

Abstract
Bacteriovorax is the halophilic genus of the obligate bacterial predators, Bdellovibrio and like organisms. The predators are known for their unique biphasic life style in which they search for and attack their prey in the free living phase; penetrate, grow, multiply and lyse the prey in the intraperiplasmic phase. Bacteriovorax isolates representing four phylogenetic clusters were selected for genomic sequencing. Only one type strain genome has been published so far from the genus Bacteriovorax. We report the genomes from non-type strains isolated from aquatic environments. Here we describe and compare the genomic features of the four strains, together with the classification and annotation.
Keywords: Predatory bacteria, Bdellovibrio and like organisms, Bacteriovorax, Marine, Gram-negative, Motile
Introduction
As a member of the highly diverse Deltaproteobacteria class, the obligate bacterial predators Bdellovibrio and like organisms possess unique ecological features that are worth exploring. They are the only known predatory bacteria that exhibit a life cycle alternating between an extracellular free-living phase and an intraperiplasmic phase and are capable of invading the periplasmic space of prey cells, resulting in the lysis of the prey and release of new progeny [1]. Based on their small size, about 1/5th that of a typical bacterium cell, BALOs have been called “the world’s smallest hunters”. Nevertheless, their genomes are larger than expected, more than 3.98 Mb in Bdellovibrio. bacteriovorus Tiberius [2], 3.78 Mb in B. bacteriovorus HD100 [1] and 3.44 Mb in Bacteriovorax marinus SJ [3]. Despite the uniqueness [4], and increasing understanding, of the potential of these organisms in various applications [5-7], their phylogeny and unique predatory features are only beginning to be understood.
Systematics has played a most important role in advancing the study of the BALOs. Based on systematic genomic molecular techniques, the original BALO genus, Bdellovibrio, has been subdivided into four genera: Bdellovibrio,Bacteriolyticum,Peredibacter, andBacteriovorax[8-10]. Being an exclusive saltwater genus, Bacteriovorax is distinct from the freshwater/terrestrial members of BALOs in many ways. It is ubiquitous in salt-water environments [10], requires at least 0.5% NaCl for growth, prefers saltwater prey [11], thrive at a lower temperature range [12] and has a lower % GC ratio of ca. 37% [13] compared to the 50.65% of the freshwater Bdellovibrio bacteriovorus HD100. Currently, Bacteriovorax marinus SJ is the only strain from the genus Bacteriovorax of which the complete genome has been sequenced and reported.
To date, variations in the 16S rRNA sequences have yielded approximately eight Bacteriovorax clusters or OTUs. The previously sequenced Bacteriovorax marinus SJT is one of the representatives that belong to phylogenetic Cluster III. This classification scheme has enabled for the first time the detection of specific Bacteriovorax strains in environmental/ecological studies. The validity of using the 16S rRNA gene was tested by comparison with the rpoB gene [10]. The results of recent studies monitoring the activities and distribution of specific phylogenetic clusters have yielded new discoveries on the distribution, predation patterns, prey preferences, and ecology of this bacterial predator [14-16].
Here we present a description of the draft genomes of Bacteriovorax isolates of four phylogenetic clusters isolated from estuarine systems, together with the description of the genomic sequencing and annotation.
Organism information
A 16S rRNA phylogenetic tree was constructed showing the phylogenetic neighborhood of the four newly sequenced Bacteriovorax strains within the family of Bdellovibrionaceae (Figure 1). As expected, Bacteriovoraxsp. strain BSW11_IV was grouped together with cluster IV, strain SEQ25 _V with cluster V, Strain DB6_IX with Cluster IX and lastly strain BAL6_X with cluster X.
Figure 1.

Phylogenetic tree highlighting (red) the position of four newly sequenced Bacteriovorax strains relative to the type strains within the family Bdellovibrionaceae and two non-type strains of each Bacteriovorax phylogenetic clusters. The tree was constructed using 16S rRNA gene sequences aligned by the RDP aligner, and was inferred using RaxML 7.25 [17] with the GTRGAMMA model of sequence evolution. The strains and their corresponding GenBank accession numbers for 16S rRNA genes were (type = T): Bacteriovorax sp. BSW11_IV; Bacteriovorax sp. SEQ25_V; Bacteriovorax sp. DB6_IX; Bacteriovorax sp. BAL6_X, Bdellovibrio bacteriovorus HD100T (BX842648); Bacteriolyticum stolpii UKi2T (AJ288899); Bacteriovorax marinus SJT (FQ312005); Peredibacter starrii A3.12T (AF084852); Bx litoralis JS5T (AF084859); Bdellovibrio exovorus JSST (EF687743); Bx sp. BB3 (DQ631715); Bx sp. OC71 (DQ536436); Bacteriovorax sp. PS23S (DQ631772); Bx sp. IHS11 (DQ631792); Bacteriovorax sp. SF11 (DQ631733); Bacteriovorax sp. ISRE1 (DQ631752); Bacteriovorax sp. GSL21 (DQ536437); Bacteriovorax sp. JDF1 (DQ631739); Bacteriovorax sp. MIA2 (DQ631695); Bacteriovorax sp. TRI41 (DQ631758); Bx sp. COCO1A (DQ631687); Bacteriovorax sp. WAIKIKKI (DQ631783); Bx sp. OC81 (DQ631721); Bacteriovorax sp. WAIKIKKI16 (DQ631770); Bacteriovorax sp. HAWAII2 (DQ631769); Bx sp. HAWAII5 (DQ631773). Deltaproteobacterium, Pelobacter carbinolicus DSM2380 (CP000142), was used as an out-group. The numbers along the branches reflect the proportion of times the groups cluster together based on 100 bootstrapped replicates. Thick branches represent those with greater than 75% bootstrap support. Phylogenetic clusters of Bacteriovorax based on 96.5% or greater 16S rRNA gene sequence similarity are denoted by brackets on the right of the tree. Clusters were numbered consistently with previous reports [9,10,18].
General features of Bacteriovoraxspp. are summarized in Table 1. Individual features of Bacteriovorax isolates have not been sufficiently explored and are largely unknown. Micrographs generated by both transmission electron microscopy and scanning microscopy (Figure 2) suggest that Bacteriovoraxspp. employ similar predation strategies as other BALO members to attack and reside in the periplasic space of its prey.
Table 1.
Classification and general features of Bacteriovorax strains according to the MIGS recommendations [19]
| MIGS ID | Property | Term | Evidence code a | |
|---|---|---|---|---|
| Current classification | Domain | Bacteria | TAS [20] | |
| Phylum | Proteobacteria | TAS [21] | ||
| Class | Deltaproteobacteria | TAS [22,23] | ||
| Order | Bdellovibrionales | TAS [24] | ||
| Family | Bacteriovoracaceae | TAS [25] | ||
| Genus | Bacteriovorax | TAS [3] | ||
| Species | Cluster IV, Cluster V, Cluster IX, Cluster X | TAS [18] | ||
| Strains: | BSW11_IV, SEQ25_V, DB6_IX, BAL6_X | IDA | ||
| Gram stain | Negative | TAS [26] | ||
| Cell shape | comma-shaped, 0.35-1.2 μm | TAS [26] | ||
| Motility | motile (one single, polar, sheathed flagellum) | TAS [26] | ||
| Sporulation | Non-sporulating | NAS | ||
| Temperature range | 10-35°C | TAS [13] | ||
| Optimum temperature | 15-30°C | TAS [3] | ||
| Carbon source | Peptides, proteins | TAS [13] | ||
| Energy source | Chemo-organotroph | TAS [13] | ||
| Terminal electron receptor | Unknown | IDA | ||
| MIGS-6 | Habitat | marine, estuarine | TAS [18] | |
| MIGS-6.3 | Salinity | >0.5% | TAS [3] | |
| MIGS-22 | Oxygen | Aerobic | NAS | |
| MIGS-15 | Biotic relationship | free living/ parasitic | TAS [3] | |
| MIGS-14 | Pathogenicity | Not reported | TAS [3] | |
| MIGS-4 | Geographic location | Breton Sound, LA (BSW11_IV); | IDA | |
| Barataria Bay, LA (SEQ25_V); | ||||
| Apalachicola Bay, FL (DB6_IX, BAL6_X) | ||||
| MIGS-5 | Sample collection time | April, 2011 (BSW11_IV); | IDA | |
| June, 2011 (SEQ25_V); | ||||
| October, 2010 (DB6_IX, , BAL6_X) | ||||
| MIGS-4.1 MIGS-4.2 | Latitude – Longitude | 29.63 -89.66 (BSW11_IV); | IDA, TAS [14] | |
| 29.38 -89.98 (SEQ25_V); | ||||
| 29.67 -85.09 (DB6_IX, BAL6_X) | ||||
| MIGS-4.3 | Depth | not reported (BSW11_IV, SEQ25_V); | NAS,TAS [14] | |
| 1.74 m (DB6_IX, BAL6_X) | ||||
| MIGS-4.4 | Altitude | not reported | IDA | |
aEvidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [27].
Figure 2.
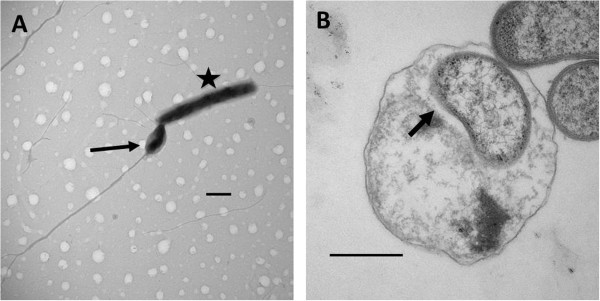
Electron micrographs showing (A) Bx sp. DB6_IX (arrow) attached to the polar end of the prey cell V. vulnificus (Star); (B) thin sections of bdelloplast, the post-BALO infection structure with the predator (arrow) residing inside the prey cell. Scale Bar represents 500 nm.
Genome sequencing information
Genome project history
The four genomes were selected for sequencing on the basis of their phylogenetic position and isolation source. Low salt Bacteriovoraxsp. BSW11_IV was isolated from Breton Sound, Louisiana (salinity 0.6 ppt; Temperature 26.4°C) and SEQ25_V was obtained from water samples of Barataria Bay, Louisiana (salinity 5.2 ppt; Temperature 19.2°C). High salt DB6_IX (Salinity 32.4 ppt; Temperature 24.1°C) and BAL6_ X (Salinity 30.9 ppt; Temperature 25.2°C) were obtained from Apalachicola Bay, Florida. The genome sequences were deposited in GenBank. Sequencing and annotation were performed at the J. Craig Venter Institute. Table 2 presents the project information and its association with MIGS version 2.0 compliance [19].
Table 2.
Genome sequencing project information
| MIGS ID | Property | BSW11_IV | SEQ25_V | DB6_IX | BAL6_X |
|---|---|---|---|---|---|
| MIGS-31 | Finishing quality | improved-high-quality draft | improved-high-quality draft | improved-high-quality draft | improved-high-quality draft |
| MIGS-28 | Libraries used | 3 KB 454 PE, 327 bp avg. insert Illumina fragment | 3 KB 454 PE, 335 bp avg. insert Illumina fragment | 3 KB 454 PE, 346 bp avg. insert Illumina fragment | 3 KB 454 PE, 316 bp avg. insert Illumina fragment |
| IGS-29 | Sequencing platforms | Illumina GAII, 454 GS FLX Titanium | Illumina GAII, 454 GS FLX Titanium | Illumina GAII, 454 GS FLX Titanium | Illumina GAII, 454 GS FLX Titanium |
| MIGS-31.2 | Fold coverage | 700× hybrid coverage | 85× hybrid coverage | 583× hybrid coverage | 81× hybrid coverage |
| MIGS-30 | Assemblers | Newbler 2.6 | CLC 5.0 | CA 7.0 | CA 7.0 |
| MIGS-32 | Gene calling method | Glimmer 3.02 | Glimmer 3.02 | Glimmer 3.02 | Glimmer 3.02 |
| Genome Database release | August 16, 2013 | August 16, 2013 | August 16, 2013 | August 5, 2013 | |
| Genbank ID | PRJNA210325 | PRJNA210326 | PRJNA210327 | PRJNA210328 | |
| Genbank Date of Release | August 16, 2013 | August 16, 2013 | August 16, 2013 | August 5, 2013 | |
| GOLD ID | Gi0051698 | Gi19265 | Gi0051699 | G0i005170 | |
| MIGS-13 | Project relevance | Environment | Environment | Environment | Environment |
Growth conditions and DNA isolation
Bacteriovorax cultures were grown separately in 70% artificial sea water (ASW) (Instant Ocean, Aquarium Systems, Inc., Mentor, Ohio) (pH 8, salinity 22 ppt.) amended with prey, Vibrio. vulnificus CMCP6 (for Bx sp.BSW11_IV and SEQ25_V), or V. parahaemolyticusRIMD 2210633 (for Bacteriovoraxsp. DB6_IX and BAL6_X). The genomes of both prey bacteria have been sequenced previously [28,29]. When cultures became clear (2–3 days after inoculation of the prey), which indicated the majority of the prey cells were lysed by the predators, 300 ml suspensions were filtered consecutively through 0.45 and 0.22 μm sterile syringe filters (Corning, NY, USA) to remove any remaining prey. Filtrates containing high concentrations of Bacteriovorax cells (ca. 4 × 108 PFU ml−1) were centrifuged at 27,485 × g for 20 min at 4°C. The pellets were then re-suspended in 1 ml of ASW respectively. To test that the concentrated Bacteriovorax suspensions were free of prey cell contamination, aliquots of 0.1 ml of the filtrate were spread-plated onto LB agar and incubated at 37°C for two days.
Subsequently, total DNA from the cell pellets were extracted using the QIAGEN Kit (QIAamp DNA Mini Kit), according to the manufacturer’s protocol. The concentration and purity of DNA was measured by a NanoDrop Spectrophotometer (ND 1000, Thermo Fisher Scientific, DE). To reconfirm the phylotype of the isolations, the DNA was PCR amplified using Bacteriovorax specific primers, Bac-676 F (5′-ATT TCG CAT GTA GGG GTA-3′) and Bac-1442R (5′-GCC ACG GCT TCA GGT AAG-3′) [30] by puReTaq Ready-To-Go PCR Beads (GE Healthcare Bio-Sciences). PCR products were purified with the QIAquick PCR-Purification Kit (QIAGEN) and sequenced with Bac-676 F primer at the DNA Sequencing Laboratory at Florida State University. 16S DNA sequences were analyzed with the Basic Local Alignment Search Tool (BLAST) server from the National Center of Biotechnology Information [31].
Genome sequencing and assembly
Genome sequencing of the four Bacteriovorax isolates was conducted at the J. Craig Venter Institute employing a combination of Illumina and 454 sequencing platforms. The 454 data consisted of a half plate of 454 FLX per genome from 3 KB mate paired libraries. The Illumina data consisted of one-quarter lane of 2 × 100 bp Illumina HiSeq data per genome. On average, 300,000 454 reads (average length trimmed 300 bp) and 10 million Illumina sequences (average length trimmed 100 bp) were generated per genome. To incorporate a hybrid assembly using both 454 and Illumina sequence libraries, one million reads were randomly sampled (with their mates) from the Illumina library using Celera [32], which was sufficient to provide high coverage in the initial assemblies.
Genome annotation
Genes were identified using GLIMMER3 [33] as part of the JCVI prokaryotic annotation pipeline followed by manual curation using the Manatee annotation-editing platform. The JCVI automated pipeline incorporates HMM3[34] searches against Pfam [35] and TIGRFAMs [36] and BLASTP against UniProt [37], JCVI’s database of experimentally characterized proteins CharProt DB [38], and PIR [39].
Genome properties
The Bacteriovoraxsp. BSW11_ IV draft genome contains 3,650,096 bp with a GC content of 37%. The hybrid assembly was scanned for contamination using BlastP and the appropriate contigs were filtered out. The final assembly comprised of 3 scafolds, 30 RNAs and 3457 CDS. For the CDSs, 2591 (75%) proteins had a BLASTP hit with an e-value of 1e-9 or better to Bacteriovorax marinus SJ, and an additional 151 (4%) CDSs had a hit within the genus Bdellovibrio.
The Bacteriovoraxsp. SEQ25_V draft genome contains 3,450,786 bp with a GC content of 37%. The sequences were assembled into 29 contigs comprised of 35 RNAs and 3,292 CDSs. Among the CDSs, 2,456 (75%) of proteins had a BLASTP hit with an e-value of 1e-9 or better to Bacteriovorax marinusSJ, and an additional 131 (4%) CDSs had a hit within the genus Bdellovibrio.
The Bacteriovoraxsp. DB6_IX draft genome contains 2,969,235 bp with a GC content of 38%. Sequences were assembled into 10 scaffolds with 30 RNAs and 3192 CDSs. Among the CDSs 2,253 (71%) proteins had a BLASTP hit with an e-value of 1e-9 or better to Bx marinus SJ, and an additional 97 (3%) CDSs had a hit within the genus Bdellovibrio.
The Bacteriovoraxsp. BAL6_ X draft genome contains 3,233,679 bp with a GC content of 36%. The reads were assembled into 9 contigs with 37 RNAs and 3,065 CDSs. Among the CDSs, 2,298 (72%) proteins had a BLASTP hit with an e-value of 1e-9 or better to Bacteriovorax marinusSJ, and an additional 92 (3%) CDSs had a hit within the genus Bdellovibrio.
It is noteworthy to point out that three phage tail fiber proteins were identified within the Bx sp. BSW11_IV genome but were absent from all the other BALO genome including the completed Bacteriovorax marinus SJ and Bdellovibrio bacteriovorus HD100 genomes. A staphylococcal phi-Mu50B-like prophage element was present in both SJ and HD 100 genomes but was not found in the genomes of the four newly sequenced Bacteriovorax isolates. The properties and the statistics of the genome are summarized in Tables 3, 4 and 5 and (Additional file 1: Table S1).
Table 3.
Summary of genomes
| Label | Size (Mb) | Topology | INSDC identifier |
|---|---|---|---|
| BSW11_IV | 3.65 | Circular | PRJNA210325 |
| SEQ25_V | 3.45 | Circular | PRJNA210326 |
| DB6_IX | 2.97 | Circular | PRJNA210327 |
| BAL6_X | 3.23 | Circular | PRJNA210328 |
Table 4.
Nucleotide content and gene count levels of the genome
| BSW11_IV | SEQ25_V | DB6_IX | BAL6_X | |||||
|---|---|---|---|---|---|---|---|---|
| Value | % of total a | Value | % of total a | Value | % of total a | Value | % of total a | |
| Genome size (bp) | 3,650,096 | 100.00% | 3,450,786 | 100.00% | 2,969,235 | 100.00% | 3,233,679 | 100.00% |
| G + C content (bp) | 1,347,908 | 36.93% | 1,243,844 | 36.05% | 1,117,420 | 37.63% | 1,179,198 | 36.47% |
| Total genes | 3,487 | 100.00% | 3,327 | 100.00% | 3,222 | 100.00% | 3,102 | 100.00% |
| RNA genes | 30 | 0.86% | 35 | 1.05% | 30 | 0.93% | 37 | 1.19% |
| Protein-coding genes | 3,457 | 99.14% | 3,292 | 98.95% | 3,192 | 99.07% | 3,065 | 98.81% |
| Proteins assigned to COGs | 2,144 | 62.02% | 2,045 | 62.12% | 1,911 | 59.87% | 1,815 | 59.22% |
| Proteins with transmembrane helices | 708 | 20.48% | 650 | 19.74% | 578 | 18.11% | 661 | 21.57% |
a)The total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome.
Table 5.
Number of genes associated with the 25 general COG functional categories
| Code | BSW11_IV | SEQ25_V | DB6_IX | BAL6_X | |||||
|---|---|---|---|---|---|---|---|---|---|
| Value | % of total a | Value | % of total | Value | % of total | Value | % of total | Description | |
| J | 166 | 4.80 | 167 | 5.07 | 137 | 4.29 | 163 | 5.31 | Translation |
| A | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | RNA processing and modification |
| K | 126 | 3.64 | 121 | 3.67 | 109 | 3.41 | 108 | 3.52 | Transcription |
| L | 115 | 3.32 | 103 | 3.12 | 100 | 3.13 | 112 | 3.65 | Replication and repair |
| B | 1 | 0.03 | 1 | 0.03 | 2 | 0.06 | 1 | 0.03 | Chromatin structure and dynamics |
| D | 24 | 0.69 | 27 | 0.82 | 23 | 0.72 | 29 | 0.94 | Cell cycle control and mitosis |
| Y | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | Nuclear structure |
| V | 45 | 1.3 | 34 | 1.03 | 31 | 0.97 | 40 | 1.30 | Defense mechanisms |
| T | 229 | 6.62 | 205 | 6.22 | 219 | 6.86 | 126 | 4.11 | Signal transduction mechanisms |
| M | 155 | 4.48 | 161 | 4.89 | 129 | 4.04 | 140 | 4.56 | Cell wall/membrane/biogenesis |
| N | 95 | 2.74 | 82 | 2.49 | 68 | 2.13 | 71 | 2.31 | Cell motility |
| Z | 2 | 0.06 | 3 | 0.09 | 2 | 0.06 | 3 | 0.10 | Cytoskeleton |
| W | 21 | 0.61 | 22 | 0.67 | 16 | 0.50 | 19 | 0.62 | Extracellular structures |
| U | 43 | 1.42 | 43 | 1.31 | 38 | 1.19 | 44 | 1.43 | Intracellular trafficking and secretion |
| O | 125 | 3.61 | 118 | 3.58 | 97 | 3.03 | 118 | 3.84 | Posttranslational modification, protein turnover, chaperones |
| C | 117 | 3.38 | 123 | 3.73 | 106 | 3.32 | 112 | 3.65 | Energy production and conversion |
| G | 72 | 2.08 | 80 | 2.43 | 67 | 2.09 | 53 | 1.72 | Carbohydrate transport and metabolism |
| E | 192 | 5.55 | 166 | 5.04 | 166 | 5.20 | 138 | 4.50 | Amino Acid transport and metabolism |
| F | 53 | 1.53s | 51 | 1.54 | 65 | 2.03 | 49 | 1.59 | Nucleotide transport and metabolism |
| H | 76 | 2.19 | 83 | 2.52 | 72 | 2.25 | 73 | 2.38 | Coenzyme transport and metabolism |
| I | 108 | 3.12 | 91 | 2.76 | 104 | 3.25 | 96 | 3.13 | Lipid transport and metabolism |
| P | 90 | 2.60 | 84 | 2.55 | 88 | 2.75 | 87 | 2.83 | Inorganic ion transport and metabolism |
| Q | 54 | 1.56 | 60 | 1.82 | 69 | 2.16 | 48 | 1.56 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 308 | 8.90 | 296 | 8.99 | 274 | 8.58 | 262 | 8.54 | General Functional Prediction only |
| S | 156 | 4.51 | 150 | 4.55 | 124 | 3.88 | 142 | 4.63 | Function Unknown |
| - | 1372 | 39.68 | 1293 | 39.27 | 1355 | 42.44 | 1244 | 40.58 | Not in COG |
aThe total is based on the total number of protein coding genes in the annotated genome.
Insights from the genome sequences
Genome Comparisons between BALO Members
Crossman et al., [40] reported that the genomic sequences of Bacteriovorax marinus SJ were unique with about one third of predicted genes over 500 bp in length having no significant hit in the databases. No genomic synteny was found between SJ and its closest whole genome sequenced relative at that time, Bdellovibrio bacteriovorus HD100.
We found that even within the genus Bacteriovorax, the genomic sequences were highly divergent with an average identity of 70%. A Venn diagram summarizing the comparison of the four Bacteriovorax isolates is presented in Figure 3. As shown in the diagram, a core of 1,513 proteins is shared by all four Bacteriovorax genomes and each encodes many proteins without orthologs in the other three (Figure 3A). When compared to the freshwater/terrestrial Bdellovibrio bacteriovorus HD 100, only a total of 843 genes were shared between all BALO members (Figure 3B). The calculated ANI [41] for BALO members (Additional file 2: Table S2) is below 75%, which is the threshold for the scores to be reliable. The AAI among the five Bacteriovorax genomes ranged between 50% to 60% (Additional file 3: Table S3), also significantly lower than the typical values found for species within a genus (73%-99.5%) [42,43]. Currently, several proposals to clarify and revise the systematics of BALOs are under consideration.
Figure 3.
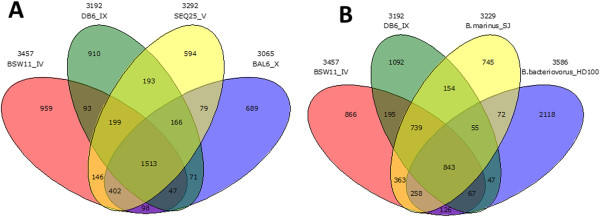
Venn diagram of shared and unique genes in (A) the four newly sequenced Bacteriovorax isolates, (B) three Bacteriovorax strains and Bdellovibrio bacteriovorus HD100. Orthology was assumed using the best reciprocal BLASTP matches (cutoff P value = 10–9).
Comparisons of BALOs and non-predatory bacteria
Phylogenetically, most genera of BALOs (including Bacteriovorax) are classified as Deltaproteobacteria. Members of this class are found in diverse environments with various lifestyles such as Myxococcus xanthus which is characterized by its gliding motility and wolf pack predatory strategy to prey on other bacteria [44], Pelobacter carbinolicus which grows by using iron and sulfur as electron acceptors [45], and the focus of this study, the obligate predators Bacteriovorax spp. which replicate within the periplasmic space of prey bacteria. Although their ecological features are distinct, the genomes of Deltaproteobacteria were found to exhibit some common characteristics. For example, most Deltaproteobacteria, including the Bdellovibrio bacteriovorus HD 100, typically possess two giant S1 ribosomal protein genes and high numbers of TonB receptors and ferric siderophore receptors which facilitate metal uptake and removal [46]. In contrast, only one giant S1 protein was found in the Bacteriovorax marinus SJ genome [40], and our study confirmed that this is the case for the other four Bacteriovorax genomes. Bacteriovorax genomes also encodes multiple TonB receptor proteins (6–11 copies) and ferric siderophore receptors (2–4 copies) that they may use for predation.
Using a reciprocal best match analysis with e-value cutoff of 10–9, 843 core genes were found to contain orthologs in all six BALO genomes including previously sequenced SJ and HD 100 genomes (see center of Figure 3B). Fifty nine of these genes (Additional file 4: Table S4) have no homologs with an E-value of 10–9 or lower to proteins from any non-predatory bacterium in the NCBI “nr” database. These genes, including periplasmic proteins, a radical activating enzyme and an outer membrane channel protein, may represent a core set of unique genes involved in the predatory process and prey interactions such as locating the prey, degradation and consumption of prey cellular content, formation of bdelloplast, synchronous nonbinary septation or release of progeny from the ghost cell.
Conclusion
The genomes of four Bacteriovorax phylogenetic clusters isolated from the environment were sequenced. The genome sizes of the four strains were comparable with Bacteriovorax SJ but were slightly smaller than the two freshwater BALOs, Bdellovibrio bacteriovorus Tiberius and B. bacteriovorus HD100. Fifty-nine genes were identified that are conserved among BALOs, but not present in other organisms, that may be responsible for their predatory life style. The unique genomic features of Bacteriovorax that are essential for their ecological function were also reported.
Abbreviations
BALOs: Bdellovibrio and Like Organisms; ANI: Average nucleotide identity; AAI: Average amino acid identity.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JHB and HNW initiated and supervised the study. HC draft the manuscript, conducted wetlab work and performed electron microscopy. HC, LMB, DSL, TLD and NG annotated the genome. HC, PM, LMB and JHB worked on genome sequencing and assembly. HC, NL, JHB, PM and HNW discussed, analyzed the data and revised the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Associated MIGS record.
Comparison of the average nucleotide identity (ANI) for the BALO genomes. ANI was calculated using ANI.pl script (https://github.com/chjp/ANI/blob/master/ANI.pl). All values are in percentages.
Percentage of average amino acid identity (AAI) between BALO genomes. AAI calculation of all two-way BLAST conserved genes was computed using AAI.rb script (http://enveomics.blogspot.com/2013/10/aairb.html).
Annotation of genes that are present in all BALO members but have no homologs from any non-predatory bacterium in NCBI’s “nr” database (E-value <10-9). Genes are listed by the protein number in BALO genomes.
Contributor Information
Huan Chen, Email: huan58.chen@gmail.com.
Lauren M Brinkac, Email: lbrinkac@jcvi.org.
Pamela Mishra, Email: pamelamishra@gmail.com.
Nan Li, Email: nli0417@gmail.com.
Despoina S Lymperopoulou, Email: dlymperop@gmail.com.
Tamar L Dickerson, Email: tayenv@gmail.com.
Nadine Gordon-Bradley, Email: gordonnadine@hotmail.com.
Henry N Williams, Email: henryneal.williams@famu.edu.
Jonathan H Badger, Email: jhbadger@gmail.com.
Acknowledgements
This work was financially supported by grants from the National Science Foundation HBCU-RISE (#0531523) and DDIG (DEB-1110620). We thank the assistance of Drs Jill W Verlander and Sharon W Matthews at the University of Florida, College of Medicine Electron Microscopy Facility; Megan Lamb at Apalachicola Bay, National Estuarine Research Reserve for research vessel operation and Dr. Aixin Hou at the Louisiana State University for assisting in sample collection and providing laboratory space for initial processing of samples.
References
- Rendulic S, Jagtap P, Rosinus A, Eppinger M, Baar C, Lanz C. A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science. 2004;303:689–92. doi: 10.1126/science.1093027. [DOI] [PubMed] [Google Scholar]
- Hobley L, Lerner TR, Williams LE, Lambert C, Till R, Milner DS. et al. Genome analysis of a simultaneously predatory and prey-independent, novel Bdellovibrio bacteriovorus from the River Tiber, supports in silico predictions of both ancient and recent lateral gene transfer from diverse bacteria. BMC Genomics. 2012;13:670. doi: 10.1186/1471-2164-13-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer ML, Ravel J, Pineiro SA, Guether-Borg D, Williams HN. Reclassification of salt-water Bdellovibrio sp. as Bacteriovorax marinus sp. nov. and Bacteriovorax litoralis sp. nov. Int J Syst Evol Microbiol. 2004;54:1011–6. doi: 10.1099/ijs.0.02458-0. [DOI] [PubMed] [Google Scholar]
- Chen H, Williams HN. Sharing of prey: coinfection of a bacterium by a virus and a prokaryotic predator. MBio. 2012;3(2):e00051–12. doi: 10.1128/mBio.00051-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atterbury RJ, Hobley L, Till R, Lambert C, Capeness MJ, Lerner TR. et al. Effects of orally administered Bdellovibrio bacteriovorus on the well-being and Salmonella colonization of young chicks. Appl Environ Microbiol. 2011;77:5794–803. doi: 10.1128/AEM.00426-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashiff A, Junka R, Libera M, Kadouri D. Predation of human pathogens by the predatory bacteria Micavibrio aeruginosavorus and Bdellovibrio bacteriovorus. J Appl Microbiol. 2011;110:431–44. doi: 10.1111/j.1365-2672.2010.04900.x. [DOI] [PubMed] [Google Scholar]
- Cao H, Hou S, He S, Lu L, Yang X. Identification of a Bacteriovorax sp. isolate as a potential biocontrol bacterium against snakehead fish‒pathogenic Aeromonas veronii. J Fish Dis. 2013;37(3):283–289. doi: 10.1111/jfd.12120. [DOI] [PubMed] [Google Scholar]
- Baer ML, Ravel J, Chun J, Hill RT, Williams HN. A proposal for the reclassification of Bdellovibrio stolpii and Bdellovibrio starrii into a new genus, Bacteriovorax gen. nov. as Bacteriovorax stolpii comb. nov. and Bacteriovorax starrii comb. nov., respectively. Int J Syst Evol Microbiol. 2000;50 Pt 1:219–24. doi: 10.1099/00207713-50-1-219. [DOI] [PubMed] [Google Scholar]
- Davidov Y, Jurkevitch E. Diversity and evolution of Bdellovibrio-and-like organisms (BALOs), reclassification of Bacteriovorax starrii as Peredibacter starrii gen. nov., comb. nov., and description of the Bacteriovorax-Peredibacter clade as Bacteriovoracaceae fam. nov. Int J Syst Evol Microbiol. 2004;54:1439–52. doi: 10.1099/ijs.0.02978-0. [DOI] [PubMed] [Google Scholar]
- Pineiro SA, Williams HN, Stine OC. Phylogenetic relationships amongst the saltwater members of the genus Bacteriovorax using rpoB sequences and reclassification of Bacteriovorax stolpii as Bacteriolyticum stolpii gen. nov., comb. nov. Int J Syst Evol Microbiol. 2008;58:1203–9. doi: 10.1099/ijs.0.65710-0. [DOI] [PubMed] [Google Scholar]
- Schoeffield AJ, Williams HN. Efficiencies of Recovery of Bdellovibrios from Brackish- Water Environments by Using Various Bacterial Species as Prey. Appl Environ Microbiol. 1990;56:230–6. doi: 10.1128/aem.56.1.230-236.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams HN. Cultural, immunologic and ecologic studies of marine bdellovibrios isolated from the Atlantic Ocean and the Chesapeake Bay. Baltimore, MD, USA: University of Maryland; 1979. [Google Scholar]
- Marbach A, Varon M, Shilo M. Properties of marine bdellovibrios. Microb Ecol. 1976;2:284–95. doi: 10.1007/BF02011648. [DOI] [PubMed] [Google Scholar]
- Chen H, Young S, Berhane T-K, Williams HN. Predatory Bacteriovorax Communities Ordered by Various Prey Species. PLoS ONE. 2012;7:e34174. doi: 10.1371/journal.pone.0034174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A, Cherrier J, Williams HN. Impact of sideways and bottom-up control factors on bacterial community succession over a tidal cycle. Proc Natl Acad Sci U S A. 2009;106:4301–6. doi: 10.1073/pnas.0809671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Athar R, Zheng G, Williams HN. Prey bacteria shape the community structure of their predators. ISME J. 2011;5:1314–22. doi: 10.1038/ismej.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 2008;57:758–71. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Pineiro S, Stine O, Chauhan A, Steyert S, Smith R, Williams H. Global survey of diversity among environmental saltwater Bacteriovoracaceae. Environ Microbiol. 2007;9:2441–50. doi: 10.1111/j.1462-2920.2007.01362.x. [DOI] [PubMed] [Google Scholar]
- Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P. et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity GM, Holt JG. In: Bergey’s Manual of Systematic Bacteriology. 2. Garrity GM, Boone DR, Castenholz RW, editor. Vol. 1. New York: Springer; 2001. The Road Map to the Manual; pp. 119–69. [Google Scholar]
- Validation List No. 107: List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2006;56:1–6. doi: 10.1099/ijs.0.64188-0. [DOI] [PubMed] [Google Scholar]
- Kuever J, Rainey FA, Widdel F. In: Bergey’s Manual of Systematic Bacteriology. Volume 2, part C. 2. Brenner DJ, Krieg NR, Staley JT, Garrity GM, editor. New York: Springer; 2005. Class VI. Deltaproteobacteria class nov. [Google Scholar]
- Garrity GM, Bell JA, Lilburn T. In: Bergey’s Manual of Systematic Bacteriology. Volume 2, Part C. 2. Brenner DJ, Krieg NR, Staley JT, Garrity GM, editor. New York: Springer; 2005. Order VII. Bdellovibrionales ord. nov; p. 1040. [Google Scholar]
- Garrity GM, Bell JA, Lilburn T. In: Bergey’s Manual of Systematic Bacteriology. Volume 2, Part C. 2. Brenner DJ, Krieg NR, Staley JT, Garrity GM, editor. New York: Springer; 2005. Family I. Bdellovibrionaceae fam. nov; pp. 1040–1. [Google Scholar]
- Williams H, Baer M, Tudor J. Bdellovibrio Stolp and Starr 1963, 243 AL. Bergey’s Manual® of Systematic Bacteriology. 2005. pp. 1041–53.
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K. et al. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet. 2003;361:743–9. doi: 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- Kim YR, Lee SE, Kim CM, Kim SY, Shin EK, Shin DH. et al. Characterization and pathogenic significance of Vibrio vulnificus antigens preferentially expressed in septicemic patients. Infect Immun. 2003;71:5461. doi: 10.1128/IAI.71.10.5461-5471.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidov Y, Friedjung A, Jurkevitch E. Structure analysis of a soil community of predatory bacteria using culture-dependent and culture-independent methods reveals a hitherto undetected diversity of Bdellovibrio-and-like organisms. Environ Microbiol. 2006;8:1667–73. doi: 10.1111/j.1462-2920.2006.01052.x. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A. et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24:2818–24. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–41. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut JD, Richter RA, Harkins D, Basu MK, Beck E. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 2013;41:D387–95. doi: 10.1093/nar/gks1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane M. UniProt Knowledgebase: a hub of integrated protein data. Database: J Biol Dat Curation. 2011;2011:.. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madupu R, Richter A, Dodson RJ, Brinkac L, Harkins D, Durkin S. et al. CharProtDB: a database of experimentally characterized protein annotations. Nucleic Acids Res. 2011;40(D1):D237. doi: 10.1093/nar/gkr1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker WC, Garavelli JS, Huang H, McGarvey PB, Orcutt BC, Srinivasarao GY. et al. The Protein Information Resource (PIR) Nucleic Acids Res. 2000;28:41–4. doi: 10.1093/nar/28.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossman LC, Chen H, Cerdeño-Tárraga A-M, Brooks K, Quail MA, Pineiro SA. et al. A small predatory core genome in the divergent marine Bacteriovorax marinus SJ and the terrestrial Bdellovibrio bacteriovorus. ISME J. 2012;7:148–60. doi: 10.1038/ismej.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102:2567–72. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis K, Tiedje J. Towards a genome-based taxonomy for prokaryotes. J Bacteriol. 2005;187:6258. doi: 10.1128/JB.187.18.6258-6264.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C, Vicente A, Souza R, Vasconcelos A, Vesth T, Alves N. et al. Genomic taxonomy of vibrios. BMC Evol Biol. 2009;9:258. doi: 10.1186/1471-2148-9-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach H. The ecology of the myxobacteria. Environ Microbiol. 1999;1:15–21. doi: 10.1046/j.1462-2920.1999.00016.x. [DOI] [PubMed] [Google Scholar]
- Lovley DR, Phillips E, Lonergan DJ, Widman PK. Fe (III) and S0 reduction by Pelobacter carbinolicus. Appl Environ Microbiol. 1995;61:2132. doi: 10.1128/aem.61.6.2132-2138.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Brocchieri L, Mrázek J, Kaiser D. Distinguishing features of δ-proteobacterial genomes. Proc Natl Acad Sci U S A. 2006;103:11352. doi: 10.1073/pnas.0604311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Associated MIGS record.
Comparison of the average nucleotide identity (ANI) for the BALO genomes. ANI was calculated using ANI.pl script (https://github.com/chjp/ANI/blob/master/ANI.pl). All values are in percentages.
Percentage of average amino acid identity (AAI) between BALO genomes. AAI calculation of all two-way BLAST conserved genes was computed using AAI.rb script (http://enveomics.blogspot.com/2013/10/aairb.html).
Annotation of genes that are present in all BALO members but have no homologs from any non-predatory bacterium in NCBI’s “nr” database (E-value <10-9). Genes are listed by the protein number in BALO genomes.