

The method of bacterial type III secretion system (T3SS)-mediated transcription activator-like effector nuclease (TALEN) protein injection and transfection of an oligonucleotide template to effectively generate precise genetic modifications in stem cells. T3SS-mediated TALEN protein delivery provides a highly efficient alternative for introducing precise gene editing within pluripotent stem cells for the purpose of disease genotype-phenotype relationship studies and cellular replacement therapies.
Keywords: Transcription activator-like effector nuclease, Type III secretion system, Embryonic stem cell, iPS cell, Genome editing
Abstract
The type III secretion system (T3SS) of Pseudomonas aeruginosa is a powerful tool for direct protein delivery into mammalian cells and has successfully been used to deliver various exogenous proteins into mammalian cells. In the present study, transcription activator-like effector nuclease (TALEN) proteins have been efficiently delivered using the P. aeruginosa T3SS into mouse embryonic stem cells (mESCs), human ESCs (hESCs), and human induced pluripotent stem cells (hiPSCs) for genome editing. This bacterial delivery system offers an alternative method of TALEN delivery that is highly efficient in cleavage of the chromosomal target and presumably safer by avoiding plasmid DNA introduction. We combined the method of bacterial T3SS-mediated TALEN protein injection and transfection of an oligonucleotide template to effectively generate precise genetic modifications in the stem cells. Initially, we efficiently edited a single-base in the gfp gene of a mESC line to silence green fluorescent protein (GFP) production. The resulting GFP-negative mESC was cloned from a single cell and subsequently mutated back to a GFP-positive mESC line. Using the same approach, the gfp gene was also effectively knocked out in hESCs. In addition, a defined single-base edition was effectively introduced into the X-chromosome-linked HPRT1 gene in hiPSCs, generating an in vitro model of Lesch-Nyhan syndrome. T3SS-mediated TALEN protein delivery provides a highly efficient alternative for introducing precise gene editing within pluripotent stem cells for the purpose of disease genotype-phenotype relationship studies and cellular replacement therapies.
Significance
The present study describes a novel and powerful tool for the delivery of the genome editing enzyme transcription activator-like effector nuclease (TALEN) directly into pluripotent stem cells (PSCs), achieving desired base changes on the genomes of PSCs with high efficiency. This novel approach uses bacteria as a protein delivery tool. It is easy to manipulate and adaptable to scaling up. This is a safe delivery system, because the delivery strains can be easily eliminated using simple antibiotic treatment. Type III secretion system (T3SS)-mediated TALEN protein delivery provides a highly efficient alternative for introducing precise gene alterations within PSCs for the purpose of disease genotype-phenotype relationship studies and cellular replacement therapies. The results of the present study also pave the way to applying the bacterial T3SS to deliver transcriptional factors into PSCs for cellular reprogramming, raising the hope of a safe technology that can be used in cell or tissue replacement therapy for human genetic diseases.
Introduction
Pseudomonas aeruginosa is a common gram-negative opportunistic human pathogen that injects proteineous exotoxins directly into host cells via a type III secretion system (T3SS) [1]. The T3SS is a complex, needle-like structure on the bacterial surface responsible for the secretion of four known exotoxins: ExoS, ExoT, ExoY, and ExoU [2]. ExoS is best characterized for its functional domains, with its N-terminal sequence serving as a signal for injection [3]. Previously, we fused various lengths of the ExoS N-termini with Cre recombinase for injection into mammalian cells and found that N-terminal 54 amino acids (ExoS54) were optimal for delivery of the exogenous Cre protein [4]. Based on this, we delivered MyoD protein, a muscle-specific master regulatory factor, into mouse embryonic fibroblasts, successfully converting them into muscle cells [5]. Furthermore, transcription activator-like effector nuclease (TALEN) proteins fused with the ExoS54 were also efficiently injected into HeLa cells, achieving site-specific DNA cleavage without the introduction of foreign genetic material [6].
TALEN is a novel gene editing tool that can specifically recognize target sequence as a dimer and introduce a double-strand DNA break (DSB) on the target site, triggering nonhomologous end joining or homologous recombination [7]. In the absence of a homologous template, the DSB activates the host DNA repair system, resulting in high-frequency gene mutations, such as nucleotide mismatches, insertions, or deletions. However, in the presence of a homologous template, the DSB triggers homologous recombination, introducing the desired DNA sequence substitutions on the target sites [8]. The current methods of TALEN delivery use the introduction of foreign genetic material, such as viral DNA/RNA, plasmid DNA, or mRNA, making it difficult to meet the safety requirements for biomedical applications. Previously, we reported on the injection of a pair of TALEN proteins targeting the Venus gene into the HeLa-Venus cell line by the T3SS of P. aeruginosa, successfully knocking out the Venus gene at the intended target site on the genome [6].
Pluripotent stem cells (PSCs), such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), can be differentiated into a wide variety of cell and tissue types in vitro; thus, gene editing in PSCs can correct the root causes and thereby eliminate the symptoms associated with genetic diseases. Accordingly, technologies capable of editing genes in PSCs are extremely important. To date, TALEN technology has been successfully applied to create disease models in many organisms, such as zebrafish, mice, rats, and hiPSCs [9–14]. However, the introduction of TALEN-encoding plasmid DNA not only resulted in low frequencies of DSBs on the target sites, but also poses a serious safety risk of insertional mutagenesis. Thus, it is critical to develop a highly efficient alternative method of introducing gene-editing enzymes into the stem cells. In the present study, we applied bacterial protein delivery technology to introduce TALEN proteins directly into mouse ESCs (mESCs), human ESCs (hESCs), and human iPSCs (hiPSCs), successfully introducing DSBs on the intended target sites. Our data show that bacterial T3SS-mediated TALEN protein delivery into the PSCs induces highly efficient target gene modifications with added benefits compared with the conventional plasmid transfection method.
Precise editing of genomic DNA in pluripotent stem cells will be essential for the studies of genotype-phenotype relationships in disease research and for therapeutic applications. However, a significant challenge of this method is the low efficiency, requiring the use of a selection marker or screening a large number of subclones [15]. Recently, precise genome editing was achieved through a combined use of the TALEN and single-stranded oligonucleotide DNA (ssODN) templates [16, 17]. In the present study, we combined the method of bacterial T3SS-mediated TALEN protein injection and ssODN template transfection, providing an effective alternative method for precise DNA modification in stem cells. Using this approach, we have not only efficiently changed a single-base in the gfp gene of a mouse ESC line to silence green fluorescent protein (GFP) production, but we have also reverted the resulting GFP-negative cells back to GFP-positive ESCs. In addition, a single-base change has been successfully introduced into the HPRT1 gene of an hiPSC line. The HPRT1 gene is encoded on the X-chromosome, and mutations in this gene cause Lesch-Nyhan syndrome (LNS), which is characterized by neurological, mental, and behavioral symptoms [18]. The HPRT1 gene encodes a hypoxanthine phosphoribosyltransferase (HPRT), which is responsible for recycling purine [19]. Natural mutations in the HPRT1 result in a low level of the HPRT enzyme for purine salvage, leading to LNS. Accordingly, LNS is observed in HPRT−/Y males, and HPRT+/− carrier females are generally asymptomatic [20]. Efficient correction of the HPRT1 mutations paves the way for building the HPRT1 disease model in medical research and future cell replacement therapy.
All the experimental results described in the present study serve as a foundation for the application of the bacterial T3SS in human disease model establishment and gene therapy in stem cells. The bacterial delivery system will be a safe and effective alternative to the currently available protein delivery methods.
Materials and Methods
Bacterial Strains and Plasmids
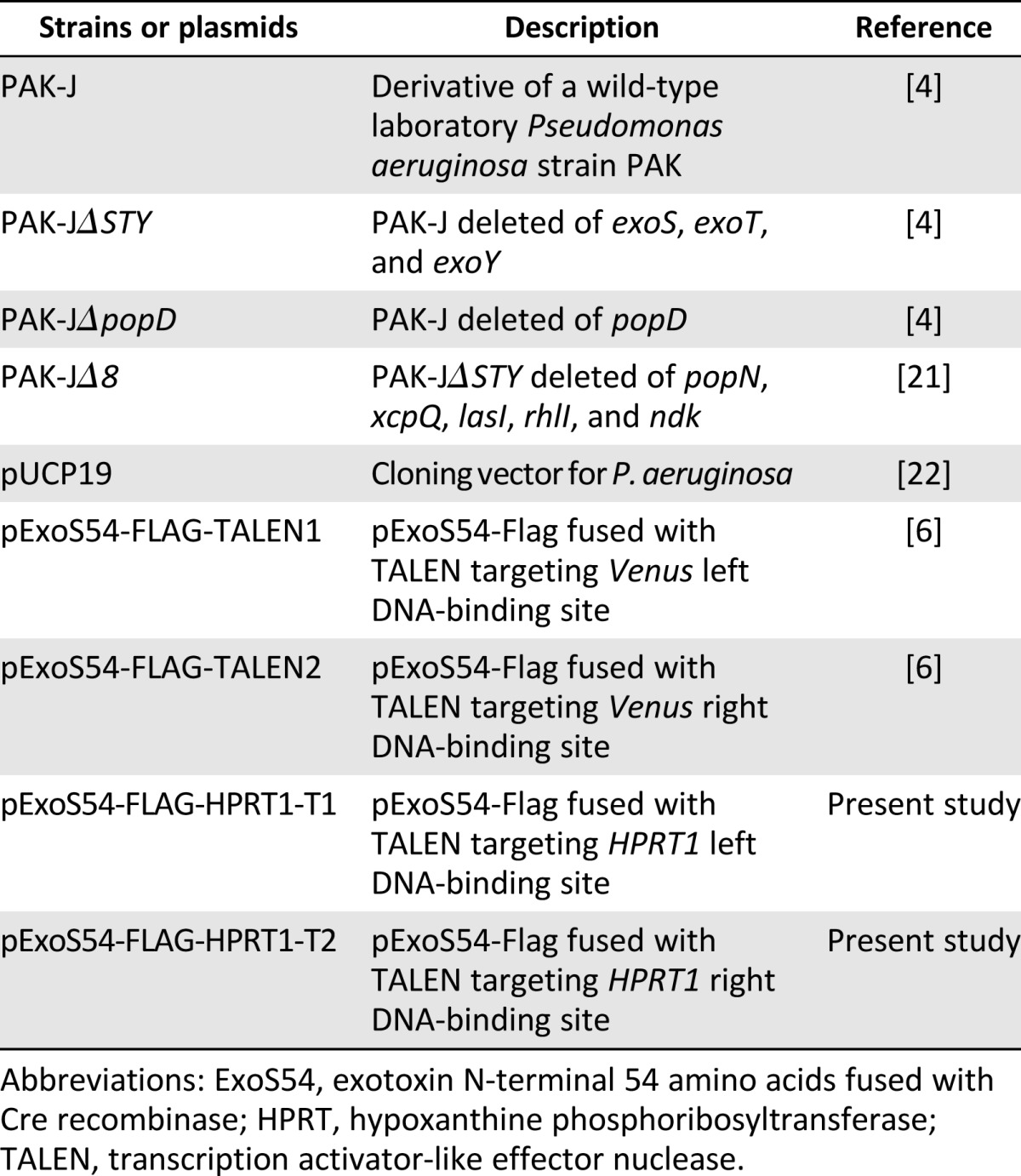
The strains and plasmids [4, 6, 21, 22] used in the present study are listed in Table 1. The P. aeruginosa strain PAK-JΔSTY is deleted of the type III secreted exotoxins (exoS, exoT, and exoY) in the background of PAK-J; PAK-JΔpopD is deleted of popD, encoding a pore-forming protein required for type III injection, in the background of PAK-J, as described previously [4]; and PAK-JΔ8 is deleted of popN, xcpQ, lasI, rhlI, and ndk in the background of PAK-JΔSTY [21]. All P. aeruginosa strains were cultured in Luria broth (LB) or L-agar (LA) plates at 37°C. Carbenicillin was used at a final concentration of 150 μg/ml for plasmid selection in P. aeruginosa.
Table 1.
Bacterial strains and plasmids
TALENs targeting the gfp and HPRT1 genes were constructed in accordance with the instructions from the Golden Gate Cloning Kit (Voytas Laboratory, University of Minnesota College of Biological Sciences, St. Paul, MN, http://www.cbs.umn.edu/research/research-cbs/faculty-labs/voytas [23]. The left and right arm sequences of TALEN targeting gfp are 5′-TTCACCGGGGTGGTGCC-3′ and 5′-CTGGACGGCGACGTAAA-3′, respectively; the left and right arm sequences of TALEN targeting HPRT1 are 5′-GTAGGACTGAACGTCTTGCTC-3′ and 5′-GATGGGAGGCCATCACATTGT-3′, respectively. The ExoS54-Flag-TALEN fusion constructs were generated by in-frame fusion of the TALEN coding sequence to the pExoS54-Flag, which has been previously described [4, 6]. The TALEN targeting region (350 base pairs [bp]) within the gfp gene was amplified using polymerase chain reaction (PCR) primers of gfp-forward: 5′-CCTACAGCTCCTGGGCAACGTGCTGG-3′; and gfp-reverse: 5′-CTGGACGTAGCCTTCGGGCATGGCGG-3′. The TALEN targeting region (625 bp) within the HPRT1 gene was amplified using PCR primers of HPRT1-forward: 5′-TTTTGAGACAAGGTCTTGCTCTATTG-3′; and HPRT1-reverse: 5′-CAGTATTGGCTTTGATGTAAAGTACT-3′. The PCR products were either subjected to digestion by restriction enzymes or directly cloned into pGEM-T Easy (Promega, Madison, WI, http://www.promega.com) vector and subjected to sequencing analysis.
The three 72-nucleotide-long single-stranded donor template DNAs used to introduce the desired nucleotide changes in either the gfp or hprt1 gene through homologous recombination were ssODN-1: 5′-AGGAGCTGTT CACCGGGGTG GTGCCCATCC TGGTCTAGCT GGACGGCGAC GTAAACGGCC ACAAGTTCAG CG-3′, ssODN-2: 5′-AGGAGCTGTT CACCGGGGTG GTGCCCATCC TGGTCGAGCT CGACGGCGAC GTAAACGGCC ACAAGTTCAG CG-3′, and ssODN-3: 5′-CCTGATTTTA TTTCTGTAGG ACTGAACGTC TTGCTTGAGA TGTGATGAAG GAGATGGGAG GCCATCACAT TG-3′.
Electroporation of P. aeruginosa
For electroporation of P. aeruginosa, 1.5 ml of an overnight culture grown in LB medium was harvested in 1.5-ml microcentrifuge tubes by centrifugation (1 minute, 16,000g) at room temperature. Each cell pellet was washed twice with 1 ml of room temperature 300 mM sucrose and resuspended in a total of 100 μl of 300 mM sucrose. For electroporation, 100 ng of pExoS54-Flag-TALEN DNA was mixed with 50 μl of electrocompetent cells and transferred into a 2-mm gap width electroporation cuvette (Bio-Rad, Hercules, CA, http://www.bio-rad.com). After applying a 2.5-kV pulse, 1 ml of LB medium was added immediately, and the cells were transferred to a culture tube (10 ml) and shaken for 1 hour at 37°C. The cells were then plated on LA plates containing 150 μg of carbenicillin per milliliter. The plates were incubated at 37°C until colonies appeared.
Cell Culture
A GFP-expressing B5 mESC line (EB5) [24] was grown on 0.1% gelatin-coated plates (EMD Millipore, Billerica, MA, http://www.emdmillipore.com) in mESC medium [25] and passaged after dissociation by 0.25% trypsin/EDTA (Thermo Scientific, Wilmington, DE, http://www.thermoscientific.com). A GFP-expressing hESC line (LT2e-H9CAGGFP) [26] and a human iPSC line originated from a male foreskin (iPS-3) [27] were grown on 5 μg/ml Vitronectin-coated plates (Life Technologies, Carlsbad, CA, http://www.lifetechnologies.com) in mTeSR E8 medium (Life Technologies) and passaged after dissociation by 0.5 mM EDTA (Life Technologies). The EB5 cell line was kindly provided by Dr. Andras Nagy (Mt. Sinai, Toronto, ON, Canada); hESC LT2e-H9CAGGFP was purchased from WiCell Research Institute (Madison, WI, http://www.wicell.org); and iPS-3 was generated from foreskin fibroblasts of a healthy individual (catalog no. CRL-2522; American Type Culture Collection, Manassas, VA, http://www.atcc.org) using Sendai virus SsVdp(KOSM)302L kindly provided by Dr. Mahito Nakanish in accordance with a published protocol [28]. The iPSCs were generated under an approved University of Florida Environmental Health and Safety biosafety approval number RD-3933. All cells were cultured at 37°C with 5% CO2 and supplemented with penicillin and streptomycin (CellGro; Corning Life Sciences, Acton, MA, http://www.cellgro.com). Ciprofloxacin was added to a final concentration of 20 μg/ml to clear the protein delivery strain of P. aeruginosa.
To isolate EB5 cell lines harboring the expected single-base mutation, GFP-negative cells collected by FACS-Sort (BD Biosciences, San Diego, CA, http://www.bdbiosciences.com) were diluted to a final cell density of 5 cells per milliliter and then plated at 100 μl per well in a 96-well plate coated with 0.1% gelatin. The single clones were checked visually about 6 days after plating and transferred to 24-well, coated plates for expended culture, 6-well plates, and finally to 60-mm plates, changing the medium every other day. Approximate 4 × 106 cells were used for genomic DNA extraction following the procedure of the Qiagen RNA/DNA Mini Kit handbook (Qiagen, Hilden, Germany, http://www.qiagen.com). A 350-bp-long gfp gene fragment was amplified from the genome by PCR and digested with BfaI restriction enzyme to detect single-base mutations.
To select human iPSC clones containing the intended single base change in the HPRT1 gene, the cells were cultured for 3 days after TALEN injection and then subjected to selection in mTeSR E8 medium containing 2.5 μg/ml 6-thio-guanime (6TG; Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com) for 6 days. Chromosomal DNA was extracted from the 6TG-resistant iPSCs using the QuickExtract DNA extract solution (Epicentre, an Illumina Company, Madison, WI, http://www.epibio.com), and then PCR was used to amplify the 625-bp fragment of the HPRT1 gene. The DNA fragments were cloned into pGEM-T Easy vector (Promega), and randomly chosen clones were subjected to DNA sequencing.
Plasmid Transfection
After the optimal transfection condition of FuGENE HD Transfection Reagent (Promega), mouse or human ESCs were seeded in 6-well plates at 70% confluence 1 day before transfection. TALEN expression plasmid DNA (2 μg), purified with the Qiagen Plasmid Kit, was diluted with cell culture medium to a final volume of 94 μl. To this DNA solution, 6 μl of FuGENE HD Transfection Reagent (Promega) was added, mixed, and incubated at room temperature for 15 minutes. The mixture was added to the cell cultures slowly with a gentle mix. The cells were incubated at 37°C with 5% CO2 for at least 4 hours before the downstream experiments. The same procedure was followed for the introduction of single-stranded oligonucleotide templates into the target cells.
Protein Injection Assay
Mouse or human ESCs were seeded in 6-well plates at approximately 70% confluence in antibiotic-free ES medium. P. aeruginosa strains were grown at 37°C in LB containing carbenicillin until the optical density (OD600) reached 1.0. Next, the bacterial cells were collected by centrifugation, washed with PBS, and diluted in ES medium without antibiotic. The ESCs were cocultured with bacterial cells at various multiplicity of infection (MOI) for the indicated period of time. Infection was terminated by washing ESCs with PBS for three times and culturing on ES medium containing 20 μg/ml ciprofloxacin.
For Western blot analysis of the injected proteins, the cells were collected at the indicated time after bacterial infection and centrifuged at 1,700 rpm for 2 minutes. The cell pellets were lysed with 40 μl PBS containing 0.25% Triton-X on ice for 10 minutes. The lysed cells were then centrifuged at 16,000 rpm for 5 minutes. The nuclear protein extracts were prepared using an extraction kit from Beyotime Biotechnology (Shanghai, China, http://www.beyotime.com) and followed the manufacturer’s instruction. The soluble fraction was collected, mixed with an equal volume of 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer and boiled for 10 minutes. Protein samples were separated on 10% SDS-PAGE, transferred onto polyvinylidene fluoride membrane, and then probed with mouse M2 monoclonal antibody against FLAG-tag (Sigma-Aldrich).
Flow Cytometry
Infected mESCs were collected by 0.25% trypsin/EDTA treatment for 5 minutes, and infected hESCs were collected by 0.5 mM EDTA treatment for 3 minutes. The cells were centrifuged at 1,700 rpm for 2 minutes and resuspended in 1 ml of ice-cold PBS. The cells were analyzed for GFP using Diva, version 6.2, on LSR-II (BD Biosciences) and FACSAria II (BD Biosciences) for flow cytometry.
Results
Bacterial T3SS-Mediated Injection of TALEN Proteins Into Mouse ESCs
A pair of TALEN constructs, targeting Venus gene, have been previously described [6]. This pair of TALENs also targets the gfp gene, encoding GFP, because they share the same target DNA sequence (Fig. 1A). In our previous report, delivery of this pair of TALEN proteins into HeLa-Venus cells by T3SS of P. aeruginosa resulted in a higher efficiency of Venus gene knockout than by transfection-mediated delivery of the TALEN-coding plasmid DNA [6]. Encouraged by our success, we delivered the same TALEN pair into a mouse ESC line (EB5) that stably expresses a gfp gene, using the bacterial T3SS delivery system. The 2 TALENs were each fused with the amino-terminal 54 amino acids of ExoS (ExoS54), which had previously been shown to be an optimal signal sequence for the delivery of exogenous proteins into mammalian cells through the T3SS of P. aeruginosa [4–6]. The plasmids encoding TALEN fusion proteins, pExoS54-FLAG-TALEN-1 and pExoS54-FLAG-TALEN-2, were each electroporated into two P. aeruginosa strains. The strain PAK-JΔSTY with a high-type III secretion capacity and reduced toxicity is due to the deletion of endogenous exotoxins [4]. Strain PAK-JΔpopD is knocked out of a gene encoding a protein required for the formation of translocon pores on the host membrane and thus is incapable of injecting effectors into the host cells [5]. The T3SS-defective strain PAK-JΔpopD was used as a negative control to verify T3SS-dependent injection of the ExoS54-FLAG-TALEN fusion proteins. The EB5 cells were infected with the resulting transformants at a MOI of 100 for 3 hours, an optimal condition based on our previous work on bacterial delivery of the TALEN proteins into HeLa cells [6]. The TALEN fusion proteins are expected to target to EB5 nuclei, because they contain nuclear localization sequences. After infection by P. aeruginosa, the nuclear proteins of EB5 cells were extracted and subjected to Western blot using anti-FLAG antibody. The TALEN fusion proteins were not only efficiently injected into the EB5 cells by P. aeruginosa in a T3SS-dependent manner, the injected TALENs were also correctly localized to the nuclei of the EB5 cells (Fig. 1B).
Figure 1.

Bacterial type III secretion system-mediated injection of TALEN proteins into mouse embryonic stem cells. (A): Scheme of TALEN binding sites on gfp gene. The left and right TALEN-binding sequences are shown in green and blue, respectively, and the spacer sequence is shown in red. (B): GFP-expressing B5 mouse embryonic stem cell line (EB5) cells were infected with the indicated strains at a multiplicity of infection (MOI) of 100 for 3 hours. For ΔSTY/TALEN1&2, the total MOI was 200. Nuclear proteins were extracted and subjected to Western blot by an anti-FLAG antibody. (C): EB5 cells were infected with PAK-JΔSTY/pExoS54-FLAG-TALEN-1 and PAK-JΔSTY/pExoS54-FLAG-TALEN-2 at an overall MOI of 200 for 3 hours. After termination of the bacterial infection (time 0 hour), nuclear proteins were extracted at the indicated time and subjected to Western blot by anti-FLAG antibody. Abbreviations: ExoS54, exotoxin S N-terminal 54 amino acids; GFP, green fluorescent protein; h, hour; TALEN, transcription activator-like effector nuclease; TALEN1&2, PAK-JΔSTY/pExoS54-FLAG-TALEN1 and PAK-JΔSTY/pExoS54-FLAG-TALEN2.
To determine the intracellular stability of injected TALEN fusion proteins, the cells were collected at various time points after the 3-hour infection at an MOI of 100. Nuclear proteins were extracted and subjected to Western blot using anti-FLAG antibody. The Western blot result showed that the injected proteins were gradually degraded in a time-dependent manner and became almost undetectable 8 hours after the termination of infection (Fig. 1C).
Functional Analysis of Bacterially Injected TALENs
To assess the function of TALEN proteins delivered by the T3SS of P. aeruginosa, GFP fluorescence of the EB5 cells was monitored. The EB5 cells were infected by one or two PAK-JΔSTY strains, each harboring one of the pExoS54-FLAG-TALEN pair, at MOI of 100 for 3 hours. The EB5 cells were then washed three times with PBS to remove floating bacterial cells. To completely eliminate the residual bacterial cells, the EB5 cells were further cultured in mESC medium supplemented by 20 μg/ml ciprofloxacin. After 3 days of culturing, fluorescence of the EB5 cell population was analyzed by flow cytometry. In parallel, the EB5 cells were transfected with eukaryotic expression plasmids encoding the gfp-targeting TALENs, following the instructions of the optimal condition for the transfection reagent and then cultured in mESC medium for 3 days. According to the fluorescence-activated cell sorting (FACS) analysis results, approximately 20% of the cells injected with the TALEN protein pair by P. aeruginosa became nonfluorescent, and 10% of the cells transfected with the plasmid pair became nonfluorescent (Fig. 2A), indicating a twofold higher gfp-targeting efficiency with the bacterial delivery of TALEN proteins than that with TALEN-coding plasmid transfection. These results are consistent with our previously reported work with HeLa cells [6]. Consistent with the FACS data, 3 days after T3SS-mediated TALEN pair injection, EB5 cell colonies that had lost their GFP expression were readily observable under the fluorescence microscope (Fig. 2B).
Figure 2.

Functional analysis of bacterially injected TALENs. (A): Fluorescence intensities of control EB5 cells (EB5), EB5 cells transfected with eukaryotic expression plasmids encoding the TALENs (transfection), EB5 cells infected by TALEN1, or EB5 cells infected by TALEN1&2. The cells were analyzed by flow cytometry 3 days after transfection or injection. (B): A representative GFP-negative EB5 cell colony (arrow) 3 days after bacterial delivery of the gfp-targeting TALEN protein pair, observed under a fluorescence microscope. (C): Sequence alignment of the TALEN-targeting region among the GFP-negative EB5 cells after bacterial delivery of the gfp-targeting TALEN protein pair. Abbreviations: EB5, GFP-expressing B5 mouse embryonic stem cell line; GFP, green fluorescent protein; TALEN, transcription activator-like effector nuclease; TALEN1, PAK-J∆STY/pExoS54-FLAG-TALEN1; TALEN1&2, PAK-J∆STY/pExoS54-FLAG-TALEN1 and PAK-J∆STY/pExoS54-FLAG-TALEN2; WT, wild type.
The GFP-negative cells were collected by FACS-Sort, and total genomic DNA was extracted. A 350-bp-long gfp fragment encompassing the TALEN target site was amplified by PCR and cloned into the TA cloning vector pGEM-T Easy (Promega). Sequence analysis of randomly chosen clones identified various mutation types around the TALEN target site (Fig. 2C), presumably resulted from the error-prone DNA repair of the DSB generated by the TALEN pair. Therefore, the T3SS of P. aeruginosa not only effectively delivered the TALENs into mouse ESCs, the injected TALENs also properly executed their biological functions, causing DSBs on the target site.
Optimization of the Bacterial TALEN Delivery Condition
To find an optimal bacterial TALEN delivery condition for the mouse ESCs, we used PAK-J∆STY expressing TALEN to infect EB5 cells for 3 hours at MOIs ranging from 20 to 800. After infection, the floating bacterial cells were removed by washing with PBS, and the surviving EB5 cells were counted. Surprisingly, the viability of the EB5 cells was the highest at an MOI of 400, with lower or higher MOIs resulting in reduced viability (Fig. 3A). The nuclear protein of each sample, derived from the same number of EB5 cells, was extracted and used to detect injected TALEN by Western blot analysis. The Western blot result revealed that the amount of injected TALEN protein increased as the MOI increased from 20 to 400 but decreased beyond an MOI of 400 (Fig. 3B). The cells infected at various MOIs were cultured for 3 days in mESC medium containing 20 μg/ml ciprofloxacin and then monitored for GFP fluorescence intensity by flow cytometry. Approximately 30% of the cells infected at an MOI of 400 lost fluorescence, illustrating additional enhancement in the efficiency of TALEN-mediated gfp gene knockout (Fig. 3C). From the cytotoxicity and flow cytometry results, we have concluded that the optimal condition for bacterial delivery of the TALEN into mESCs is a 3-hour infection at an overall MOI of 400 (200 for each of the TALEN pair).
Figure 3.

Factors influencing the bacterial delivery of TALEN proteins. (A): The number of green fluorescent protein-expressing B5 mouse embryonic stem cell line (EB5) cells surviving infection of PAK-J∆STY/pExoS54-FLAG-TALEN1 and PAK-J∆STY/pExoS54-FLAG-TALEN2 (1:1 ratio) at the indicated MOI for 3 hours. The number of surviving cells was compared with that of the no infection control using a two-sample t test. ∗, p < .05; ∗∗, p < .001; ∗∗∗, p < .0001. Error bars represent SD of triplicate assays. (B): TALEN proteins injected into EB5 cells after infection at the indicated MOI for 3 hours. Nuclear protein extracts from the same number of surviving cells were prepared and subjected to Western blot analysis by anti-FLAG antibody. (C): Fluorescent-activated cell sorting (FACS) analysis results of EB5 cells 3 days after TALEN injection at the indicated MOI for 3 hours. The FACS data of the infected cell populations were compared with those of the no injection control using a two-sample t test. ∗, p < .05; ∗∗, p < .001; ∗∗∗, p < .0001. Error bars represent SD of triplicate assays. Abbreviations: MOI, multiplicity of infection; TALEN, transcription activator-like effector nuclease.
TALEN-Mediated Single-Base Change on Genomic DNA
Targeted gene correction in PSCs will be much more important than simple gene knockout, as described above. A 72-base-long single-stranded oligonucleotide DNA (ssODN-1) was designed as a template for homologous recombination in the gfp gene. This ssODN introduces a single nucleotide change, converting a GAG into a stop codon TAG in the GFP open reading frame, which also generates a new BfaI restriction enzyme recognition site (CTAG) (Fig. 4A).
Figure 4.


TALEN-mediated single-base change of gfp gene on genomic DNA. (A): Strategy of single-base modification in gfp gene. The ssODN template with a single base change from the wild-type sequence introduces a stop codon and a BfaI restriction enzyme digestion site. The second ssODN removes the stop codon and adds a new SacI restriction enzyme digestion site. (B): Fluorescent-activated cell sorting (FACS) analysis of fluorescence cell population 3 days after either transfection of ssODN-1 and eukaryotic expression plasmids encoding the TALEN pair (transfection; BII) or transfection of ssODN-1 followed by injection of TALEN proteins by P. aeruginosa (transfection-injection; BIII). As a control, an untreated EB5 cell is shown (BI). Percentages of the GFP-negative cells in the whole population are shown. (C): A 350-bp fragment encompassing the TALEN-targeting region was amplified by polymerase chain reaction (PCR) from GFP-negative EB5 cells that were FACS-sorted after either transfection of ssODN-1 and TALEN coding plasmids or ssODN-1 transfection followed by TALEN protein injection. The PCR products were subjected to 2% agarose electrophoresis with (+) or without (−) digestion by BfaI restriction enzyme. An uninfected EB5 cell was used as the control. M represents the DNA marker. The percentage of mutation was calculated using ImageJ. (D): Single cell cloning of EB5 with desired single-base change in the gfp gene. The gfp fragments were PCR amplified from 12 cell lines obtained by single cell cloning and subjected to 2% agarose electrophoresis after digestion by the BfaI restriction enzyme. Two desired cell lines, numbers 4 and 6, were obtained. (E): FACS analysis of fluorescence cell population 3 days after transfection of ssODN-2 and injection of TALEN proteins by P. aeruginosa. As a control, gfp-silenced EB5 cells (EB5-Mut1) were injected with the TALEN proteins only. The percentage of the GFP-positive cells in the whole population are shown. (F): After transfection of ssODN-2 and TALEN protein injection into EB5-Mut1, GFP-positive cells were FACS-sorted, and a 350-bp fragment encompassing the TALEN-targeting region was amplified by PCR. The PCR products were digested with (+) or without (−) the SacI restriction enzyme and subjected to 2% agarose electrophoresis. An uninfected EB5 cell and a gfp-silenced EB5 cell (EB5-Mut1) were used as controls. The percentage of mutation was calculated using ImageJ. Abbreviations: bp, base pair; EB5, GFP-expressing B5 mouse embryonic stem cell line; GFP, green fluorescent protein; ssODN, single-stranded oligonucleotide DNA; TALEN, transcription activator-like effector nuclease.
First, EB5 cells were transfected with the ssODN-1. At 4 hours after transfection, the EB5 cells were infected with a 1:1 mix of 2 PAK-J∆STY strains, each expressing 1 of the 2 ExoS54-TALEN fusions, at an overall MOI of 400 for 3 hours. After injection, floating bacterial cells were cleared by washing with PBS, and the EB5 cells were cultured in mESC medium containing 20 μg/ml ciprofloxacin for 3 days and then subjected to flow cytometry analysis. As a control, the EB5 cells were transfected with a 1:1 mix of the TALEN pair expressing plasmids, together with the ssODN-1 template. Consistent with the gfp gene knockout experiment shown in Figure 2A, EB5 cells transfected with TALEN-expressing plasmids resulted in approximately 10% GFP-negative cells, and bacterial delivery of the TALEN proteins resulted in almost 20% GFP-negative cells (Fig. 4B), indicating that pretransfection of the ssODN-1 template had no negative effect on the overall gfp gene knockout efficiency. The GFP-negative cells were sorted by FACS in each of the EB5 cell groups, and their genomic DNA was extracted. The 350-bp TALEN targeting gfp region was amplified by PCR and subjected to digestion by BfaI enzyme. The digestion results showed that both plasmid transfection and TALEN injection by T3SS produced the desired single-base change in the genome, resulting in 2 DNA fragments with sizes of 230 and 120 bp (Fig. 4C). Quantitative analysis of the DNA bands by ImageJ (NIH, Bethesda, MD, http://imagej.nih.gov/ij) revealed that approximately 25% of GFP-negative EB5 cells from plasmid transfected and 35% of GFP-negative EB5 cells from TALEN injection had acquired the new BfaI restriction site. Considering the 20% rate of GFP-negative EB5 cells by T3SS-mediated TALEN injection, of which 35% had the expected single nucleotide change, the overall rate of desired single nucleotide change in the EB5 cell population was 7.0% (20% × 35%). However, in the case of plasmid transfection-mediated TALEN delivery, the overall efficiency was 2.5% (10% × 25%). Thus, the new combination of template ssODN transfection with bacterial injection of the TALEN pair into mESCs resulted in an almost threefold higher efficiency of target gene modification than the conventional transfection approach.
The EB5 cell line with single-base gfp mutation was further subjected to single cell cloning. The GFP-negative cells obtained by FACS-Sort were diluted and reseeded into a 96-well plate for single cell cloning. Each putative cell clone was expanded through 24-well plate, 6-well plate, and, finally, 60-mm culture plates. Approximately 4 × 106 cells of each clone were harvested for genomic DNA extraction, and their gfp gene fragment was amplified and subjected to digestion by the BfaI restriction enzyme. As the DNA digestion results show (Fig. 4D), 2 clones (clones 4 and 6) of 12 screened had the new BfaI site, and 1 (clone 1) had a mixture of the 2 cell types. Sequence analysis of the PCR products of clones 4 and 6 confirmed the presence of correct single-base mutations.
The clone 4 GFP-negative cell line (EB5-Mut1) containing the correct single-base mutation was further reverted back to a GFP-positive cell line. A 72-base-long ssODN-2 template was designed that introduces 2 single nucleotide changes, 1 reverting the stop codon TAG back to GAG and 1 introducing a new SacI restriction enzyme site (GAGCTC) without changing the amino acid sequence (Fig. 4A). The EB5-Mut1 cell line was transfected with the ssODN-2, then infected with a 1:1 mix of the 2 ExoS54-TALEN delivery strains 4 hours later, at an overall MOI of 400 for 3 hours. After the infection, the floating bacterial cells were cleared by washing with PBS, and the cells were cultured in mESC medium containing 20 μg/ml ciprofloxacin for 3 days and then subjected to flow cytometry analysis. According to the FACS analysis results (Fig. 4E), approximately 11% cells had reverted back to GFP positive. The GFP-positive cells were sorted by FACS, and their genomic DNA was extracted. A 350-bp fragment of the TALEN targeting gfp region was amplified by PCR and subjected to digestion by SacI enzyme. The 350-bp PCR fragments obtained from EB5 and EB5-Mut1 were used as controls. The digestion results showed that TALEN injection by T3SS produced the desired single base change in the genome, resulting in 2 DNA fragments sized 230 bp and 120 bp (Fig. 4F). Quantitative analysis of the DNA bands shown in Figure 4F revealed that almost 100% of GFP-positive EB5 cells (EB5-Mut2) acquired the new SacI restriction site.
T3SS-Mediated Injection of TALEN Proteins Into Human ESCs and iPSCs
We also tested the use of the P. aeruginosa strain to inject TALEN proteins into hESCs and hiPSCs. During the initial trials, we found that hESCs and hiPSCs were much more sensitive to the bacterial cytotoxicity than were the mouse ESCs. To decrease the bacterial cytotoxicity, we chose the P. aeruginosa strain PAK-JΔ8 as the delivery strain. PAK-JΔ8 is deleted of five additional genes from the original delivery strain PAK-JΔSTY, including an inhibitor for the type III secretion system (popN), a structural gene for the type II secretion system (xcpQ), genes for quorum sensing synthesis (lasI and rhlI), and a nucleoside diphosphate kinase (ndk), which also displays toxicity against eukaryotic cells [21]. The PAK-JΔ8 shows much lower toxicity than PAK-JΔSTY yet maintains a high type III secretion capacity. The hESC line LT2e-H9CAGGFP was seeded at 70% confluence and infected by the 2 TALEN delivery strains at various MOI for 3 hours. After TALEN injection, the cells were cultured in hESC medium containing 20 μg/ml ciprofloxacin for 3 days and then monitored for GFP fluorescence by flow cytometry. As a control, eukaryotic expression vector plasmids encoding the TALEN pair were delivered by transfection. According to the FACS results, 3 hours of infection at an MOI of 100 was optimal for TALEN delivery into the hESCs or hiPSCs (Fig. 5A). Compared with the control of plasmid transfection, approximately 10% more GFP-negative cells were obtained by bacterial delivery under an overall MOI of 100 (Fig. 5B). As expected, nonfluorescent cell clusters of hESCs were easily spotted under the fluorescent microscope after bacterial injection of the TALEN pair (Fig. 5C).
Figure 5.


Type III secretion system (T3SS)-mediated injection of TALEN proteins into human ESCs and iPSCs. (A): Percentage of reduction of GFP-positive LT2e-H9CAGGFP cells after infection by the PAK-J∆8/pExoS54-FLAG-TALEN1 and PAK-J∆8/pExoS54-FLAG-TALEN2 (1:1 ratio) at the indicated MOI for 3 hours. The data were compared with those of untreated controls using a two-sample t test. ∗∗, p < .001. Error bars represent SD of triplicate assays. (B): Fluorescence intensity of LT2e-H9CAGGFP cells transfected by eukaryotic expression plasmids encoding gfp-targeting TALEN pair or infected by a 1:1 mixture of PAK-J∆8/pExoS54-FLAG-TALEN1 and PAK-J∆8/pExoS54-FLAG-TALEN2. Cells were analyzed by flow cytometry 3 days after treatment. The data were compared with those from untreated control using a two-sample t test. ∗∗∗, p < .0001. Error bars represent SD of triplicate assays. (C): A representative GFP-negative LT2e-H9CAGGFP cell cluster after bacterial delivery of gfp-targeting TALEN protein pair, observed under fluorescence microscope. (D): Schematic representation of TALEN binding sites on HPRT1 gene. The left and right TALEN binding sequences are shown in green and blue, respectively, and the spacer sequence is shown in red. (E): Sequence changes in the HPRT1 target site among iPSCs surviving the 6-thio-guanime (6TG) selection after P. aeruginosa-mediated TALEN delivery. (F): Strategy of single-base modification in the HPRT1 gene. The 72-base-pair-long ssODN-3 introduces a stop codon in the HPRT open reading frame and eliminates an XhoI restriction enzyme recognition site. (G): A polymerase chain reaction fragment of the HPRT1 gene was amplified from iPSCs surviving 6TG selection after gene modification by the ssODN-3 and P. aeruginosa-mediated TALEN delivery. The DNA fragments were digested (+) or undigested (−) with XhoI before being subjected to electrophoresis on a 0.8% agarose gel. Untreated iPSCs and iPSCs injected with the TALEN but without the ssODN-3 template were used as negative controls. The percentage of mutation was calculated using ImageJ. (H): Sequence changes in the HPRT1 target site among iPSCs surviving 6TG selection after P. aeruginosa-mediated TALEN delivery and ssODN-3 transfection. Abbreviations: ESCs, embryonic stem cells; ExoS54, exotoxin S N-terminal 54 amino acids fused with Cre recombinase; GFP, green fluorescent protein; hESCs, human embryonic stem cells; iPSCs, induced pluripotent stem cell; MOI, multiplicity of infection; TALEN, transcription activator-like effector nuclease; WT, wild type.
To apply the bacterial TALEN delivery technology in disease model establishment and therapy, we generated a pair of TALEN constructs that target exon 2 of the human HPRT1 gene (Fig. 5D) [9]. The HPRT1 gene encodes HPRT, which is responsible for recycling purine. Naturally occurring mutations in the HPRT1 gene cause decreased levels of HPRT for purine salvage, leading to neurological and behavioral problems [19]. The HPRT1 gene is located on the X chromosome; thus, its mutations cause sex-linked diseases. Cells lacking HPRT activity are resistant to a toxic nucleotide analog 6TG, which is metabolized by HPRT and integrated into the DNA, resulting in cell death; thus, cells with a functional HPRT enzyme are poisoned by 6TG. We injected the HPRT1-targeting TALENs into a male-originated iPSC line at 70% confluence under the optimal condition (MOI of 100 for 3 hours). After injection, the bacterial cells were washed off with PBS, and the cells were cultured in the iPSC medium containing 20 μg/ml ciprofloxacin. The cells were cultured for 3 days to allow phenotypic expression before drug selection. After 3 days of culture, the cells were selected on iPSC medium containing 2.5 μg/ml 6TG for 6 days. During the 6TG selection period, most of the uninfected control cells gradually died, although many cells injected with the TALEN proteins by P. aeruginosa T3SS survived and formed visible colonies. Assuming each colony had arisen from a single cell, the overall efficiency of the HPRT1 gene mutation was approximately 1%. The clones were pooled, and chromosomal DNA was extracted and PCR-amplified a 625-bp fragment encompassing the TALEN-targeting region of the HPRT1 gene. The PCR product was cloned into pGEM-T Easy vector (Promega), and 10 clones were randomly chosen for sequence analysis. The sequencing results revealed various types of alternations around the TALEN cleavage site (Fig. 5E), indicating that the injected TALENs efficiently introduced double-stranded DNA breaks, triggering error-prone DNA repair, which resulted in the observed HPRT1 gene mutations on the chromosomes of the iPSCs.
To generate the desired nucleotide change in the HPRT1 gene of human iPSCs, a 72-base-long ssODN-3 was designed as a template for homologous recombination. The ssODN-3 introduces a single nucleotide change, converting a CGA into a stop codon TGA in the HPRT1 open reading frame, which also destroys an XhoI enzyme digest site (CTCGAG) (Fig. 5F). First, the iPSCs were transfected with the ssODN-3. Next, 4 hours later, the cells were infected with a 1:1 mix of 2 PAK-J∆8 strains, each expressing one of the TALEN pair, at an overall MOI of 100 for 3 hours. After injection, floating bacterial cells were washed off with PBS, and the iPSCs were cultured in iPSC medium containing 20 μg/ml ciprofloxacin for 3 days to allow phenotypic expression. The cells were then selected in iPSC medium containing 2.5 μg/ml 6TG for 6 days, and the emerging 6TG-resistant colonies were used for genomic DNA extraction. The 625-bp HPRT1 target sequence was amplified by PCR, and the resulting fragment was subjected to digestion by XhoI enzyme. The wild-type HPRT1 fragment can be digested by XhoI enzyme into two similar size DNA fragments (313 bp and 312 bp). However, the correct single nucleotide change by homologous recombination and some nonhomologous end-joining (NHEJ) are expected to lose the XhoI recognition site. The digestion result of the “no template” control (Fig. 5G) showed that TALEN injection alone resulted in 20% DNA losing their XhoI site, presumably through mutations during NHEJ. In the experimental sample in which both TALEN and ssODN-3 were delivered, approximately 45% DNA lost their XhoI site (Fig. 5G). The 650-bp HPRT1 fragment insensitive to the XhoI enzyme digestion was gel purified and cloned into the TA cloning vector pGEM-T Easy (Promega). Sequence analysis of eight randomly chosen clones identified five with the expected single base change and three nonspecific deletions around the XhoI site (Fig. 5H). Apparently, a combination of template DNA transfection with bacterial injection of TALEN into iPSCs resulted in a high efficiency target gene modification.
Discussion
P. aeruginosa is naturally able to deliver a series of proteins into host cells via its T3SS, which proved to be a very useful tool for the delivery of exogenous proteins. Previously, we successfully used this system to inject various nuclear proteins into eukaryotic cells, such as Cre recombinase, MyoD transcription factor, and TALEN proteins, to realize cellular reprograming or genome editing [4–6]. In the present study, the ExoS54-TALEN fusion proteins were efficiently injected using the P. aeruginosa T3SS into mESCs, hESCs, and hiPSCs, achieving target gene modifications (Fig. 6). According to our data, the bacterial delivery of gfp-targeting TALEN proteins resulted in almost 20% stem cells that lost their GFP activity; the plasmid transfection method under the optimal condition resulted in approximately 10% GFP-negative cells, demonstrating that the bacterial delivery method results in higher efficiency of gene editing than the plasmid transfection method in stem cells. From our previous study, the T3SS seems to be able to inject uniformly among the target cells, with almost all the cells being injected with the protein, which might account for the higher targeting efficiency. However, the overall efficiency of target gene modification was much lower than the observed percentage of cells injected with the proteins, possibly owing to the relatively short intracellular half-life of the injected TALEN proteins. Previously, we delivered the same TALEN fusion proteins into HeLa-Venus cells, successfully knocking out the Venus gene on the chromosome, with almost 10% efficiency [6]. Our present study has shown that the efficiency of target gene knockout in stem cells is twofold higher than that in the HeLa-Venus cell line, indicating that this pair of TALEN proteins either is injected more into stem cells or has higher enzymatic activity in stem cells than it does in HeLa cells. On the basis of the intracellular stability of the injected TALEN proteins, the half-life within the ESCs [6] compared with that of HeLa cells did not seem significantly different; thus, the longer intracellular half-life is not a likely reason for the observed differences.
Figure 6.

Summary of T3SS-mediated genome editing. ExoS54-TALEN fusion proteins are produced inside bacterial cells and directly injected into the host cytosol through the bacterial T3SS needle. The injected ExoS54-TALEN proteins target to nucleus, find their target sequences on the chromosome, and introduce DSBs. In the presence of the ssDNA template (delivered by transfection), the DSB triggers homologous recombination, resulting in the desired base changes on the chromosomally encoded gfp or hprt1 gene. Abbreviations: DSB, double-strand break; ExoS54, exotoxin S N-terminal 54 amino acids fused with Cre recombinase; GFP, green fluorescent protein; HR, homologous recombination; ssDNA, single-strand DNA; T3SS, type III secretion system; TALEN, transcription activator-like effector nuclease.
The ability to alter a single nucleotide on the genome is very important for various applications, such as reversing inborn errors by correcting mutations, establishing clinical disease models, and establishing cause and effect relationships for single nucleotide polymorphisms tightly linked to specific phenotypes. Single nucleotide mutations can be repaired by introducing a single-stranded oligonucleotide DNA (ssODN) template into the target cells. The efficiency of the corrective change depends on several of factors (i.e., the length of the ssODN, the proliferative cell cycle, and the presence of DNA breaks in the host genome) [29, 30]. Recently, a single base change on the genome has been achieved through a combined use of the TALEN and ssODN templates [16, 17]. In the present study, we applied the TALEN protein delivery system for a specific base change in two systems (Fig. 6). First, TALEN delivery by T3SS of P. aeruginosa was combined with an ssODN template introduction by transfection to efficiently change a specific base of the gfp gene in mESCs, silencing the gfp gene. Through single cell cloning, the GFP-negative mESC line (EB5-Mut1) was isolated and further reverted back to GFP positive through another round of targeted gene editing (EB5-Mut2), distinguished from the original GFP-positive EB5 by having a new SacI restriction site. Compared with the conventional methods, this approach not only enhanced the efficiency of single-base editing, but also shortened the experimental period and eliminated the need to screen a large number of subclones. Second, we applied the TALEN/ssODN delivery system to human iPSCs, efficiently silencing the HPRT1 gene on the X chromosome, which is relevant to LNS [19]. The overall efficiency of target gene knockout was significantly lower than that in the targeting gfp gene, most likely reflecting differences in the targeting efficiency of the TALEN molecules, because a similar efficiency for TALEN protein injection was observed when targeting gfp and hprt1. Our success in introducing single base changes in the targeted genes with high frequency opens the door for a broad application of the new technology to many other disease model systems.
While searching for the optimal condition for bacterial delivery of the TALEN, an unexpected phenomenon was observed. After the 3 hours of bacterial infection, more cell deaths were observed at low MOIs, especially an MOI of 20 and the least cell deaths occurred at an MOI of 400 (Fig. 3A). This indicates that the bacteria caused the least cytotoxicity at an MOI 400. It is surprising that the highest TALEN injection was also observed at an MOI 400. Because it seems to be a cell density-dependent phenomenon, we speculate that the quorum sensing system of P. aeruginosa might regulate the expression of one or more exotoxins. Quorum sensing is an important mechanism of bacterial gene regulation, involving the bacteria detecting certain chemicals to perceive the density of the surrounding bacterial population and to coordinately respond to this information by regulating various gene expressions. Efforts are underway to test the possibility that one or more virulence factors of P. aeruginosa might be regulated by the quorum sensing system and be suppressed under high bacterial cell density. Such efforts will help us to discover additional exotoxins, enabling us to construct P. aeruginosa strains with much lower cytotoxicity for broad application of the bacterial protein delivery system.
ESC and iPSC lines are useful tools in the study of cellular and organ development, disease modeling, and replacement therapy. Many properties are shared among mESCs, hESCs, and hiPSCs, such as surface markers and the transcriptional factors expressed in them; however, mESCs and hESCs/hiPSCs differ in their responses to extrinsic signals and in the expression of various markers [31]. Our observation that hESCs/hiPSCs are much more sensitive to PAK-J∆STY than mouse ESCs reveals another difference in the features between them. It will be interesting to study the underlying mechanisms for such profound differences in sensitivity to various bacterial strains. Owing to the high sensitivity of hESCs/hiPSCs to the cytotoxic effects of PAK-JΔSTY, we chose a much less cytotoxic P. aeruginosa strain (PAK-JΔ8) to deliver the TALEN proteins. Strain PAK-JΔ8 is a derivative of PAK-JΔSTY, with additional deletion of genes, including a negative regulator for T3SS (popN), the type II secretion system (xcpQ), quorum sensing (IasI and rhlI), and a nucleoside diphosphate kinase [21]. The relative contribution of each deleted gene on the overall cytotoxicity of P. aeruginosa remains to be determined. Nonetheless, depending on the target cell types, both strains are useful tools for the delivery of exogenous proteins, not only for genome editing, but also for many other applications, such as cellular reprogramming. As predicted, the efficiency of genome editing can be adjusted by several bacterial injection parameters, such as MOI, infection time, and the number of repeated infections. The T3SS of P. aeruginosa is an easy to use, efficient, and robust method of protein delivery for various applications.
Recently, Sun and Zhao constructed a single-chain TALEN for genome editing that is beneficial for reducing protein load, simplifying the design, and decreasing production costs [32]. Additionally, the CRISPR/Cas9 system has emerged as a potential alternative to the TALEN system [33, 34]. CRISPR/Cas9 recognizes the targeted DNA sequence through a short guiding RNA and introduces double-strand DNA breaks. More recently, in an optimized CRISPR/Cas9 system, Cas9 with a D10A mutation triggered single-stranded DNA cleavage, significantly reducing the unintended mutation types, especially the off-target effects [35, 36]. We expect to apply P. aeruginosa T3SS with these advanced gene editing tools, raising hope for the development of additional improved methods for genome editing.
Conclusion
Our study describes a powerful tool for the delivery of TALEN proteins into PSCs, offering an alternative method for gene editing in PSCs with high efficiency. The results of the present study also pave the way to applying the P. aeruginosa T3SS to deliver transcriptional factors into PSCs for cellular reprogramming, raising the hope for safe technology that can be used for cell or tissue replacement therapy for human genetic diseases. The ability of this bacterial delivery system has been demonstrated for injection of various functional proteins into PSCs to exert their biological functions. Although many shortcomings need to be overcome, such as bacterial cytotoxicity, we are confident that the system can be improved further through continued efforts to lower the cytotoxicity and enhance the delivery capacity for the application of the bacterial protein delivery system in various biomedical applications.
Acknowledgments
We thank Drs. Craig G. Moneypenny, Neal Benson, and Lixia Yang for assistance in FACS analysis and Dr. Christopher Cassano for critical reading of our report. This work was supported by NIH/National Center for Advancing Translational Sciences Clinical and Translational Science Award to the University of Florida (Grant UL1 TR000064), Florida Department of Health Biomedical Research Programs (Grant 3KB04), National Science Foundation of China (Grants 31170128, 31370167, and 81327801), National Basic Research Program of China (Grant 2012CB518700), International Science and Technology Cooperation Program of China (Grant 2015DFG32500), Tianjin Municipal Science and Technology Commission, China (Grants 14JCYBJC28500 and 15JCZDJC33000), and National Research Foundation of Korea (Grant 2014K1A3A1A20034794).
Author Contributions
J.J., F.B., and Y.J.: performance of experiments; K.E.S., U.-H.H., and D.W.: provision of study material or patients; W.W.: conception and design, data analysis and interpretation; N.T. and S.J.: conception and design, data analysis and interpretation, manuscript writing.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
References
- 1.Gellatly SL, Hancock RE. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog Dis. 2013;67:159–173. doi: 10.1111/2049-632X.12033. [DOI] [PubMed] [Google Scholar]
- 2.Hauser AR. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat Rev Microbiol. 2009;7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rangel SM, Logan LK, Hauser AR. The ADP-ribosyltransferase domain of the effector protein ExoS inhibits phagocytosis of Pseudomonas aeruginosa during pneumonia. MBio. 2014;5:e01080-e14. doi: 10.1128/mBio.01080-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bichsel C, Neeld DK, Hamazaki T, et al. Bacterial delivery of nuclear proteins into pluripotent and differentiated cells. PLoS One. 2011;6:e16465. doi: 10.1371/journal.pone.0016465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bichsel C, Neeld D, Hamazaki T, et al. Direct reprogramming of fibroblasts to myocytes via bacterial injection of MyoD protein. Cell Reprogram. 2013;15:117–125. doi: 10.1089/cell.2012.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia J, Jin Y, Bian T, et al. Bacterial delivery of TALEN proteins for human genome editing. PLoS One. 2014;9:e91547. doi: 10.1371/journal.pone.0091547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright DA, Li T, Yang B, et al. TALEN-mediated genome editing: Prospects and perspectives. Biochem J. 2014;462:15–24. doi: 10.1042/BJ20140295. [DOI] [PubMed] [Google Scholar]
- 8.Gaj T, Gersbach CA, Barbas CF., III ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zu Y, Tong X, Wang Z, et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat Methods. 2013;10:329–331. doi: 10.1038/nmeth.2374. [DOI] [PubMed] [Google Scholar]
- 10.Huang P, Xiao A, Zhou M, et al. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol. 2011;29:699–700. doi: 10.1038/nbt.1939. [DOI] [PubMed] [Google Scholar]
- 11.Sung YH, Baek IJ, Kim DH, et al. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol. 2013;31:23–24. doi: 10.1038/nbt.2477. [DOI] [PubMed] [Google Scholar]
- 12.Tesson L, Usal C, Ménoret S, et al. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol. 2011;29:695–696. doi: 10.1038/nbt.1940. [DOI] [PubMed] [Google Scholar]
- 13.Hockemeyer D, Wang H, Kiani S, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding Q, Lee YK, Schaefer EA, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyaoka Y, Chan AH, Judge LM, et al. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods. 2014;11:291–293. doi: 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Wang Y, Huang H, et al. Precise gene modification mediated by TALEN and single-stranded oligodeoxynucleotides in human cells. PLoS One. 2014;9:e93575. doi: 10.1371/journal.pone.0093575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivera-Torres N, Strouse B, Bialk P, et al. The position of DNA cleavage by TALENs and cell synchronization influences the frequency of gene editing directed by single-stranded oligonucleotides. PLoS One. 2014;9:e96483. doi: 10.1371/journal.pone.0096483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jinnah HA. Lesch-Nyhan disease: From mechanism to model and back again. Dis Model Mech. 2009;2:116–121. doi: 10.1242/dmm.002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cristini S, Navone S, Canzi L, et al. Human neural stem cells: A model system for the study of Lesch-Nyhan disease neurological aspects. Hum Mol Genet. 2010;19:1939–1950. doi: 10.1093/hmg/ddq072. [DOI] [PubMed] [Google Scholar]
- 20.Mekhoubad S, Bock C, de Boer AS, et al. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell. 2012;10:595–609. doi: 10.1016/j.stem.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neeld D, Jin Y, Bichsel C, et al. Pseudomonas aeruginosa injects NDK into host cells through a type III secretion system. Microbiology. 2014;160:1417–1426. doi: 10.1099/mic.0.078139-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Visca P, Ciervo A, Orsi N. Cloning and nucleotide sequence of the pvdA gene encoding the pyoverdin biosynthetic enzyme L-ornithine N5-oxygenase in Pseudomonas aeruginosa. J Bacteriol. 1994;176:1128–1140. doi: 10.1128/jb.176.4.1128-1140.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cermak T, Doyle EL, Christian M, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hadjantonakis AK, Gertsenstein M, Ikawa M, et al. Generating green fluorescent mice by germline transmission of green fluorescent ES cells. Mech Dev. 1998;76:79–90. doi: 10.1016/s0925-4773(98)00093-8. [DOI] [PubMed] [Google Scholar]
- 25.Marx CJ. Development of a broad-host-range sacB-based vector for unmarked allelic exchange. BMC Res Notes. 2008;1:1. doi: 10.1186/1756-0500-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macarthur CC, Xue H, Van Hoof D, et al. Chromatin insulator elements block transgene silencing in engineered human embryonic stem cell lines at a defined chromosome 13 locus. Stem Cells Dev. 2012;21:191–205. doi: 10.1089/scd.2011.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 28.Nishimura K, Sano M, Ohtaka M, et al. Development of defective and persistent Sendai virus vector: A unique gene delivery/expression system ideal for cell reprogramming. J Biol Chem. 2011;286:4760–4771. doi: 10.1074/jbc.M110.183780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parekh-Olmedo H, Kmiec EB. Progress and prospects: Targeted gene alteration (TGA) Gene Ther. 2007;14:1675–1680. doi: 10.1038/sj.gt.3303053. [DOI] [PubMed] [Google Scholar]
- 30.Radecke F, Peter I, Radecke S, et al. Targeted chromosomal gene modification in human cells by single-stranded oligodeoxynucleotides in the presence of a DNA double-strand break. Mol Ther. 2006;14:798–808. doi: 10.1016/j.ymthe.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Koestenbauer S, Zech NH, Juch H, et al. Embryonic stem cells: Similarities and differences between human and murine embryonic stem cells. Am J Reprod Immunol. 2006;55:169–180. doi: 10.1111/j.1600-0897.2005.00354.x. [DOI] [PubMed] [Google Scholar]
- 32.Sun N, Zhao H. A single-chain TALEN architecture for genome engineering. Mol Biosyst. 2014;10:446–453. doi: 10.1039/c3mb70412b. [DOI] [PubMed] [Google Scholar]
- 33.Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu Y, Sander JD, Reyon D, et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]