

Abstract
Results of behavioral genetic and molecular genetic studies have converged to suggest that both genes contribute to the development of ADHD. Although prior linkage studies have produced intriguing results, their results have been inconsistent, with no clear pattern of results emerging across studies. We genotyped 5,980 SNPs across the genome in 1,187 individuals from families with children diagnosed with ADHD. We then performed two nonparametric linkage analyses on ADHD families: (1) an affected sibling pair linkage analysis on 217 families with 601 siblings diagnosed with ADHD and (2) a variance components linkage analysis using the number of ADHD symptoms as the phenotype on 260 families with 1,100 phenotyped siblings. The affection status linkage analysis had a maximum LOD score of 1.85 on chromosome 8 at 54.2 cM. The maximum LOD score in the variance components linkage analysis was 0.8 on chromosome 8 at 93.4 cM. The absence of regions of significant or suggestive linkage in these data suggest that there are no genes of large effect contributing to the ADHD phenotype.
Keywords: ADHD, linkage analysis, SNPs
INTRODUCTION
With a prevalence of eight to twelve percent worldwide [Faraone et al., 2003], attention deficit/hyperactivity disorder (ADHD) is among the most common childhood psychiatric disorders. Its name reflects the range of possible clinical presentations, which may include hyperactivity as well as inattention and impulsivity [Faraone, 2005]. In spite of this heterogeneity, and some shift in diagnostic criteria [American Psychiatric Association, 1987], it is also among the best-validated childhood diagnoses from both clinical, longitudinal and neurobiological perspectives [Faraone and Biederman, 1998; Faraone et al., 2000, 2006; Faraone, 2004a]. This feature, along with early observations that family members of children with ADHD were at elevated risk for ADHD [Morrison and Stewart, 1971], and that the heritability of ADHD is about 76%, made ADHD an attractive target for genetic studies [Faraone, 2004b; Faraone et al., 2005].
In an attempt to find regions of chromosomes which might harbor genes for ADHD, several groups have conducted genome-wide linkage scans. A study of 126 American affected sib-pairs found three regions showing some evidence of linkage (LOD scores >1.5): 5p12, 10q26, 12q23, and 16p13 [Fisher et al., 2002]. An expanded sample of 203 families found stronger evidence for the 16p13 region, previously implicated in autism, with a maximum LOD score of 4 [Smalley et al., 2002]. A study of 164 Dutch affected sib-pairs also identified a peak previously noted in autism, at 15q15, with a peak LOD score of 3.5 [Bakker et al., 2003]. Two other peaks, at 7p13 and 9q33, yielded LOD scores of 3.0 and 2.1, respectively. A genome-wide scan of families from a genetically isolated community in Columbia implicated 8q12, 11q23, 4q13, 17p11, 12q23, and 8p23 [Arcos-Burgos et al., 2004a]. A study of 155 sib-pairs from Germany reported a maximum LOD score of 2.59 for chromosome 5p at 17cM. They also reported nominal evidence for linkage to chromosomes 6q, 7p, 9q, 11q, 12q and 17p, which had also been identified in previous scans.
Table I summarizes the maximum LOD scores from the prior linkage studies. Although there is some overlap in nominally significant linkage peaks, there is no evidence for the replication of a genome-wide significant finding using strict criteria [Lander and Kruglyak, 1995]. Nevertheless, given that these studies show some overlap for peaks at lower LOD scores and that each, individually had low power to detect linkage to genes of small effect, these regions remain of interest for replication studies and for fine mapping efforts. For ADHD, the lack of replication across the available studies completed so far suggests that genes of moderately large effect are unlikely to exist. Thus, eventually, pooled data sets and for this reason, further studies are may be needed to increase the power of available linkage data.
TABLE I.
Maximum LOD Scores From Prior Genome Scans of ADHD
| Study | Region | LOD |
|---|---|---|
| USAa | 16p13 | 3.7 |
| USAa | 17p11 | 3.0 |
| USAa | 12p13 | 2.6 |
| USAa | 5p13 | 2.6 |
| USAa | 6q12 | 3.3 |
| The Netherlandsb | 15q15 | 3.5 |
| The Netherlandsb | 7p13 | 3.0 |
| The Netherlandsb | 9q33 | 2.1 |
| Columbiac | 17p11 | 2.8 |
| Columbiac | 11q22 | 2.5 |
| Columbiac | 5q33.3 | 2.4 |
| Germanyd | 5p13 | 2.6 |
Fisher et al. [Fisher et al., 2002], Ogdie et al. [Ogdie et al., 2002, Ogdie et al., 2003, Ogdie et al., 2004].
Bakker et al. [Bakker et al., 2003].
Arcos-Burgos et al. [Arcos-Burgos et al., 2004b].
Hebebrand et al. [Hebebrand et al., 2006].
METHODS
Subjects
For a family to be included in the study, we required two or more full biological siblings with a lifetime diagnosis of DSM-IV ADHD who were willing to provide blood samples for DNA extraction and had at least one participating parent. Families were predominantly ascertained through the Clinical and Research Program in Pediatric Psychopharmacology within the child psychiatry unit at the Massachusetts General Hospital (49%). Remaining families were referred from local private child psychiatry and pediatric practices (24%), newspaper and support group advertisements (18%), the outpatient psychiatry unit at Children’s Hospital in Boston (5%) and an outpatient child psychiatry clinic at the University of Nebraska (4%). No ethnic or racial groups were excluded. Potential subjects were excluded if they had been adopted, if their nuclear family was not available for study or if they did not wish to provide a blood sample. We also excluded youth with major sensorimotor handicaps, active psychosis, inadequate command of the English language, or suspected IQ less than 70. Parents provided written informed consent for themselves and their children; children and adolescents provided written assent to participate.
A two-stage procedure selected the final sample. The first stage confirmed the diagnosis of the sibling pair with the primary caregiver or adult sibling pair using a telephone questionnaire that included ADHD and exclusion criteria. The second stage was a structured interview (see below). Families with sibling pairs receiving positive diagnoses at both stages were included in the study.
The Schedule for Affective Disorders-Child Epidemiological Version (Kiddie—SADS-E) was used to assess lifetime and current DSM-IV diagnoses of ADHD and a range of internalizing and externalizing disorders in youth. The K-SADS-E is a widely used semi-structured psychiatric diagnostic interview, with established psychometric properties [Orvaschel, 1994]. It was designed for use in clinical and epidemiologic research with children and adolescents age 6–17. For all youth, psychiatric data were collected from the mother or primary caregiver. In addition, youth 12 and older were directly interviewed. Children younger than 12 were not interviewed directly because studies suggest limited reliability of such interviews [Edelbrock et al., 1985; Achenbach and McConaughy, 1987; Schwab-Stone et al., 1994; Breton et al., 1995]. Subjects 18 and older received the Structured Clinical Interview for DSM IV (SCID) [First et al., 1997] with a supplement from the K-SADS-E to assess child psychiatric disorders.
All interviews were conducted by personnel in the Clinical and Research Program in Pediatric Psychopharmacology at the Massachusetts General Hospital. Subjects were interviewed in person, with the exception of families ascertained from the University of Nebraska who were interviewed over the telephone. Interviewers had Master’s or Bachelor’s degrees in psychology or a related field. They underwent a 3-month training program that included mastery of DSM-IV criteria, observation of experienced raters, and achievement of high levels of inter-rater reliability with senior raters. Kappa coefficients assessed diagnostic agreement between randomly audiotaped interviews and board certified child psychiatrists and psychologists for 173 subjects. The Kappa for ADHD was 0.99. Raters were blind to the ascertainment status of subjects because they were conducting comparable psychiatric interviews for other family studies at MGH. Final diagnoses were made after blind review of interview data by psychiatrists and clinical psychologists. The committee made a Best Estimate diagnosis as described by Leckman et al. [1982]. Definite diagnoses were assigned to subjects who met all diagnostic criteria and warranted clinical concern due to the nature of the symptoms and impairment. To combine discrepant parent–offspring reports, the most severe diagnosis was used, unless it was felt to be unreliable. Symptoms were coded as present if endorsed by either rater.
Information on all 18 DSM-IV ADHD symptoms was collected. The inattentive symptoms include: (1) inability to pay attention to details; (2) difficulty with sustained attention in tasks or play activities; (3) listening problems; (4) difficulty following instructions; (5) problems organizing tasks and activities; (6) avoidance or dislike of tasks that require mental effort; (7) tendency to lose things like toys, notebooks, or homework; (8) distractibility; and (9) forgetfulness in daily activities. Hyperactive-impulsivity symptoms include: (1) fidgeting or squirming; (2) difficulty remaining seated; (3) restlessness; (4) difficulty playing quietly; (5) always seeming to be “on the go”; (6) excessive talking; (7) blurting out answers before hearing the full question; (8) difficulty waiting for a turn or in line; and (9) problems with interrupting or intruding. The total number of these symptoms that each individual exhibited was also computed.
Genotyping
Genotyping services were provided by the Center for Inherited Disease Research (CIDR) using 5,980 single nucleotide polymorphisms (SNPs) spaced at an average of rv121 kb intervals, following CIDR’s standard genotyping procedures (http://www.cidr.jhmi.edu/human_snp.html). In brief, CIDR performs whole genome SNP linkage scan genotyping using Illumina’s BeadArray™ technology on a BeadLab system. CIDR uses the Illumina® Linkage IVb Marker Panel. Using this Illumina platform a total of 6,008 total SNP assays were attempted for genotyping. Illumina’s Gentrain software was used to evaluate all genotypes using a quantitative quality score called GenCall score. A GenCall score ranges from 0 to 1 and reflects the proximity within a cluster plot of the intensities of that genotype to the centroid of the nearest cluster. Genotypes were dropped from the analysis if any of several problems occurred: (1) GenCall scores less than 0.25; (2) poorly defined clusters; (3) excessive replicate and/or Mendelian errors; or (4) more than 50% missing data, unless otherwise noted. A total of 5,980 SNPs passed these quality control specifications. Pedigree and Mendelian inconsistencies were checked using GAS, version 2.0 [Young, 1995], GRR [Abecasis et al., 2001], and PEDCHECK [O’Connell and Weeks, 1998]. Descriptive summaries of quality of the data were also evaluated using PLINK [Purcell et al., 2007]. All monozygotic twins and SNPs with Hardy–Weinberg equilibrium P-values less than 0.01 were removed from the analysis.
Linkage Analysis
Two multipoint nonparametric genetic linkage analyses were preformed on these data. In the first analysis, Kong and Cox [1997] LOD scores were calculated in an affected sibling pairs linkage analysis. In the second analysis, variance components linkage analysis was performed using the total number of ADHD symptoms as the quantitative trait of interest. Both Analyses were performed using Merlin [Abecasis et al., 2002]. Allele frequencies were calculated in Merlin using all available individuals. In both analyses, interpolated DeCode genetic distances were used to map the SNPs [Kong et al., 2002]. Analyses were done with adjusting for the possible linkage disequilibrium between SNPs with a correlation of 0.30 or greater [Abecasis and Wigginton, 2005]. The X chromosome was also analyzed in Merlin using the minx option.
Empirical simulation to establish genomewide significance were performed if a LOD score greater than 3.0 was achieved in either linkage analysis. Such simulations were performed in MERLIN [Abecasis et al., 2002]. MERLIN generates simulated chromosomes that have the original data structure with regard to marker informativeness, marker spacing, and missing data patterns. Data are generated under the null hypothesis of no linkage and no association to our phenotype, ADHD. One thousand simulations were generated to assess genome-wide significance.
RESULTS
After cleaning procedures, 5,980 SNPs were genotyped and released for 1,187 individuals. The missing genotype rate after removing Mendelian inconsistencies was 0.56% over the entire sample. The discordance rate for duplicate genotypes assayed on the same plates as the study genotypes was 0.002%. Descriptive statistics on the individuals used in the linkage analyses are provided in Table II.
TABLE II.
Descriptive Statistics for the Linkage Sample
| A: Overall number sample size | |
| Number of families | 271 |
| Number of genotyped individuals | 1,170 |
| B: Descriptive information for the affection status linkage analysis | |
| Number of families | 217 |
| Gender distribution | |
| Male | 358 (59.6%) |
| Female | 243 (40.4%) |
| Number of affected siblings | 601 |
| Number of siblings per family | |
| 2 | 98 |
| 3 | 82 |
| 4 | 30 |
| 5 | 5 |
| 6 | 1 |
| 8 | 1 |
| C: Descriptive information for the ADHD symptom count linkage analysis | |
| Number of families | 260 |
| Number of phenotyped siblings | 1,100 |
| Number of affected siblings per family | |
| 2 | 9 |
| 3 | 52 |
| 4 | 108 |
| 5 | 64 |
| 6 | 20 |
| 7 | 4 |
| 8 | 1 |
| 9 | 2 |
| Gender distribution | |
| Male | 577 (52.5%) |
| Female | 523 (47.6%) |
| Average number of the total ADHD symptoms (standard deviation) | 9.19 (6.06) |
The results of both linkage scans are summarized in Figure 1. The affection status linkage analysis had no LOD scores greater than greater than 2, indicating that there was no convincing evidence for linkage using this phenotype. The maximum LOD scores for this analysis were 1.85 on chromosome 8 at 54.2 cM and 1.81 on chromosome 15 at 51.7 cM. In the variance components linkage analysis, there were no regions with LOD scores greater than 1.0. The maximum LOD score was 0.8 on chromosome 8 at 93.4 cM. Because there were no LOD scores above 3, no empirical P-values were generated.
Fig. 1.
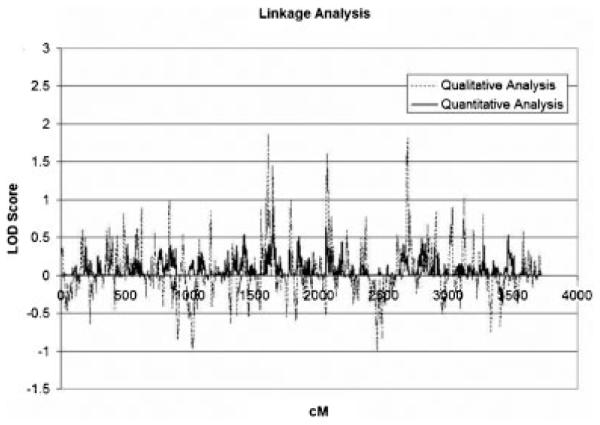
Linkage analysis results for the qualitative and quantitative traits.
DISCUSSION
Although prior linkage studies of ADHD (Table I) have produced suggestive evidence for linkage, our linkage scan of 271 sib-pair families was negative. The maximum LOD scores for both the diagnostic and quantitative phenotypes were small, and the regions implicated by those weak findings do not overlap with the suggestive peaks reported by prior studies. Our results clearly rule out the possibility that major single genes are involved in the etiology of ADHD, but given that linkage analysis has low power to detect genes of small effect, such susceptibility loci cannot be ruled out.
In comparing our work with prior studies, we can see no obvious reason why our study produced uniformly negative findings whereas the others reported suggestive evidence for linkage. Our sample was as large or larger than the other sib-pair samples. Additionally, our methods of assessment and diagnosis, although not identical, should have defined a similar ADHD phenotype. Indeed, distribution of ethnicity and rates of comorbidity were not dramatically different than found in other studies. Moreover, even with phenotypic differences among studies, such differences should not have dramatically affected the degree of genetic involvement given that our review of 20 twin studies shows high heritability for ADHD using several different methods of diagnosis.
Because the current scan used a different method of genotyping (i.e., Illumina’s SNP panel rather than microsatellites), it is possible that the improved data quality translated into fewer false positive signals in the current sample compared to others. Given that the method of linkage analysis is known to have low power for genes of small effect [Risch and Merikangas, 1996], it is feasible that the variable pattern of linkage results across previous studies has been caused by the difficulty that low powered studies have in distinguishing true linkage signals from false positive signals. This hypothesis suggests that to clarify linkage analysis results for ADHD will require either an extremely large sample or a collaborative meta-analysis which pools linkage analysis data sets, as has been done for schizophrenia [Lewis et al., 2003] and bipolar disorder [McQueen et al., 2005].
The only firm conclusion we can draw from this report is that the effects of ADHD genes cannot be very large. This, in turn, suggests that the discovery of ADHD genes will need to rely on the method of association applied to either targeted candidate genes [e.g., Brookes et al., 2006; Qian et al., 2006; Wang et al., 2006; Li et al., 2006a,b] or a full genome-wide association scan.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01HD37694, R01HD37999 and R01MH66877 to S. Faraone. CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, Contract Number N01-HG-65403. We are grateful for the contributions of the following colleagues from the MGH data collection site: Kim Hannah-Lise Schofield, Kimberley Mullen, Christine Roe RN, Deborah LaMonica MD and from the Nebraska data collection site: David Bylund Ph.D., Jean Deupree Ph.D., Cynthia Ellis M.D., Shelley Smith Ph.D.
Grant sponsor: NIH; Grant numbers: R01HD37694, R01HD37999, R01MH66877.
Footnotes
ELECTRONIC DATABASE INFORMATION
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=181500.
REFERENCES
- Abecasis GR, Wigginton JE. Handling marker-marker linkage disequilibrium: Pedigree analysis with clustered markers. Am J Hum Genet. 2005;77:754–767. doi: 10.1086/497345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. GRR: Graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Achenbach TM, McConaughy SH. Empirically based assessment of child and adolescent psychopathology: Practical applications. Vol. 13. Sage Publications, Inc; Newbury Park: 1987. [Google Scholar]
- American Psychiatric Association . DSM-III-R. American Psychiatric Association; Washington, DC: 1987. [Google Scholar]
- Arcos-Burgos M, Castellanos FX, Konecki D, et al. Pedigree disequilibrium test (PDT) replicates association and linkage between DRD4 and ADHD in multigenerational and extended pedigrees from a genetic isolate. Mol Psychiatry. 2004a;9:252–259. doi: 10.1038/sj.mp.4001396. [DOI] [PubMed] [Google Scholar]
- Arcos-Burgos M, Castellanos FX, Pineda D, et al. Attention-deficit/hyperactivity disorder in a population isolate: Linkage to Loci at 4q13.2, 5q33.3, 11q22, and 17p11. Am J Hum Genet. 2004b;75:998–1014. doi: 10.1086/426154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker SC, van der Meulen EM, Buitelaar JK, et al. A whole-genome scan in 164 Dutch sib pairs with attention-deficit/hyperactivity disorder: Suggestive evidence for linkage on chromosomes 7p and 15q. Am J Hum Genet. 2003;72:1251–1260. doi: 10.1086/375143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton J, Bergeron L, Valla J, Lepine S, Houde L, Gaudet N. Do children aged 9 through 11 years understand the DISC version 2.25 questions? J Am Acad Child Adolesc Psychiatry. 1995;34:946–956. doi: 10.1097/00004583-199507000-00019. [DOI] [PubMed] [Google Scholar]
- Brookes K, Xu X, Chen W, et al. The analysis of 51 genes in DSM-IV combined type attention deficit hyperactivity disorder: Association signals in DRD4, DAT1 and 16 other genes. Mol Psychiatry. 2006 doi: 10.1038/sj.mp.4001869. [DOI] [PubMed] [Google Scholar]
- Edelbrock C, Costello AJ, Dulcan MK, Kalas R, Conover NC. Age differences in the reliability of the psychiatric interview of the child. Child Dev. 1985;56:265–275. [PubMed] [Google Scholar]
- Faraone SV. Etiology and pathophysiology of adult attention deficit hyperactivity disorder. Primary Psychiatry. 2004a;11:28–40. [Google Scholar]
- Faraone SV. Genetics of adult attention-deficit/hyperactivity disorder. Psychiatr Clin North Am. 2004b;27:303–321. doi: 10.1016/S0193-953X(03)00090-X. [DOI] [PubMed] [Google Scholar]
- Faraone SV. The scientific foundation for understanding attention-deficit/hyperactivity disorder as a valid psychiatric disorder. Eur Child Adolesc Psychiatry. 2005;14:1–10. doi: 10.1007/s00787-005-0429-z. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Biederman J. Neurobiology of attention-deficit hyper-activity disorder. Biological Psychiatry. 1998;44:951–958. doi: 10.1016/s0006-3223(98)00240-6. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Biederman J, Spencer T, et al. Attention deficit hyperactivity disorder in adults: An overview. Biol Psychiatry. 2000;48:9–20. doi: 10.1016/s0006-3223(00)00889-1. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Sergeant J, Gillberg C, Biederman J. The worldwide prevalence of ADHD: Is it an American condition? World Psychiatry. 2003;2:104–113. [PMC free article] [PubMed] [Google Scholar]
- Faraone SV, Perlis RH, Doyle AE, et al. Molecular genetics of attention deficit hyperactivity disorder. Biol Psychiatry. 2005;57:1313–1323. doi: 10.1016/j.biopsych.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Faraone S, Biederman J, Mick E. The age dependent decline of attention-deficit/hyperactivity disorder: A meta-analysis of follow-up studies. Psychol Med. 2006;36:159–165. doi: 10.1017/S003329170500471X. [DOI] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JBW. Structured clinical interview for DSM-IV axis I disorders-clinician version (SCID-CV) American Psychiatric Press; Washington, DC: 1997. [Google Scholar]
- Fisher SE, Francks C, McCracken JT, et al. A genomewide scan for loci involved in attention-deficit/hyperactivity disorder. Am J Hum Genet. 2002;70:1183–1196. doi: 10.1086/340112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebebrand J, Dempfle A, Saar K, et al. A genome-wide scan for attention-deficit/hyperactivity disorder in 155 German sib-pairs. Mol Psychiatry. 2006;11:196–205. doi: 10.1038/sj.mp.4001761. [DOI] [PubMed] [Google Scholar]
- Kong A, Cox NJ. Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- Lander E, Kruglyak L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- Leckman JF, Sholomskas D, Thompson D, Belanger A, Weissman MM. Best estimate of lifetime psychiatric diagnosis: A methodological study. Arch Gen Psychiatry. 1982;39:879–883. doi: 10.1001/archpsyc.1982.04290080001001. [DOI] [PubMed] [Google Scholar]
- Lewis CM, Levinson DF, Wise LH, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder. Part II. Schizophrenia. Am J Hum Genet. 2003;73:34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang Y, Zhou R, et al. Association between polymorphisms in serotonin 2C receptor gene and attention-deficit/hyperactivity disorder in Han Chinese subjects. Neurosci Lett. 2006a;407(2):107–111. doi: 10.1016/j.neulet.2006.08.022. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang X, Wang Y, et al. The serotonin 5-HT1D receptor gene and attention-deficit hyperactivity disorder in Chinese Han subjects. Am J Med Genet B Neuropsychiatr Genet. 2006b;141B:874–876. doi: 10.1002/ajmg.b.30364. [DOI] [PubMed] [Google Scholar]
- McQueen MB, Devlin B, Faraone SV, et al. Combined analysis from eleven linkage studies of bipolar disorder provides strong evidence of susceptibility loci on chromosomes 6q and 8q. Am J Hum Genet 77. 2005 doi: 10.1086/491603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JR, Stewart MA. A family study of the hyperactive child syndrome. Biol Psychiatry. 1971;3:189–195. [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogdie M, Macphie I, Minassian S, et al. A genomewide scan for attention-deficit/hyperactivity disorder in an extended sample: Suggestive linkage on 17p11. Am J Hum Genet. 2002;72:1268–1279. doi: 10.1086/375139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogdie MN, Macphie IL, Minassian SL, et al. A genomewide scan for attention-deficit/hyperactivity disorder in an extended sample: Suggestive linkage on 17p11. Am J Hum Genet. 2003;72:1268–1279. doi: 10.1086/375139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogdie MN, Fisher SE, Yang M, et al. Attention deficit hyperactivity disorder: Fine mapping supports linkage to 5p13, 6q12, 16p13, and 17p11. Am J Hum Genet. 2004;75:661–668. doi: 10.1086/424387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvaschel H. Schedule for affective disorder and schizophrenia for school-age children epidemiologic version. 5th Nova Southeastern University, Center for Psychological Studies; Ft. Lauderdale: 1994. [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Q, Wang Y, Li J, et al. Evaluation of potential gene–gene interactions for attention deficit hyperactivity disorder in the Han Chinese population. Am J Med Genet B Neuropsychiatr. 2006 doi: 10.1002/ajmg.b.30422. [DOI] [PubMed] [Google Scholar]
- Genet Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Schwab-Stone M, Fallon T, Briggs M, Crowther B. Reliability of diagnostic reporting for children aged 6–11 years: A test-retest study of the Diagnostic Interview Schedule for Children—Revised. Am J Psychiatry. 1994;151:1048–1054. doi: 10.1176/ajp.151.7.1048. [DOI] [PubMed] [Google Scholar]
- Smalley SL, Kustanovich V, Minassian SL, et al. Genetic linkage of attention-deficit/hyperactivity disorder on chromosome 16p13, in a region implicated in autism. Am J Hum Genet. 2002;71:959–963. doi: 10.1086/342732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Wang Y, Zhou R, et al. Possible association of the alpha-2A adrenergic receptor gene (ADRA2A) with symptoms of attention-deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:130–134. doi: 10.1002/ajmg.b.30258. [DOI] [PubMed] [Google Scholar]
- Young A. Genetic analysis system. (2.0) 1995 [Google Scholar]