was used as a negative control. RRV was activated for 30 min by treatment with 2 μg/mL of trypsin. At day 15 post-seeding the cells were deprived of FCS and washed with DMEM without serum three times, before infection at a multiplicity of infection (MOI) of 5 focus-forming units (ffu)/cell. After 45 min the inoculum was removed, the monolayers were washed twice, new DMEM without FCS was added, and the cells were incubated for the indicated times before analysis.
Preparation of a fraction containing MV
To obtain a fraction that contained MV we used a previously described method to isolate exosomes (32), with minor modifications. Supernatants of Caco-2 cells were collected at different times after RRV infection or mock treatment, and filtered using 0.22-μm filters (F1). Filtered supernatants were ultracentrifuged at 100,000 × g in a 70-Ti rotor (Beckman Coulter, Inc., Fullerton, CA) for 90 min. Supernatants were collected (F2), and the pellets were resuspended in PBS (pH 7.4), and ultracentrifuged at 100,000 × g for 90 min. The pellets were resuspended in PBS (F3) in a volume 1/200th that of F2. All fractions were analyzed by Western blot (WB), as described below. F3 from RV-infected cells were tittered, and a control preparation containing the same amounts of purified RRV was used in the selected experiments described in the text.
A preparation similar to the Caco-2 F3 was obtained from 10% stool suspensions in PBS from children with GE. The stool suspensions were centrifuged at 8000 × g and then filtered using 0.22-μm filters, followed by ultracentrifugation at 100,000 × g. The pellet was resuspended in PBS and analyzed for the presence of CD63 by WB. Stool samples with relatively high levels of CD63 were subject to immunoprecipitation (IP), as described below.
To compare F3s released from infected and non-infected Caco-2 cells, identical volumes of the preparations were used to quantify the exosome markers CD63 and AChE activity. In functional studies to compare F3 from infected and non-infected cells, the quantity of proteins present in F3 was determined using the method of Bradford, and the quantities of F3 containing the same amount of protein were used for comparison. In other experiments the quantity of AChE, a membrane vesicle marker, was quantified in the F3, and amounts of F3 containing the same quantities of AChE were used for comparison. This latter strategy permits more accurate comparison of MV in F3 from infected and non-infected cells, because viral proteins in F3 of infected cells increased their relative content of protein.
Western blots
Western blots were performed as previously described, with minor modifications (33). All samples were resuspended in reducing Laemmli buffer (DTT 0.1 M), and evaluated by WB, except for samples for CD63 identification, which were resuspended in non-reducing (without DTT) Laemmli buffer, as previously described (7). Proteins were separated by SDS-PAGE (10% gels), and transferred to PVDF membranes (BioRad, Hercules, CA). The membranes were blocked with Tris-HCl (pH 7.5) containing 5% skim milk and 0.05% Tween 20 for 1 h, and then incubated with appropriately titered primary antibodies for 1 h (mouse mAb anti-HSC70, clone B-6 from Santa Cruz Biotechnology, Santa Cruz, CA; mouse mAb anti-HSP70, clone C92F3A-5 from StressGen Biotechnologies, Victoria, B.C., Canada; mouse mAb anti-CD63, clone H5C6 from BD Pharmingen, San Diego, CA; anti-MFG-E8 rabbit polyclonal antibody, SC-33545 from Santa Cruz Biotechnology; anti-calnexin rabbit polyclonal antibody SPA-860 from Stressgen Biotechnologies; and mouse mAb anti-RV VP6, clone 1026, a generous gift from E. Kohli, Université de Dijon, Dijon, France). After washing, the blots were incubated for 50 min with ImmunoPure peroxidase-conjugated goat anti-mouse IgG or anti-rabbit IgG (Pierce Biotechnology, Inc., Rockford, IL). The blots were developed using the chemiluminescent Supersignal West Dura Extended Duration substrate (Pierce Biotechnology), and CL-XPosure films (Pierce Biotechnology). As a positive control for the detection of HSC70, a recombinant bovine HSC70 (SPP-751) from StressGen Biotechnologies was used. Of note, the mAb available to identify CD63 only recognizes the non-reduced form of the protein, producing a smeared band on WB (7).
IP of vesicles that express CD63
For IP of CD63, M-450 sheep anti-mouse IgG Dynabeads (Dynal-Biotech ASA, Oslo, Norway) were incubated overnight with a mAb anti-CD63 (clone H5C6; BD Pharmingen), or an isotype control mAb (clone MOPC-21; BD Pharmingen) (21). Magnetic beads were washed twice with PBS-BSA 0.1%. Then 25 μL of F3 was mixed with the beads and incubated with agitation for 2 h at room temperature. After magnetic separation the supernatants with unbound free proteins were saved, and the magnetic beads were washed six times with PBS-BSA 0.1%. Both free proteins and proteins bound to the beads were resuspended in non-reducing and reducing (with DTT) Laemmli buffer and evaluated by WB. For stool samples, IP was performed as for culture supernatants, but the beads were incubated with the samples overnight. Evaluation of VP6 in the immunoprecipitated proteins was evaluated by WB as described above, and/or by ELISA. When using this latter method, the beads were treated with deoxycholate (0.1% w/v for 1 h at room temperature), in order to solubilize exosome-like vesicles (34). The VP6 ELISA we used was similar to one previously described for RV antigen (3), but the plates were initially coated with the anti-VP6 1026 mAb.
Flotation of vesicles on sucrose gradients
Flotation of vesicles released by Caco-2 cells on continuous sucrose gradients was performed as previously described (35). The F3s were resuspended in 3 mL of 2.5 M sucrose, in 20 mM HEPES/NaOH (pH 7.2). A linear sucrose gradient (2–0.25 M sucrose, in 20 mM HEPES/NaOH [pH 7.2]) was layered on top of the MV suspension in an ultracentrifuge tube. The samples were centrifuged at 100,000 × g for 15 h (SW41 rotor; Beckman Coulter). Gradient fractions of 1 mL were collected from the bottom of each tube. The density of each fraction was determined using a refractometer (aus Jena, Jena, Germany). The fractions were diluted in PBS and ultracentrifuged at 100,000 × g. The pellets were resuspended in PBS and in non-reducing or reducing Laemmli buffer for WB analysis.
Analysis of T-cell proliferation and viability
CD4+ T cells were purified by negative selection (Miltenyi Biotec, Auburn, CA), using PBMCs that were isolated from healthy donors using Ficoll-Hypaque gradients. The purity of CD4+ T cells (>86%) was determined by flow cytometry. Single-cell suspensions of CD4+ T cells (5 × 106/mL) were labeled with 2 μM CFSE (Molecular Probes, Eugene, OR) for 8 min at room temperature. The cells were washed twice with RPMI 1640 containing 10% AB human serum. Then 1 × 106 CFSE-labeled T cells were stimulated polyclonally with 1.25 μg/mL of the superantigen staphylococcal enterotoxin B (SEB; Sigma-Aldrich), and 0.5 μg/mL of anti-CD28 and anti-CD49d mAbs (BD Pharmingen). Simultaneously, T cells were treated or not with different quantities of F3 preparations. Cells not stimulated with SEB or stimulated with SEB and treated with cesium-chloride-purified RRV (TLPs) were used as controls. After 5 d, the T cells were stained with a LIVE/DEAD® Violet Viability stain (Invitrogen), following the manufacturer's protocol (36), and anti-CD4 PercP Cy5.5 mAb (BD Pharmingen), and over 105 cells were acquired on a FACSAria I, using FlowJo software for analysis. Proliferation was evaluated on viable (violet-negative) cells excluding doublets. In some experiments, the CD4+ T cells were treated with 10 μM of the TGF-β receptor inhibitor ALK5i (SB431542; Sigma-Aldrich) for 30 min at 37°C before stimulation.
Measurement of AChE and TGF-β1
Supernatants from mock-treated and RRV-infected Transwell cultures were ultracentrifuged 90 min at 100,000 × g. The pellets were resuspended in PBS, and AChE activity was measured using the Amplex® Red assay kit (Invitrogen), following the manufacturer's protocol. The samples were read in a Tecan fluorometer (Tecan GENios; Phoenix Research Products, Hayward, CA). The AChE present in F3 and fractions of 1.10–1.18 g/mL and >1.24 g/mL were measured using this same kit. The detection limit was 2 mU/mL. Total (active plus latent) TGF-β1 was measured via ELISA, as previously described (16), by using a DuoSet® kit (detection limit 31.2 pg/mL; R&D Systems). The TGF-β1 measured most likely corresponds to the part of the cytokine expressed on the surface of MV, since the fractions were not solubilized for these measurements.
Immunofluorescence microscopy
Caco-2 cells were seeded onto 12-mm polycarbonate tissue culture inserts with 0.4-μm pores (Costar Transwell filters; Corning Inc.) at a density of 105 cells/cm2, and supplemented with fresh medium every other day for 7 d, and then daily for a total of 21 d. Integrity of the cellular monolayer was verified by measuring the transepithelial resistance at day 21 (Millicell; Millipore, Bedford, MA), as previously described (16). Polarized cells were infected with activated RRV at a MOI of 10 for 16 h, fixed with 2% paraformaldehyde in 100 mM phosphate buffer (pH 7.4) for 15 min, and permeabilized in PBS with 1% saponin and 3% bovine serum albumin. The cells were incubated with a 1:2000 dilution of 1E11, an mAb anti-VP6, and a 1:100 dilution of commercial rabbit anti-CD63 antibody (LAMP-3; Santa Cruz Biotechnology), or anti-EEA1 antibody (early endosome antigen 1; Abcam, Cambridge, U.K.), and 1:250 dilutions of Alexa Fluor 647 anti-mouse and Alex Fluor 488 anti-rabbit secondary antibodies (Invitrogen), and a 1:20 dilution of Alexa Fluor 594 phalloidin (Invitrogen). The samples were then mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA), and imaged with a confocal microscope (LSM510; Zeiss, Jena, Germany) using 0.2-μm sections. Z-stacks were reconstructed into three dimensions using Volocity software (Improvision; PerkinElmer Co., Coventry, England). In some experiments rabbit polyclonal antibodies against CD26 and CD9 were used (SC-9153 and SC-9148; Santa Cruz Biotechnology).
Electron microscopy
F3 of RRV-infected or mock-treated Caco-2 cells were studied by electron microscopy. The samples were resuspended in 4% paraformaldehyde and placed on Formvar-carbon-coated electron microscopy grids. Contrast medium consisted of uranyl-oxalate and methylcellulose. The vesicles were visualized with a Zeiss EM 109 electron microscope.
Statistical analysis
Statistical analysis was performed with SPSS software version 12.0 (SPSS Inc., Chicago, IL), and Graph Pad Prism software version 5.0 (Graph Pad Software Inc., San Diego, CA), using the non-parametric Wilcoxon, Mann-Whitney U, and Fisher's exact tests. Significance was set at p < 0.05. Data are shown as medians unless otherwise noted.
Results
RV infection of Caco-2 cells enhances the release of HSPs, TGF-β, and exosome markers associated with MV
We have shown that heat shock proteins HSC70 and HSP70 and TGF-β1 are released from Caco-2 cells infected with RRV (16), and that these proteins can be released free or associated with exosomes and other MV (15,18,32). To determine if these proteins are released with MV, supernatants (F1) of Caco-2 cells 24 h after treatment with a control preparation (mock), or infected with RRV, were ultracentrifuged at 100,000 × g, and the supernatants (F2, that should contain the soluble form of the proteins), and pellets (F3, potentially containing MV), were recovered and characterized (18,19,37). As shown in Fig. 1A, HSC70 and HSP70 proteins were present in all three fractions. Although the HSPs are enriched in F3, it seems that HSPs are mainly released in a soluble form, since the final volume of F2 is much greater (200 times) than that of F3. In addition, the F3 contained TGF-β1 and the exosome proteins CD63 (a tetraspanin protein) (7,38–40), MFG-E8 (lactadherin) (41), and AChE (20,28) (Fig. 1A, B, and C). The F3 of infected cells also contained RV VP6 (Fig. 1A), but F3 contained neither calnexin (an ER protein that should not be associated with exosomes) (19,20), nor the histone H2B (a nuclear protein enriched in vesicles from apoptotic cells, and also absent in exosomes) (32) (Fig. 1A and data not shown).
FIG. 1.


RV infection induces release of larger quantities of proteins associated with MV. (A) Supernatants collected from RV-infected cells (MOI 5) or mock-infected Caco-2 cells for 24 h were filtered with 0.22-μm filters (F1), and ultracentrifuged at 100,000 × g for 90 min. The supernatant (F2) and pellet (F3) were recovered after ultracentrifugation. HSC70, HSP70, VP6, the ER protein calnexin, the exosome marker CD63, and lactadherin (MFG-E8, showing a 30-kDa intracellular protein form, and one 46 kDa in size associated with MV fractions) were evaluated in all fractions by Western blot. As a positive control we used 20 μg of cell lysate (Cx+), and equal volumes of each fraction were loaded in the gels. A representative Western blot of five independent experiments with similar results is shown. Western blots for CD63 identification were done in non-reducing conditions, producing a smeared band. (B) TGF-β1 was measured in five independent F3 preparations by ELISA. In three of these preparations AChE was simultaneously measured. The quantity of TGF-β1 is reported relative to the quantity of producing cells (p = 0.062 by Wilcoxon test for the comparison between mock- and RRV-infected F3), or to the AChE activity. The bars represent medians. (C) AChE activity was measured at 0, 6, 16, 24 and 48 h post-infection in apical and basolateral supernatants of RRV- and mock-infected Caco-2 cells, and the means from three independent experiments are shown.
The F3 of RV-infected cells contained comparably more HSC70, TGF-β1, and other exosome proteins than those of control-treated cells (Fig. 1A and B and data not shown). Although other explanations are possible, these results suggest that RV infection caused an increase in MV release. Of note, the content of TGF-β1 per MV seems similar in MV of mock- or RV-infected cells, since in experiments in which the results were normalized by the F3 content of AChE, the corresponding F3 had the same quantity of this cytokine (Fig. 1B).
We have previously observed that at 24 h post-infection (hpi), the integrity of the Caco-2 monolayer is lost, accompanied by cell death (16). To determine if Caco-2 cells released F3 vesicles before cell death caused by the virus infection, we evaluated the presence of CD63 at 6 hpi, and apical and basolateral AChE activity at 6, 16, 24, and 48 hpi in polarized cells grown in a Transwell system (16). As can be seen in Fig. 1C and Supplementary Fig. 1 (see online supplementary material at http://www.liebertonline.com), both CD63 and AChE are released at higher levels by RRV-infected cells at 6 hpi, before cellular viability is compromised, suggesting that they are actively released. AChE activity gradually increased apically from 6 to 48 hpi, and to a lesser degree in the basolateral compartment at 16 hpi. The increased detection in the basal compartment coincides with the loss of cellular viability and monolayer integrity observed during RV infection (16).
RV VP6 is associated with vesicles expressing CD63 in vitro
All RV proteins are expected to be found in the F3 of infected cells, because most of the infectious and non-infectious virion particles released by the infected cells are present in this fraction. Our interest was to establish if RV antigens could associate with MV, and for this purpose the presence of VP6 was studied in F3. To determine if RV VP6 is associated with vesicles expressing CD63, the F3s were subject to IP using magnetic beads coupled with antibodies against CD63 (21), or an isotype control mAb and CD63 and VP6 were detected by WB. We chose to detect VP6 because it is an abundant RV protein for which specific antibodies are available. However, it is probable that the detection of an association of CD63 with VP6 may also reflect the association of other RV antigens with the MV. We observed that a small fraction of VP6 co-immunoprecipitated (co-IP) with CD63 (Fig. 2A). We could observe this association as early as 12 hpi (data not shown), when cellular damage caused by RRV infection was very low (16). This association of CD63 and VP6 in the F3 is not an artifact produced during the ultracentrifugation step used to obtain this fraction, since the same results were obtained directly using F1 (Fig. 2A). We determined that the mAb against CD63 (clone H5C6) could neither recognize RV proteins by WB, using lysates from RRV-infected Caco-2, nor immunoprecipitate RV proteins, using purified RV double-layer particles (data not shown).
FIG. 2.

RV VP6 is associated with vesicles that express CD63 in vitro. (A) At 24 hpi F1 and F3 from RRV-infected Caco-2 cells were subject to IP with magnetic beads coupled to anti-CD63 or an isotype control mAb. Proteins captured by the beads (Beads), or remaining in the supernatant (Free), were detected by WB using mAbs anti-CD63 and anti-VP6 (mouse mAb 1026). Representative WBs detecting CD63 and VP6 of two independent experiments performed with F1, and six using F3 with similar results, are shown. (B) Confocal microscopy of infected Caco-2 cells at 16 hpi with some co-localization of VP6 (mAb 1E11), and CD63 (a CD63 [green], b VP6 [red], and c merge), but no co-localization of VP6 with the EEA1 (green), used as a control (d EEA-1, e VP6, and f merge). Blue staining corresponds to actin. The white arrows illustrate a co-localization example that is enlarged in the upper right images. Each panel is composed of an xz section (top), and an xy section (bottom).
To determine if the association of CD63 and VP6 occurred intracellularly, these two proteins were studied in RV-infected Caco-2 cells by confocal microscopy. Using a polyclonal antibody against CD63 we found co-localization of both proteins, especially on the apical pole of the cells (Fig. 2B, panels a, b, and c). Similar results were obtained using an anti-CD63 mAb (the same clone as that used for the IP experiments shown in Fig. 3), coupled to FITC (data not shown), confirming the specificity of the immunostaining. In agreement with previous results, VP6 also co-localized with CD26 (the dipeptidyl peptidase IV) (42), and CD9 (another tetraspanin), both present in exosomes (8) (data not shown). VP6 did not co-localize with EEA1, which is a marker of early endosomes (Fig. 2B, panel d–f). Taken together, these results suggest that RV VP6 is associated with CD63-expressing vesicles, and that this association occurs intracellularly.
FIG. 3.

RV VP6 is associated with vesicles that express CD63 in vivo. F3 from stool samples from a child infected with RV were subject to IP with magnetic beads coupled to an anti-CD63 or to an isotype control (Iso C) mAb. The presence of free (localized in the supernatants; Free), or bead bound (Bead), VP6 co-IP from the stool sample was evaluated by WB (A) or by ELISA (B). In B, the open bar represents the ELISA optical density (OD) for VP6 detection after IP with an isotype control, and the solid bar with anti-CD63 mAb. The dotted line shows the cutoff value of the assay. Results from one child are shown and similar results were observed in two other children.
RV VP6 is associated with vesicles that express CD63 in vivo
To determine if CD63-expressing vesicles could be playing a role in RV GE, stool samples from children with GE due or not due to RV were studied (Supplementary Table 1; see online supplementary material at http://www.liebertonline.com). To this end, we evaluated by WB the presence of this tetraspanin in preparations from stool samples similar to F3 from Caco-2 cell supernatants. CD63 is normally only found associated with lipid membranes (43), and therefore the presence of this marker in stool samples may reflect MV release in vivo. CD63 was detectable at similar frequencies in samples from children with GE due to RV (15/24), and not due to RV (5/12) (data not shown, p = 0.29 by Fisher's exact test). However, diarrhea itself may be inducing the release of these vesicles, since there was a trend toward the detection of CD63 more frequently (7/8) in samples from children taken less than 3 days after the onset of diarrhea (DAOD), than in samples taken from children (13/28) 3 or more DAOD (data not shown, p = 0.053 by Fisher's exact test). To determine if RV antigens are associated with CD63-expressing vesicles in vivo, the F3-like preparations from a subset of stool samples from RV-infected children that contained CD63 were subject to CD63 IP, and the presence of VP6 in the immunoprecipitated proteins was evaluated by WB and ELISA (Fig. 3). Successful IP of CD63 was seen in 8/11 stool samples independent of DAOD. From samples with successful CD63 IP, VP6 co-IP with CD63 in 3/8 stool samples (Fig. 3A and B).
These results suggest that the association CD63-VP6 observed in F3 of supernatants of Caco-2 cells infected with RRV can also occur in vivo in the intestine during RV infection.
F3 from infected and non-infected cells inhibit T-cell viability
It has been shown that MV from tumors can induce T-cell death (14) or inhibit the proliferation of activated T cells (15). To determine if the F3s have these activities, we incubated purified CD4+ T cells stained with CFSE and activated with a polyclonal stimulus (SEB/α-CD49d/α-CD28) for 5 d in the presence or absence of 5 μg of F3 from mock- or RRV-infected Caco-2 cells. Both F3 preparations (mock and RRV) induced cell death and inhibited proliferation of purified CD4+ T cells (Fig. 4A and B); the RRV F3 had a greater effect than the mock F3. As expected, when we tested quantities of F3 from mock- and RV-infected cells normalized for their content of AChE, those from RV-infected cells had a higher effect than those from mock-infected cells (Fig. 4B). Similar quantities (compared to the virus detected in F3 of infected cells) of purified RV TLPs had no effect on cell viability or proliferation of CD4+ T cells (data not shown), indicating that the effect of RRV F3 on both T-cell functions are modulated by the MV present in these preparations.
FIG. 4.


F3 from RV-infected cells inhibits T-cell function more than F3 from non-infected cells. CD4+ T cells purified by magnetic beads were stained with CFSE and simultaneously exposed to a polyclonal stimulus (SEB: SEB/α-CD49d/α-CD28), and F3 from mock-infected (SEB + Mock), or RRV-infected cells (SEB + RRV), or media. After 5 d the cells were stained with violet viability dye and anti-CD3 and 1 × 105 cells were acquired and analyzed with a FACS Aria. (A) The percentages of viable cells are indicated in the upper panels, and the percentage of proliferating cells (CFSE) are indicated in the lower panels. (B) The percentages of viable cells and proliferating cells of 14 independent experiments using 7 different F3 preparations and lymphocytes from 8 different donors are shown, comparing 5 μg or equivalent quantities of AChE activity (mU) of MV mock and RRV (median 1.07 μg, range 0.6–1.3 of mock protein per 5 μg of RRV protein). Medians are depicted and significant differences between groups were established with the Wilcoxon or Mann-Whitney U test (*p < 0.05; **p < 0.0002).
The effect of MV from tumors on T-cell proliferation has been attributed to their content of TGF-β1 (15), which we found present in the F3 (Fig. 1C). To determine the role of the TGF-β1 in our model, we studied the effect of the F3s on CD4+ T-cell viability and proliferation in the presence of the TGF-β-receptor inhibitor ALK5i (SB431542). The decrease in viability and inhibition of CD4+ T-cell proliferation induced by both the mock and RRV F3s was partially reversed in three of three experiments when the cells were pretreated with the inhibitor (Fig. 5A and B), suggesting that TGF-β is at least partially involved in mediating these functions.
FIG. 5.

The TGF-β-receptor inhibitor ALK5i restores viability and proliferation of T cells. CD4+ T cells purified by magnetic beads were stained with CFSE and treated with ALK5i before exposure to the polyclonal stimulus (SEB/α-CD49d/α-CD28), and the F3 of mock-infected or RRV-infected cells or media. After 5 d the cells were stained with violet viability dye and 1 × 105 cells were acquired and analyzed with a FACS Aria. (A) Percentages of viable CD4+ T cells are shown. (B) Percentages of proliferating CD4+ T cells (CFSElow) are shown. Bars represent medians of three independent experiments.
Mock and RRV F3 contain a heterogeneous mixture of MV
To further characterize MV from mock and RRV-treated cells, F3s were examined by electron microscopy. Typical round exosome-like vesicles 30–90 nm in size (37), and other MV of irregular shape and size, were found in both preparations (Fig. 6A). The F3s were also studied by flotation on a linear sucrose gradient. We found at least two different kinds of MV in the mock and RRV preparations at 12 and 24 hpi (Fig. 6B): vesicles with a typical flotation density described for exosomes (between 1.10 and 1.18 g/mL, or 24–40% of sucrose), and denser vesicles (>1.24 g/mL, or 51.5–65% of sucrose) (32). CD63, AChE, HSC70, and VP6 were present in both types of MV, and in general the presence of one molecule was associated with the presence of the others (Fig. 6B). Although no clear differences in the gradients of MV from mock- or RRV-infected cells were evident (Fig. 6B, 12 hpi), F3 from mock-treated cells appeared to contain less dense MV in some preparations (Fig. 6B, 24 hpi). These MV fractions have a complex mixture of proteins (Fig. 6C).
FIG. 6.

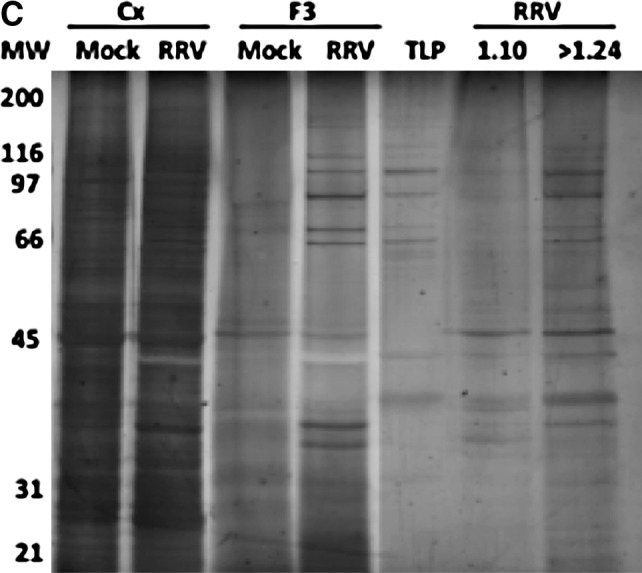
MV from RV-infected and mock-infected cells are heterogeneous. (A) The morphology of the vesicles in the F3 of RRV- or mock-treated cells at 24 hpi was examined by electron microscopy. The scale bars in the mock and RRV images are 0.2 and 0.5 μm, respectively. The thick arrows indicate MV with typical exosome morphology, and the thin arrow indicates a free virus particle. (B) F3 from 12 (top) or 24 hpi (bottom) supernatants of mock- and RRV-infected Caco-2 were ultracentrifuged on a continuous sucrose gradient (2.0–0.5 M) at 100,000 × g for 15 h. Then 1-mL fractions were collected and pelleted by ultracentrifugation. The pellets were evaluated for CD63, VP6, and HSC70, by WB and AChE activity as described in the text. Representative WBs of 2–5 independent experiments with similar results are shown. (C) SDS PAGE of mock and RRV F3s, and pooled fractions between 1.10–1.18 and >1.24 g/mL are shown. Cellular lysates (Cx) and purified virus (TLPs) were also included as a control. For each assay, 2.5 μg of protein was loaded and the proteins were visualized by silver stain (Plus One Silver Stain Kit; GE Healthcare, Uppsala, Sweden).
To determine if high- and low-density fractions differentially inhibit T-cell function, we tested the effect of different quantities of 1.10–1.18 g/mL and >1.24 g/mL fractions on T cells. Four independent experiments with different quantities of each fraction (according to the AChE activity recovered) were assayed for their capacity to inhibit viability/proliferation of polyclonally stimulated T CD4+ CFSE-stained cells (Fig. 7, Table 1, and data not shown). Both fractions from cells infected with RV had a greater effect than those from mock-infected cells (Fig. 7, Table 1, and data not shown). Thus RV infection differentially produces exosomes and denser vesicles with greater T-cell inhibitory capacity.
FIG. 7.

Exosomes and denser vesicles from RV-infected cells had an increased capacity to inhibit T-cell function. F3 from 24 hpi RRV- or mock-infected cells were run on a continuous sucrose gradient and three fractions of different densities (1.04, 1.10–1.18, and >1.24 g/mL) were recovered and ultracentrifuged at 100,000 × g for 90 min. Pellets resuspended in PBS were evaluated for their AChE activity. The results obtained for 4 mU of AChE activity are shown. A comparable volume of the 1.04-g/mL fraction was used as a negative control. After 5 d the cells were stained with violet viability dye, and 1 × 105 cells were acquired and analyzed with a FACS Aria. The percentages of viable cells are indicated in the upper panels, and the percentage of proliferating cells (CFSE) are indicated in the lower panels. A representative experiment of four performed is shown.
Table 1.
High- and Low-Density Membrane Vesicles from Rotavirus-Infected and Non-Infected Cells Have Differential T-Cell Inhibitory Capacities
| % Viable cells | % CFSElow CD4+T cells | |||
|---|---|---|---|---|
| Fraction (mU AChE) | Mock | RRV | Mock | RRV |
| Control 1.04 | 96.84 | 91.79 | 39.08 | 30.49 |
| 1.10–1.18 | ||||
| (1 mU) | 93.95 | 88.05 | 33.88 | 27.84 |
| (4 mU) | 91.52 | 76.25 | 30.36 | 12.64 |
| (8 mU) | 93.33 | 66.34 | 31.54 | 6.10 |
| >1.24 | ||||
| (1 mU) | 89.07 | 77.67 | 29.10 | 9.93 |
| (4 mU) | 76.79 | 72.55 | 23.56 | 6.27 |
| (8 mU) | 79.02 | 68.89 | 21.68 | 6.53 |
F3s from 24 hpi RRV-infected or mock-infected cells were run on a continuous sucrose gradient, and three fractions of different densities (1.04, 1.10–1.18, and >1.24 g/mL) were recovered and ultracentrifuged at 100,000 × g for 90 min, and the pellets resuspended in PBS were evaluated for their AChE activity. Quantities of each fraction containing 1, 4, and 8 mU of AChE were tested for their capacity to inhibit viability/proliferation of polyclonally-stimulated T CD4+ CFSE-stained cells. A comparable volume (relative to the highest volume used) of the 1.04-g/mL fraction was used as a negative control. Percentages of viable and proliferating cells (CFSElow) treated with the different fractions are shown. Three additional experiments, in which a single dose of MV was tested, showed similar results.
Discussion
We have shown that after infection of Caco-2 cells with RRV that TGF-β1 and HSP are released, in part associated with MV that also express markers of exosomes, but not of the endoplasmic reticulum (ER) or nuclei. MV from RV-infected and non-infected cells were heterogeneous, with morphologies and densities comparable to and not comparable to those of exosomes. Both types of MV from RV-infected cells were more efficient at inhibiting T-cell function than those from non-infected cells.
We previously showed that polarized Caco-2 cells infected with RV release both HSC70 and HSP70 (16). These HSPs are associated with exosomes in some cells (18–21). HSPs secreted by RRV-infected Caco-2 cells were present in the F3s containing MV expressing CD63, AChE, and lactadherin (MFG-E8), which have previously been reported to be markers of exosomes (7,28,32,41), but not the ER protein (calnexin) (19,20,32), or a nucleus protein (histone H2B) (15,32) (Fig. 1A and C and data not shown). However, HSPs are predominantly released in a soluble form (F2 fraction), since this fraction has a volume 200 times greater than that of the F3. Free HSPs (44), or those associated with MV (13), could potentially participate in the transfer of viral antigen to dendritic cells, favoring stimulation of specific T cells.
VP6 is also associated with CD63+ MV (Fig. 2). CD63 (LAMP-3) is a component of the late endosomal and lysosomal membranes, and it is found associated with internal membranes of multivesicular bodies and exosomes (6,45,46). Although exosome biogenesis has been proposed to involve early endosomes (20), we believe that VP6 interactions with MV occur within late endosomes, because VP6 co-localized with CD63, but not with EEA1. In agreement with the co-IP experiments (Fig. 2A), co-localization between VP6 and CD63 was a relatively rare event (Fig. 2B). Our interest was to establish if RV antigens could associate with MV, and for this purpose the presence of VP6 was studied in F3. Other virion-associated proteins were not examined. However, in preliminary experiments we have found that a small (<1%) fraction of infectious virus also co-immunoprecipitates with CD63 (data not shown). The relevance of these associations to the viral replication cycle or the existence of other viral proteins in MV remains to be determined.
The association of RV antigen with MV occurs before disruption of infected cells (Fig. 2B), and seems to occur in vivo, since small amounts of VP6 were co-immunoprecipitated with CD63 from stool samples from a subgroup of children infected with RV (Fig. 3). These vesicles could be released by RV-infected enterocytes, or come from breast milk (38). However, this latter possibility is unlikely because we found that stool samples from children breast-fed or not breast-fed contained these vesicles (data not shown). Further studies are needed to determine which factors could be related to the formation of CD63+ vesicles associated with VP6 in children.
Exosomes from tumors have been shown to directly interact with activated T cells, affecting their viability (14) or capacity to proliferate (15). TGF-β1 present in these exosomes is two logs more potent than soluble TGF-β1 at inhibiting T-cell proliferation (15). We analyzed these two T-cell functions and showed that F3s from Caco-2 cells infected or non-infected with RV affected the viability and decreased the proliferation of CD4+ T cells in response to a polyclonal stimulus. Both these effects were more pronounced for F3s of RV-infected than non-infected cells (Fig. 4B). When F3s that contained equal quantities of AChE activity were compared, fractions from infected cells were more active (Fig. 4B). The effect of the F3s is in part due to TGF-β (Figs. 1B and 5). TGF-β could be responsible for induction of both the apoptosis (47) and proliferation inhibition attributed to MV (48,49). However, since the MV contain a complex mixture of proteins (Fig. 6C), other described mechanisms [e.g., FasL (50,51) and TRAIL (14)] could also be mediating cell death. TGF-β has also been associated with the generation and expansion of regulatory T cells (52), and such cells have been shown to mediate the inhibitory activity of exosomes (15). Future studies are needed to better understand this effect.
MV analyzed by electron microscopy and density sucrose gradients were found to be heterogeneous, with a subset of MV having flotation densities typical of exosomes (1.10–1.18 g/mL), and others with densities of apoptotic vesicles (>1.24 g/mL). Exosomes and denser vesicles from RV-infected cells showed a higher inhibitory capacity than those from non-infected cells (Fig. 7 and Table 1). Therefore, RV infection induces a heterogeneous population of MV that have a greater effect on T-cell viability and proliferation than those from non-infected cells. Further studies are necessary to identify the molecules and mechanisms involved in these functions.
Tolerogenic MV have been described as a mechanism used by tumors to evade the immune response (14,15). However, exosomes may also modulate immune responses during pregnancy, (53) and in normal tissues. For example, MV similar to exosomes of thymic or ocular ciliary epithelial origin have been shown to mediate tolerance by a mechanism associated with the induction of regulatory T cells partially dependent on TGF-β (54) and FasL expression (55), respectively. In the gut, a typically tolerogenic environment (56), exosomes may promote immune responses under certain circumstances (8,12), or tolerance (11) for dietary antigens. Although in most studies, exosomes from IEC have been from epithelial cell lines, there is also evidence that these MV exist in vivo under normal physiological conditions (9,10). It is thus probable that MV-associated regulatory mechanisms are present in certain normal tissues, and are enhanced during tumorigenesis or infection. In vitro apical release of MV seems to predominate (Fig. 1C), and these MV may affect other enterocytes or dendritic cells that have access to the intestinal lumen (57). Moreover, basolateral release of MV also occurs (Fig. 1C), and may also participate in establishing a tolerogenic environment during RV infection. In our in vitro system, basolateral MV seem to come from the apical compartment after monolayer integrity is lost. However, in vivo apical MV may traverse the intestinal barrier due to an increase in intestinal barrier permeability caused by RV infection (58), permitting their contact with lamina propria T cells. In addition, specialized dendritic cells that have dendrites that protrude to the luminal side of the intestine could also capture the MV (57), and modulate the immune response (6).
Other virus and/or viral antigens have been reported, as we show here for RV, to be associated with exosomes (59–63). In the HIV and hepatitis C virus model, exosomes have been proposed to favor virus dissemination (61,62). In the Epstein-Barr virus model, exosomes have been proposed to have an inhibitory effect on T-cell proliferation that depends on their expression of an effector viral protein (60). MVs released by HIV- or CMV-infected cells have also been suggested to have immunomodulatory functions (63,64). Altogether our results suggest that RV may be exploiting the basal tolerogenic mechanism of MV release associated with the gut environment to modulate the immune response. Future studies are needed to determine if this is indeed the case.
In agreement with previous reports showing that RV causes apoptosis in Caco-2 cells (65), we found that RV infection disrupts monolayer integrity and induces cell death (16). Thus, although the protocol used to purify the MV eliminates large apoptotic vesicles, it is probable that the F3 preparations contain some apoptotic vesicles. Apoptotic bodies have been shown to contain histones (32,66) and AChE (67), and to have high densities on sucrose gradients (>1.24 g/mL) (32). In our F3s we found AChE and CD63 (the latter not previously reported to our knowledge to be present in apoptotic bodies), but not the histone H2B (Fig. 1 and data not shown). The former proteins were also present in our sucrose density gradient fractions with densities >1.24 g/mL. Thus our F3 and high-density sucrose gradient fractions may contain a heterogeneous group of MV that includes apoptotic bodies. High-density MV released by non-infected cells had a more pronounced effect on T-cell viability and proliferation than exosomes liberated by those same cells (Fig. 7). Future studies are necessary to better characterize these different MV, and the function of high-density MV released by RV-infected cells.
Supplementary Material
Acknowledgments
This work was supported by funds from Pontificia Universidad Javeriana and Colciencias grant 1203-04-16466. Luz-Stella Rodriguez is funded by Colciencias. We would like to thank Ladys Sarmiento and Maria Leonor Caldas for electron microscopy analysis (Laboratorio de Microscopía y Análisis de Imágenes, Instituto Nacional de Salud, Bogotá, Colombia), Martha Mesa for helping with the enrollment of the children, Lina Margarita Gutiérrez for purifying the virus, and Daniel Herrera for helping with the art work.
Author Disclosure Statement
The authors do not have conflict of interests.
References
- 1.Angel J. Franco MA. Greenberg HB. Rotavirus vaccines: recent developments and future considerations. Nat Rev Microbiol. 2007;5:529–539. doi: 10.1038/nrmicro1692. [DOI] [PubMed] [Google Scholar]
- 2.Coulson BS. Grimwood K. Hudson IL, et al. Role of coproantibody in clinical protection of children during reinfection with rotavirus. J Clin Microbiol. 1992;30:1678–1684. doi: 10.1128/jcm.30.7.1678-1684.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaimes MC. Rojas OL. Gonzalez AM, et al. Frequencies of virus-specific CD4(+) and CD8(+) T lymphocytes secreting gamma interferon after acute natural rotavirus infection in children and adults. J Virol. 2002;76:4741–4749. doi: 10.1128/JVI.76.10.4741-4749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rojas OL. Gonzalez AM. Gonzalez R, et al. Human rotavirus specific T cells: quantification by ELISPOT and expression of homing receptors on CD4+ T cells. Virology. 2003;314:671–679. doi: 10.1016/s0042-6822(03)00507-5. [DOI] [PubMed] [Google Scholar]
- 5.Mesa MC. Gutierrez L. Duarte-Rey C, et al. A TGF-beta mediated regulatory mechanism modulates the T cell immune response to rotavirus in adults but not in children. Virology. 2010;399:77–86. doi: 10.1016/j.virol.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 6.Thery C. Ostrowski M. Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 7.van Niel G. Raposo G. Candalh C, et al. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology. 2001;121:337–349. doi: 10.1053/gast.2001.26263. [DOI] [PubMed] [Google Scholar]
- 8.Van Niel G. Mallegol J. Bevilacqua C, et al. Intestinal epithelial exosomes carry MHC class II/peptides able to inform the immune system in mice. Gut. 2003;52:1690–1697. doi: 10.1136/gut.52.12.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin XP. Almqvist N. Telemo E. Human small intestinal epithelial cells constitutively express the key elements for antigen processing and the production of exosomes. Blood Cells Mol Dis. 2005;35:122–128. doi: 10.1016/j.bcmd.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 10.Buning J. von Smolinski D. Tafazzoli K, et al. Multivesicular bodies in intestinal epithelial cells: responsible for MHC class II-restricted antigen processing and origin of exosomes. Immunology. 2008;125:510–521. doi: 10.1111/j.1365-2567.2008.02864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karlsson M. Lundin S. Dahlgren U, et al. “Tolerosomes” are produced by intestinal epithelial cells. Eur J Immunol. 2001;31:2892–2900. doi: 10.1002/1521-4141(2001010)31:10<2892::aid-immu2892>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 12.Mallegol J. Van Niel G. Lebreton C, et al. T84-intestinal epithelial exosomes bear MHC class II/peptide complexes potentiating antigen presentation by dendritic cells. Gastroenterology. 2007;132:1866–1876. doi: 10.1053/j.gastro.2007.02.043. [DOI] [PubMed] [Google Scholar]
- 13.Andre F. Schartz NE. Movassagh M, et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet. 2002;360:295–305. doi: 10.1016/S0140-6736(02)09552-1. [DOI] [PubMed] [Google Scholar]
- 14.Huber V. Fais S. Iero M, et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterology. 2005;128:1796–1804. doi: 10.1053/j.gastro.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 15.Clayton A. Mitchell JP. Court J, et al. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007;67:7458–7466. doi: 10.1158/0008-5472.CAN-06-3456. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez LS. Barreto A. Franco MA. Angel J. Immunomodulators released during rotavirus infection of polarized caco-2 cells. Viral Immunol. 2009;22:163–172. doi: 10.1089/vim.2008.0110. [DOI] [PubMed] [Google Scholar]
- 17.Simons M. Raposo G. Exosomes—vesicular carriers for intercellular communication. Curr Opin Cell Biol. 2009;21:575–581. doi: 10.1016/j.ceb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 18.Thery C. Regnault A. Garin J, et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol. 1999;147:599–610. doi: 10.1083/jcb.147.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bausero MA. Gastpar R. Multhoff G. Asea A. Alternative mechanism by which IFN-gamma enhances tumor recognition: active release of heat shock protein 72. J Immunol. 2005;175:2900–2912. doi: 10.4049/jimmunol.175.5.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gastpar R. Gehrmann M. Bausero MA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005;65:5238–5247. doi: 10.1158/0008-5472.CAN-04-3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clayton A. Turkes A. Navabi H, et al. Induction of heat shock proteins in B-cell exosomes. J Cell Sci. 2005;118:3631–3638. doi: 10.1242/jcs.02494. [DOI] [PubMed] [Google Scholar]
- 22.Liu S. Stolz DB. Sappington PL, et al. HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix-induced hyperpermeability of Caco-2 monolayers. Am J Physiol Cell Physiol. 2006;290:C990–C999. doi: 10.1152/ajpcell.00308.2005. [DOI] [PubMed] [Google Scholar]
- 23.Marzesco AM. Janich P. Wilsch-Brauninger M, et al. Release of extracellular membrane particles carrying the stem cell marker prominin-1 (CD133) from neural progenitors and other epithelial cells. J Cell Sci. 2005;118:2849–2858. doi: 10.1242/jcs.02439. [DOI] [PubMed] [Google Scholar]
- 24.Cocucci E. Racchetti G. Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 25.Huang FP. Platt N. Wykes M, et al. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J Exp Med. 2000;191:435–444. doi: 10.1084/jem.191.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleeton MN. Contractor N. Leon F, et al. Peyer's patch dendritic cells process viral antigen from apoptotic epithelial cells in the intestine of reovirus-infected mice. J Exp Med. 2004;200:235–245. doi: 10.1084/jem.20041132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thery C. Duban L. Segura E, et al. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat Immunol. 2002;3:1156–1162. doi: 10.1038/ni854. [DOI] [PubMed] [Google Scholar]
- 28.Savina A. Furlan M. Vidal M. Colombo MI. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Biol Chem. 2003;278:20083–20090. doi: 10.1074/jbc.M301642200. [DOI] [PubMed] [Google Scholar]
- 29.Johnstone RM. Adam M. Hammond JR, et al. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes) J Biol Chem. 1987;262:9412–9420. [PubMed] [Google Scholar]
- 30.Mesa MC. Rodriguez LS. Franco MA. Angel J. Interaction of rotavirus with human peripheral blood mononuclear cells: plasmacytoid dendritic cells play a role in stimulating memory rotavirus specific T cells in vitro. Virology. 2007;366:174–184. doi: 10.1016/j.virol.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Narvaez CF. Angel J. Franco MA. Interaction of rotavirus with human myeloid dendritic cells. J Virol. 2005;79:14526–14535. doi: 10.1128/JVI.79.23.14526-14535.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thery C. Boussac M. Veron P, et al. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol. 2001;166:7309–7318. doi: 10.4049/jimmunol.166.12.7309. [DOI] [PubMed] [Google Scholar]
- 33.Barreto A. Gonzalez JM. Kabingu E, et al. Stress-induced release of HSC70 from human tumors. Cell Immunol. 2003;222:97–104. doi: 10.1016/s0008-8749(03)00115-1. [DOI] [PubMed] [Google Scholar]
- 34.MacKay PA. Leibundgut-Landmann S. Koch N, et al. Circulating, soluble forms of major histocompatability complex antigens are not exosome-associated. Eur J Immunol. 2006;36:2875–2884. doi: 10.1002/eji.200636041. [DOI] [PubMed] [Google Scholar]
- 35.Raposo G. Nijman HW. Stoorvogel W, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183:1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perfetto SP. Chattopadhyay PK. Lamoreaux L, et al. Amine reactive dyes: an effective tool to discriminate live and dead cells in polychromatic flow cytometry. J Immunol Methods. 2006;313:199–208. doi: 10.1016/j.jim.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 37.Thery C. Amigorena S. Raposo G. Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 doi: 10.1002/0471143030.cb0322s30. Chapter 3:Unit 3 22. [DOI] [PubMed] [Google Scholar]
- 38.Admyre C. Johansson SM. Qazi KR, et al. Exosomes with immune modulatory features are present in human breast milk. J Immunol. 2007;179:1969–1978. doi: 10.4049/jimmunol.179.3.1969. [DOI] [PubMed] [Google Scholar]
- 39.Caby MP. Lankar D. Vincendeau-Scherrer C, et al. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005;17:879–887. doi: 10.1093/intimm/dxh267. [DOI] [PubMed] [Google Scholar]
- 40.Wiley RD. Gummuluru S. Immature dendritic cell-derived exosomes can mediate HIV-1 trans infection. Proc Natl Acad Sci USA. 2006;103:738–743. doi: 10.1073/pnas.0507995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veron P. Segura E. Sugano G, et al. Accumulation of MFG-E8/lactadherin on exosomes from immature dendritic cells. Blood Cells Mol Dis. 2005;35:81–88. doi: 10.1016/j.bcmd.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 42.Jourdan N. Maurice M. Delautier D, et al. Rotavirus is released from the apical surface of cultured human intestinal cells through nonconventional vesicular transport that bypasses the Golgi apparatus. J Virol. 1997;71:8268–8278. doi: 10.1128/jvi.71.11.8268-8278.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hemler ME. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu Rev Cell Dev Biol. 2003;19:397–422. doi: 10.1146/annurev.cellbio.19.111301.153609. [DOI] [PubMed] [Google Scholar]
- 44.Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- 45.Escola JM. Kleijmeer MJ. Stoorvogel W, et al. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem. 1998;273:20121–20127. doi: 10.1074/jbc.273.32.20121. [DOI] [PubMed] [Google Scholar]
- 46.Kobayashi T. Vischer UM. Rosnoblet C, et al. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol Biol Cell. 2000;11:1829–1843. doi: 10.1091/mbc.11.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ito M. Minamiya Y. Kawai H, et al. Tumor-derived TGFbeta-1 induces dendritic cell apoptosis in the sentinel lymph node. J Immunol. 2006;176:5637–5643. doi: 10.4049/jimmunol.176.9.5637. [DOI] [PubMed] [Google Scholar]
- 48.Kehrl JH. Wakefield LM. Roberts AB, et al. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. 1986;163:1037–1050. doi: 10.1084/jem.163.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Datto MB. Li Y. Panus JF, et al. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monleon I. Martinez-Lorenzo MJ. Monteagudo L, et al. Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J Immunol. 2001;167:6736–6744. doi: 10.4049/jimmunol.167.12.6736. [DOI] [PubMed] [Google Scholar]
- 51.Andreola G. Rivoltini L. Castelli C, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med. 2002;195:1303–1316. doi: 10.1084/jem.20011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamagiwa S. Gray JD. Hashimoto S. Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+ CD25+ regulatory T cells from human peripheral blood. J Immunol. 2001;166:7282–7289. doi: 10.4049/jimmunol.166.12.7282. [DOI] [PubMed] [Google Scholar]
- 53.Taylor DD. Akyol S. Gercel-Taylor C. Pregnancy-associated exosomes and their modulation of T cell signaling. J Immunol. 2006;176:1534–1542. doi: 10.4049/jimmunol.176.3.1534. [DOI] [PubMed] [Google Scholar]
- 54.Wang GJ. Liu Y. Qin A, et al. Thymus exosome-like particles induce regulatory T cells. J Immunol. 2008;181:5242–5248. doi: 10.4049/jimmunol.181.8.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKechnie NM. King BC. Fletcher E. Braun G. Fas-ligand is stored in secretory lysosomes of ocular barrier epithelia and released with microvesicles. Exp Eye Res. 2006;83:304–314. doi: 10.1016/j.exer.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 56.Izcue A. Powrie F. Special regulatory T-cell review: Regulatory T cells and the intestinal tract–patrolling the frontier. Immunology. 2008;123:6–10. doi: 10.1111/j.1365-2567.2007.02778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rescigno M. Urbano M. Valzasina B, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 58.Dickman KG. Hempson SJ. Anderson J, et al. Rotavirus alters paracellular permeability and energy metabolism in Caco-2 cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:G757–G766. doi: 10.1152/ajpgi.2000.279.4.G757. [DOI] [PubMed] [Google Scholar]
- 59.Flanagan J. Middeldorp J. Sculley T. Localization of the Epstein-Barr virus protein LMP 1 to exosomes. J Gen Virol. 2003;84:1871–1879. doi: 10.1099/vir.0.18944-0. [DOI] [PubMed] [Google Scholar]
- 60.Keryer-Bibens C. Pioche-Durieu C. Villemant C, et al. Exosomes released by EBV-infected nasopharyngeal carcinoma cells convey the viral latent membrane protein 1 and the immunomodulatory protein galectin 9. BMC Cancer. 2006;6:283. doi: 10.1186/1471-2407-6-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Masciopinto F. Giovani C. Campagnoli S, et al. Association of hepatitis C virus envelope proteins with exosomes. Eur J Immunol. 2004;34:2834–2842. doi: 10.1002/eji.200424887. [DOI] [PubMed] [Google Scholar]
- 62.Nguyen DG. Booth A. Gould SJ. Hildreth JE. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J Biol Chem. 2003;278:52347–52354. doi: 10.1074/jbc.M309009200. [DOI] [PubMed] [Google Scholar]
- 63.Walker JD. Maier Cl. Pober JS. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J Immunol. 2009;182:1548–1559. doi: 10.4049/jimmunol.182.3.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Esser MT. Graham DR. Coren LV, et al. Differential incorporation of CD45, CD80 (B7-1), CD86 (B7-2), and major histocompatibility complex class I and II molecules into human immunodeficiency virus type 1 virions and microvesicles: implications for viral pathogenesis and immune regulation. J Virol. 2001;75:6173–6182. doi: 10.1128/JVI.75.13.6173-6182.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chaibi C. Cotte-Laffitte J. Sandre C, et al. Rotavirus induces apoptosis in fully differentiated human intestinal Caco-2 cells. Virology. 2005;332:480–490. doi: 10.1016/j.virol.2004.11.039. [DOI] [PubMed] [Google Scholar]
- 66.Schiller M. Bekeredjian-Ding I. Heyder P, et al. Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ. 2008;15:183–191. doi: 10.1038/sj.cdd.4402239. [DOI] [PubMed] [Google Scholar]
- 67.Zhang XJ. Yang L. Zhao Q, et al. Induction of acetylcholinesterase expression during apoptosis in various cell types. Cell Death Differ. 2002;9:790–800. doi: 10.1038/sj.cdd.4401034. [DOI] [PubMed] [Google Scholar]
- 68.Rojas OL. Caicedo L. Guzman C, et al. Evaluation of circulating intestinally committed memory B cells in children vaccinated with attenuated human rotavirus vaccine. Viral Immunol. 2007;20:300–311. doi: 10.1089/vim.2006.0105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.