

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease, which begins with isolated steatosis and advances to nonalcoholic steatohepatitis (NASH), steatofibrosis, and cirrhosis. The pathways involved in disease progression are not understood. Loss-of-function mutations in Wnt coreceptor LDL receptor-related protein 6 (LRP6) underlie early-onset atherosclerosis, metabolic risk factors, and NAFLD in humans by unknown mechanisms. We generated mice with the human disease-associated LRP6R611C mutation and phenotypically characterized their liver. Homozygote LRP6R611C (LRP6mut/mut) mice exhibited both steatohepatitis and steatofibrosis. These traits were associated with increased activity of the noncanonical Wnt/Ras homolog family member A, Rho-associated protein kinase 2, and PKC-α/-μ pathways. Accordingly, there was increased TGF-β1 activity, coupled with enhanced expression of smooth muscle α-actin and vimentin that colocalized with albumin in LRP6mut/mut mouse liver. LRP6 knockdown reprogramed HepG2 cells to express both these markers, linking impaired Wnt signaling with hepatocyte transdifferentiation. The causal link between altered Wnt signaling and NASH was established by normalization of the disease pathways and rescue of the liver traits by Wnt3a administration to LRP6mut/mut mice. Thus, this study identifies diverse disease pathways that underlie a spectrum of NASH-related liver diseases and are linked by a single human genetic variant. LRP6 and noncanonical Wnt pathways are important potential therapeutic targets against NASH.—Wang, S., Song, K., Srivastava, R., Dong, C., Go, G.-W., Li, N., Iwakiri, Y., Mani, A. Nonalcoholic fatty liver disease induced by noncanonical Wnt and its rescue by Wnt3a.
Keywords: LRP6, PKC, hepatic steatosis, RhoA, ROCK
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease, affecting close to one-third of the adult population in developed countries (1). It encompasses a spectrum of liver disorders from simple deposition of triglyceride (TG)-rich lipid droplets in the cytoplasm known as hepatic steatosis to the inflammatory stage of nonalcoholic steatohepatitis (NASH), steatofibrosis, advanced hepatic cirrhosis, and hepatocellular carcinoma (2). The mechanisms underlying progression of disease from early to late stages are not understood. The evolution of NASH from NAFLD and its progression to end-stage liver disease often span several decades, a factor that has limited the examination of its pathophysiology in humans. Animal models have been used to assess the contribution of single genes or dietary factors to the disease process (3), but most either did not replicate the entire spectrum of human disease or did not model a disease-related variant in human.
A large body of evidence suggests strong associations between NAFLD and various components of metabolic syndrome (MetS) (4) or other coronary artery disease (CAD) risk factors (5). Accordingly, NAFLD has been linked to increased risk for CAD and type 2 diabetes (6). Nonconservative loss-of-function mutations in the gene encoding Wnt coreceptor LDL receptor-related protein 6 (LRP6) (Online Mendelian Inheritance in Man and CAD, autosomal-dominant 2) have been associated with autosomal-dominant early-onset CAD and MetS, including elevated plasma TG and LDL-C levels, diabetes, and hypertension (7, 8). Most recent studies have shown that adult LRP6 mutation careers, including those with LRP6R611C mutation, have NAFLD and NASH, assayed by liver and spleen computed tomography attenuation values and increased plasma alanine aminotransferase (Supplemental Fig. S1). Fatty liver disease is one of the traits we previously reported as part of the phenotypic characterization of homozygote LRP6R611C (referred to as LRP6mut/mut from now on in the manuscript) mice on a high-fat diet (9). The spectrum of liver disease in LRP6mut/mut mice and its mechanisms had not been investigated. LRP6mut/mut mice exhibited a broad range of NASH-related liver disorders, providing a unique opportunity to examine the mechanisms underlying progression of NAFLD to advanced stages in a single model of a human disease variant. Pathways that link altered Wnt signaling with steatohepatitis and steatofibrosis are explored and described in the current study.
MATERIALS AND METHODS
Generation of LRP6mut/mut mice
As described previously (9), mice with LRP6R611C (Lrp6R611C/+) mutation on C57BL/6 background were generated by manipulating mouse endogenous LRP6 through homologous recombination. Briefly, constructs containing mouse homologous DNA with 2 nucleotide mutations at the positions 100,443–100,445, which results in R593C (mouse equivalent of human R611C mutation) substitution, were generated by the Vegalab limited liability company (Wilmington, DE, USA). C57BL/6 embryonic stem cells were injected with the constructs and targeted into the BALB/c blasts to generate chimeras. Chimeras were crossed with B6 mice, and genotype-positive mice were backcrossed for 12 generations prior to experimentation. Lrp6R611C/+ mice were intercrossed to obtain homozygous offspring in the expected Mendelian ratios. The viable offspring were genotyped by PCR. LRP6mut/mut mice showed normal reproductive activities and had normal size and growth. Southern blot analysis of a limited number of mice confirmed the presence of LRP6R611C allele only. The mutant LRP6 protein was expressed in most tissues, including liver, heart, skeletal muscle, and adipose tissue, at normal levels. Mice were fed ad libitum and at age 6–8 weeks, were fed with either a normal chow diet (9% kcal from fat) or a high-cholesterol diet (HCD) for 7 months (40% kcal from fat, 1.25% cholesterol, and 0.5% cholic acid; D12109; Research Diets, Inc., New Brunswick, NJ, USA). Experiments were carried out after overnight fasting. Each experiment was carried out in 6–7 mice. After mice were killed, tissues were snap frozen, and plasma was separated, followed by storage at −80°C for further analysis. All procedures were approved by the Institutional Animal Care and Use Committee at Yale University.
Antibodies
Antibodies to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), β-actin, lamin B1, LRP6, p-LRP6 (S1490), Ras homolog family member A (RhoA), Rho-associated protein kinase (ROCK) 2, p-β-catenin (Ser33/37-Thr41), IL-6, IL-1β, vimentin, p-calcium/calmodulin-dependent protein kinase (CAMK)-II, glycogen synthase kinase 3β, TGF-β1, TGF-β1-RI, TGF-β1RII, p-PKC pan, PKC α, p-PKC α, PKC δ(T505), p-PKC δ(Y311), PKC ε(22B10), p-PKC δ/θ (Ser643/676), Smad2, p-Smad2, Smad3, p-Smad3, and cyclin D1 were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies to β-catenin, p-RhoA (Ser188), TGF-β1, transcription factor 7-like 2 (TCF7L2), and albumin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to F4/80, patatin-like phospholipase domain-containing protein 3 (PNPLA3), smooth muscle α-actin (α-SMA), hydroxyproline, and p-PKC-μ (S738 plus S742) were purchased from Abcam Inc. (Cambridge, MA, USA); antibody to p-ROCK2 (Ser1366) and antibodies to CD68 were purchased from Thermo Fisher Scientific (Waltham, MA, USA) and BioLegend (San Diego, CA, USA), respectively.
Histologic analysis
Nine-month-old wild-type (WT) and LRP6mut/mut mice fed an HCD for 7 months were killed for an in vivo function test. Liver tissues were embedded in Tissue-Tek optimum cutting temperature (OCT) cryostat molds (Leica Microsystems, Buffalo Grove, IL, USA) and frozen at −80°C. These tissues were used to generate 5-μm-thick sections in a cryostat.
Formalin-fixed paraffin-embedded liver samples were cut to 5-μm-thick sections. The sections were stained with hematoxylin and eosin (H&E) for structural evaluation, and stained with Masson’s trichrome, Sirius Red, and desmin for fibrosis analysis.
For immunohistochemistry, the sections were rehydrated, and antigen retrieval was performed. Primary antibodies used were rabbit anti-myeloperoxidase (MPO) serum (1761A, 1:1000; a kind gift from William C. Sessa, Yale University) and anti-IL-6 (1:500; Abcam Inc.). The sections were incubated with secondary antibodies conjugated with horseradish peroxidase (HRP; 1:500; EMD Millipore, Billerica, MA, USA) at room temperature for 1 hour. Counterstaining was performed using Mayer’s hematoxylin.
For immunofluorescence, the liver was embedded in OCT compound and frozen in dry ice-acetone. Five-micrometer-thick frozen sections were stained with antibodies against CD68, PNPLA3, vimentin, α-SMA, albumin, RhoA, ROCK2, β-catenin, JNK, CAMK-II, RAC, TCF7L2, and fluorochrome-conjugated secondary antibodies: 1:500 Alexa Fluor donkey anti-rabbit 488, donkey anti-rabbit 594, donkey anti-goat 488, donkey anti-goat 594, chicken anti-rabbit 594, and goat anti-rabbit 488 (Invitrogen, Life Technologies, Carlsbad, CA, USA). The sections were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen, Life Technologies, Grand Island, NY, USA). The sections incubated only with secondary antibodies served as negative controls. Images were taken with a light microscope (Eclipse 80i; Nikon, Tokyo, Japan). ImageJ 1.45 (National Institutes of Health, Bethesda, MD, USA) was used for image analysis of the entire liver section. At least 20 images per liver section were randomly taken and used for the analyses.
Generation of stable LRP6-knockdown cells
HepG2 cells were bought from American Type Culture Collection (Manassas, VA, USA). For LRP6 knockdown in HepG2 cells, cells were maintained in DMEM containing 10% heat-inactivated fetal bovine plasma and 1× penicillin-streptomycin at 37°C in a humidified O2/CO2 (19:1) atmosphere. Lentivirus particles containing LRP6 targeting small hairpin RNA (shRNA; 5′-CGGCGAATTGAAAGCAGTGAT-3′) were purchased from Santa Cruz Biotechnology and transduced into HepG2 cells. Transduced cells were selected using 1 mg/ml puromycin.
PKC and TGF-β inhibition in LRP6-knockdown cells
PKC inhibitor Go 6983 and TGF-β inhibitor SB431542 were obtained from Sigma-Aldrich (St. Louis, MO, USA). LRP6-knockdown HepG2 cells were cotreated for 12 hours with DMSO (solvent control), Go 6983 (5 μm), or SB431542 (10 μm). At the end of the treatment time, cytosolic protein and nuclear protein were isolated for immunoblotting.
Quantitative RT-PCR
Total RNA was isolated from liver tissues using the RNeasy Plus Mini Kit (Qiagen, Germantown, MD, USA), and cDNA was synthesized from 0.5 μg total RNA using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA). Real-time PCR amplification was performed using specific primers (Supplemental Table S1) and iQ SYBR Green Supermix (Bio-Rad Laboratories). Reactions were performed in quadruple with GAPDH and β-actin internal control. Quantification of mRNA levels was expressed as fold increase relative to the control.
Immunoblotting
Cytoplasmic proteins were prepared using a cytoplasmic extraction kit (#78833; Thermo Fisher Scientific). Cell lysates were processed and applied to SDS-PAGE and were immunoblotted using target primary antibodies followed by appropriate HRP-conjugated secondary antibodies. ECL reagents were applied to develop the blots, and blots were quantified with Image Lab (Bio-Rad Laboratories).
Recombinant mouse Wnt3a treatment
Isolated primary hepatocytes were treated with 100 ng/ml recombinant mouse Wnt3a (rmWnt3a; R&D Systems, Minneapolis, MN, USA) for 12 hours, and the lysates were immunoblotted to examine protein and phosphoprotein expressions. Mice were injected intraperitoneally with 25 μg/kg rmWnt3a every other day for 3 weeks. Mice were killed, and explanted liver was cut into smaller pieces, formalin fixed, and embedded in paraffin and, subsequently, cut to 5-μm-thick sections. Other liver tissues were harvested for immunoblotting.
Statistical analysis
All mouse studies included 6 mice in each group and were repeated at least twice. All in vitro studies were carried out in quadruple. Comparisons between 2 groups were performed using the Student’s t test. For multiple comparisons, the least significant difference test in conjunction with ANOVA was carried out. Data are means ± sd, and statistical significance is defined as P < 0.05.
RESULTS
LRP6mut/mut mice exhibit steatohepatitis
Mice with LRP6R611C mutation on C57BL/6 background were generated by manipulating mouse endogenous LRP6 through homologous recombination (see Materials and Methods). Nine-month-old homozygote LRP6mut/mut mice on chow diet exhibited grossly normal hepatic structure. However, there was significant infiltration of the liver with inflammatory cells compared to WT mice (Fig. 1A). Two-month-old WT and LRP6mut/mut mice were fed with an HCD (40% kcal from fat, 1.25% cholesterol, and 0.5% cholic acid; D12109) for 7 months and, subsequently, examined for liver disease. The hepatic fat content was significantly greater in LRP6mut/mut versus WT mice on an HCD (Fig. 1B, first column), as previously reported (9). Further investigations revealed considerable infiltration of inflammatory cells in LRP6mut/mut mouse liver, whereas none was detectable in WT mice (Fig. 1B, columns 2–4). In addition, the hepatic tissues surrounding portal and central veins were significantly altered (Fig. 1B). Immunohistochemical/immunofluorescent analysis of the liver sections revealed significantly greater expressions of MPO, IL-6 (Fig. 1C), and CD68 (Fig. 1D) in LRP6mut/mut mouse liver compared to WT mice. These changes partially correlated with an increase in MPO, IL-6, and CD68 transcription in LRP6mut/mut mouse liver compared to WT mice as shown by RT-PCR (Fig. 1E). Albumin and CD68 were expressed in different cells and did not colocalize, excluding the possibility of hepatocyte differentiation into CD68-positive cells (Fig. 1D). Western blot analysis showed increased protein levels of inflammatory cell markers CD68, MPO, and F4/80 and the cytokine IL-6 in LRP6mut/mut mouse liver compared to WT mice (Fig. 1F). There was also increased expression of PNPLA3, a membrane-bound sterol regulatory element-binding protein (SREBP) 1c-regulated triacylglycerol (TAG) lipase (10) with a genetic variant associated with fatty liver disease (11), in LRP6mut/mut mouse liver as compared to WT mice by Western blot analysis (Fig. 1F). PNPLA3 overexpression may be the consequence of increased SREBP1c activity. Alternatively, it may further contribute to increased liver fat in LRP6mut/mut mice via acyltransferase activities. Taken together, these results indicated the induction of steatohepatitis by R611C mutation.
Figure 1.

Steatohepatitis in Lrp6mut/mut mice. A) H&E staining shows significant infiltration of inflammatory cells in 9-month-old homozygote Lrp6mut/mut mice on chow diet. B) Oil Red O (ORO) and H&E staining of Lrp6mut/mut mouse liver on an HCD shows increased hepatic fat and considerable infiltration of inflammatory cells in portal vein (PV) and central vein (CV) areas. Immunohistochemical (IHC)/immunofluorescent analysis of the liver sections demonstrates greater expressions of MPO, IL-6 (C), and CD68 (D) in Lrp6mut/mut mouse liver compared to WT mice. There is a lack of coexpression of albumin and CD68. mRNA (E) and protein (F) expression of the hepatic CD68, F4/80, MPO, PNPLA3, and IL-6 in LRP6mut/mut and WT mice is presented. The relative intensities by densitometry are shown. Yellow arrows (A–C) point to positive staining for antigens. Error bars represent sd. *P < 0.05; **P < 0.01.
Liver steatohepatitis in LRP6mut/mut mice is triggered by activation of the noncanonical Wnt pathways
We next focused on the investigation of the pathogenesis of steatohepatitis in LRP6mut/mut mice. LRP6 is a coreceptor for Wnt ligands. Upon phosphorylation, LRP6 initiates a cascade of events that leads to activation of canonical Wnt signaling. Lrp6R611C mutation, as we had previously shown (9), results in a significant reduction in LRP6 phosphorylation (Fig. 2B) but only modest impairment of canonical Wnt signaling (i.e., increased β-catenin phosphorylation; Supplemental Fig. S2) in LRP6mut/mut mouse liver compared to WT mice (Supplemental Figs. S2 and S3). Recent studies have shown that the normal activity of LRP6 is equally critical for inhibition of noncanonical Wnt pathways, including Ras-related C3 botulinum toxin substrate 1 (Rac1), PKC, CAMK-II, RhoA, and ROCK (12). Activation of the noncanonical Wnt5a (13) and its downstream targets Rac1, RhoA, and ROCK has been implicated in inflammatory processes, in part through enhanced activity of ILs (14, 15). Furthermore, noncanonical Wnt involving JNK activation had been implicated in fatty liver disease, but not liver inflammation or fibrosis (16). To investigate links between LRP6 mutation and liver steatosis and inflammation, the expression levels of Rac1, JNK, RhoA/ROCK2, CAMK-II, and PKC were examined in LRP6mut/mut and WT mouse liver. There were increased expressions of RhoA and ROCK2 mRNA and protein and their phosphorylated forms in LRP6mut/mut mouse liver compared to WT (Fig. 2A, B). The hepatic expression levels of Rac1, JNK, and CAMK-II were not significantly changed in LRP6mut/mut mouse liver compared to WT mice (Supplemental Figs. S2 and S3).
Figure 2.
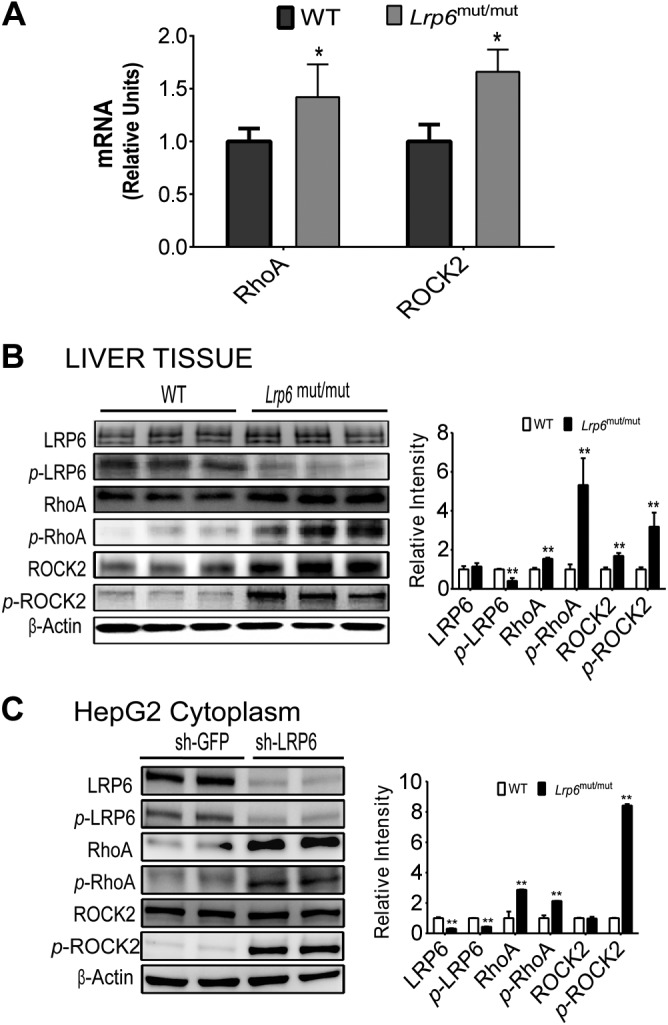
Hepatic inflammation in Lrp6mut/mut mice is triggered by the activation of noncanonical Wnt. A) mRNA expression levels of hepatic RhoA and ROCK2 were significantly higher in Lrp6mut/mut compared to WT mice. Western blot analysis demonstrates the greater activities of the RhoA/ROCK2 pathway in LRP6mut/mut liver compared to WT mice (B) and after LRP6 knockdown in HepG2 cells (C). The relative intensities by densitometry are shown. sh-GFP, GFP shRNA; sh-LRP6, LRP6-specific shRNA. Error bars represent sd. *P < 0.05; **P < 0.01.
To establish a causal link between impaired LRP6 function and increased RhoA/ROCK2 activation, LRP6 was knocked down in HepG2 cells by using an LRP6-specific shRNA (17). Correspondingly, LRP6 knockdown led to increased expression of the noncanonical Wnt proteins RhoA and ROCK2 and their phosphorylated forms (Fig. 2C). These findings associated loss of function of LRP6 with increased activation of the noncanonical pathway involving RhoA/ROCK2.
The noncanonical Wnt effectors CAMK-II and PKC had been implicated in vascular inflammation (18). In our model, RhoA/ROCK2 are the predominant noncanonical Wnt proteins that play a proinflammatory role in the LRP6mut/mut mouse liver. These differences may be due to tissue-specific activities of Wnt signaling or caused explicitly by this unique LRP6 mutation. Regardless, these findings implicate activation of noncanonical Wnt in liver steatosis and inflammation and identify the Wnt/RhoA/ROCK2 axis as a potential target for drug development against liver inflammation.
LRP6mut/mut mice develop steatofibrosis
Trichrome, Sirius Red, and desmin staining of liver sections showed greater staining throughout the liver in 9-month-old LRP6mut/mut mice fed an HCD for 7 months compared to WT mice (Fig. 3A–C) (P < 0.05). This finding indicated the development of liver fibrosis in LRP6mut/mut versus WT mice.
Figure 3.

Lrp6mut/mut mice develop steatofibrosis. Trichrome (A), Sirius Red (B), and desmin (C) staining of liver sections and their quantification showed the presence of perivascular liver fibrosis in 9-month-old Lrp6mut/mut compared to WT mice. PV, portal vein; CV, central vein. D) Immunofluorescence staining demonstrated greater vimentin expression in Lrp6mut/mut mouse liver compared to WT mice. E) mRNA expression levels of hepatic α-SMA, collagen-1α, and vimentin were significantly higher in Lrp6mut/mut compared to WT mice. Error bars represent sd. *P < 0.05; **P < 0.01.
Myofibroblasts are the primary source of extracellular matrix and the major substrate for fibrous scars and the development of liver fibrosis (19, 20). Myofibroblasts are immunophenotypically characterized by the expression of fibrotic genes such as α-SMA and vimentin and abundant pericellular matrix (20). Vimentin is a type III intermediate filament protein that is cleaved by caspases during apoptotic cell death. Hence, its increased expression in the liver is associated with both hepatic fibrosis and apoptosis. We first explored the expression of vimentin in LRP6mut/mut mouse liver by immunofluorescent staining. The expression of vimentin was considerably higher in LRP6mut/mut mouse liver compared to WT mice (Fig. 3D). These effects were mainly at the posttranscriptional level because vimentin mRNA expression levels were not significantly greater in LRP6mut/mut liver as compared to WT mice (Fig. 3D). In contrast, α-SMA and collagen-1α mRNA expression levels were significantly higher in LRP6mut/mut compared to WT mice (Fig. 3E). Accordingly, there were increased protein levels of α-SMA, vimentin, and hydroxyproline in LRP6mut/mut liver compared to WT littermates assayed by Western blot analysis (Fig. 4A).
Figure 4.

Increased PKC-dependent activation of canonical TGF-β1 underlies liver fibrosis in LRP6mut/mut mice. A) Western blot analysis demonstrates greater activities of α-SMA, vimentin, hydroxyproline, and PKC/TGF-β pathway in Lrp6mut/mut liver compared to WT mice. Similar results are shown in HepG2 cells after LRP6 knockdown. The relative intensities by densitometry are shown. B) Higher expression of nuclear p-Smad3 in HepG2 cells after LRP6 knockdown is presented. C) After inhibition of PKC and TGF-β by Go 6983 and SB431542, respectively, the LRP6-knockdown HepG2 cells showed lower expression of α-SMA and vimentin vs. controls. D) Colocalization of albumin and a-SMA in mice liver are shown. The insets show 2-fold magnification and arrows indicate the presence of colocalization. The relative intensities by densitometry are shown. Error bars represent SD. *P < 0.05; **P < 0.01. D) The colocalization of albumin with α-SMA in Lrp6mut/mut liver and its comparison with WT mice are shown.
TGF-β is a growth factor that has been shown to be a major trigger of fibrosis in the liver by increasing α-SMA expression, and secretion of extracellular matrix proteins fibronectin and collagen types I and II (21, 22). Accordingly, there were increased expressions of TGF-β1 and its receptors TGF-β1-RI and TGF-β1RII in LRP6mut/mut compared to WT hepatocytes (Fig. 4A). Consistent with this finding, there was increased activity of the canonical TGF-β signaling in LRP6mut/mut hepatocytes as shown by higher Smad2 and Smad3 phosphorylation compared to WT mice. TGF-β1 synthesis can be induced by PKC (23), a G-protein-linked protein that constitutes a major component of the noncanonical Wnt pathways. Correspondingly, there was increased phosphorylation of PKC-α and PKC-μ (Fig. 4A), but not other PKC subtypes (data not shown) in LRP6mut/mut versus WT mouse liver. Taken together, increased activities of RhoA/ROCK2, and PKC, demonstrate the important role of noncanonical Wnt in the pathogenesis of NAFLD in LRP6R611C mice.
To examine the role of LRP6 in the regulation of the hepatic cell differentiation, LRP6 was knocked down in HepG2 cells. Strikingly, this led to increased expression of α-SMA and vimentin in HepG2 cells expressing LRP6-specific shRNA compared to sham shRNA (Fig. 4A). Accordingly, expressions of TGF-β1 and its receptors TGF-β1-RI and TGF-β1RII were significantly greater in HepG2 cells expressing LRP6-shRNA compared to sham shRNA (Fig. 4A). Consequently, cytoplasmic and nuclear extracts of LRP6-knockdown HepG2 cells showed increased activation of Smad3 compared to sham shRNA (Fig. 4B). To establish the relationship between PKC and TGF-β1 pathway and the fibrosis, we pharmacologically inhibited them in HepG2 cells after knockdown of LRP6 and measured the protein expression of α-SMA and vimentin and the activity of Smad3. The pharmacologic inhibition of TGF-β1 with SB431542 strongly inhibited the expression of α-SMA and vimentin and reduced the phosphorylation of nuclear Smad3. Inhibition of PKC with Go 6983 also reduced the expression of α-SMA and vimentin, but to a lesser degree.
Previous studies have suggested that stimuli such as TGF-β may induce phenotypic and genotypic transdifferentiation of stellate cells and possibly hepatocytes into myofibroblasts in the liver (24). The existence of this process, known as epithelial-to-mesenchymal transition (EMT), in the liver has been contested (25). Nevertheless, the coexpression of the mesenchymal markers α-SMA, and vimentin, with the hepatocyte markers, has been verified in clinical and experimental samples of liver fibrosis (26). Interestingly, our studies in LRP6mut/mut mice have implicated the transdifferentiation of vascular smooth muscle cell as the key event in CAD development (unpublished data). This prompted us to examine the potential role of hepatocyte transdifferentiation in NAFLD. We first examined the colocalization of albumin with α-SMA in LRP6mut/mut and WT mouse liver. These studies showed significant colocalization of α-SMA and albumin in LRP6mut/mut mouse liver (Fig. 4D).
Taken together, these findings suggest that Lrp6R611C stimulation of PKC triggers TGF-β activation and consequently provides a substrate for cellular transdifferentiation and fibrogenicity in LRP6mut/mut mouse liver. The widespread expression of α-SMA in the liver suggests that the transdifferentiation occurs in hepatocytes, which are the most abundant cells. However, this concept can be only verified by fate mapping.
The steatohepatitis in LRP6mut/mut mouse liver is rescued by rmWnt3a
To establish a causal link between altered Wnt signaling and NAFLD, a rescue study was carried out. rmWnt3a (i.p. 25 μg/kg; every other day for 3 weeks) was administered to LRP6mut/mut mice and WT littermates, and its effect on hepatic inflammation was investigated. The Wnt3a administration rescued the fatty liver of LRP6mut/mut mice as reported earlier (9). Most strikingly, the immunofluorescence analysis of LRP6mut/mut liver revealed a dramatic reduction of CD68 expression after rmWnt3a treatment (Fig. 5A). rmWnt3a treatment also led to a reduced expression of PNPLA3 in LRP6mut/mut mouse liver (Fig. 5B). Interestingly, intraperitoneal administration of Wnt3a, a Wnt molecule generally considered as a canonical Wnt, led to reduced activation of the canonical Wnt proteins ROCK2 and RhoA, assayed by immunofluorescence studies (Fig. 5C, D). These results were confirmed by Western blot analysis, which also showed reduced expressions of MPO, IL-6, and CD68 in LRP6mut/mut mouse liver after rmWnt3a administration (Fig. 5E). These findings provide a robust link between Lrp6R611C mutation, increased activation of RhoA/ROCK2 and steatohepatitis in vivo. Our findings are consistent with earlier studies in macrophages implying an anti-inflammatory function for Wnt3a and proinflammatory functions for the noncanonical Wnt5a (27). The anti-inflammatory function of Wnt3a has been attributed in part to adhering monocytes to endothelial cells and inhibiting their migration through endothelial layers (28).
Figure 5.

rmWnt3a rescue of hepatic inflammation. A) Immunofluorescence analysis reveals hepatic CD68 expression before and after rmWnt3a treatment. B) Reduced expression of PNPLA3 in LRP6mut/mut and WT hepatocytes after rmWnt3a treatment is shown. C and D) Intraperitoneal administration of Wnt3a reduces activation of ROCK2 and RhoA. E) Western blot analysis demonstrates reduced expressions of MPO, F4/80, and IL-6 in LRP6mut/mut mouse liver after rmWnt3a administration. The relative intensities by densitometry are shown. Error bars represent sd. *P < 0.05; **P < 0.01.
Reduction of the hepatic fibrosis in liver with Wnt3a activation
Strikingly, trichrome staining of LRP6mut/mut mouse liver after rmWnt3a administration revealed a drastic reduction of liver fibrosis (Fig. 6A). Accordingly, immunofluorescent staining for α-SMA and vimentin showed reduced intensity in LRP6mut/mut and WT liver after rmWnt3a treatment (Fig. 6B, C). Western blot analysis confirmed lower liver α-SMA and vimentin protein content in both LRP6mut/mut and WT mice after rmWnt3a treatment compared to pretreatment (Fig. 6D). Correspondingly, rmWnt3a treatment dramatically decreased TGF-β expression, providing further support for its regulation by Wnt signaling (Fig. 6D).
Figure 6.

Reduction of hepatic fibrosis with Wnt3a activation. Liver fibrosis in Lrp6mut/mut mice was reduced after rmWnt3a administration, assayed by trichrome staining (A) and by immunofluorescence staining of α-SMA (B) and vimentin (C). D) Protein expression of the hepatic fibrosis inducer TGF-β1 was significantly rescued by rmWnt3a. The relative intensities by densitometry are shown. Error bars represent sd. *P < 0.05; **P < 0.01.
DISCUSSION
NAFLD is the most common cause of liver inflammation and fibrosis (1, 29). The mechanisms of disease progression from simple hepatic steatosis to advanced liver disease are not known. We had earlier reported the development of fatty liver disease as part of LRP6mut/mut mice phenotyping (9). The mechanism underlying fatty liver disease in these mice was not explored. In the current study, we have explored the entire spectrum of NAFLD in LRP6mut/mut mice and have dissected the underlying mechanisms. LRP6mut/mut mice fed with an HCD for a prolonged period (7 months) exhibit progression of fatty liver to NASH and liver fibrosis.
A major function of Wnt signaling is the inhibition of adipocyte differentiation (30). Accordingly, its inactivation is required for enhanced adipogenesis, and its impaired function may trigger lipotoxicity (31). Lipotoxicity of the liver is coupled with an increased burden of fatty acids in hepatocytes, oxidative stress, and lipid peroxidation, which are known inducers of hepatic inflammation and fibrosis (32). Thus, altered Wnt signaling and the ensuing steatosis in LRP6mut/mut mice may have partially accounted for hepatic inflammation and fibrosis. A remarkable finding, however, was the presence of liver inflammation in LRP6mut/mut mice on chow diet and in the absence of fatty liver. This finding suggested that hepatic inflammation in LRP6mut/mut mice is an independent process, which is less influenced by environmental factors such as diet compared to fatty liver disease. This prompted further investigation into pathways that link altered Wnt signaling with steatohepatitis in LRP6mut/mut mice.
Our investigations suggested that impaired LRP6 phosphorylation in LRP6mut/mut mouse liver is leading to enhanced activation of the noncanonical Wnt pathways involving RhoA/ROCK2 and PKC. The noncanonical Wnts (13) and their downstream targets Rac1 and RhoA/ROCK have been implicated in vascular inflammation (14, 15). The association between noncanonical Wnt and liver inflammation and fibrosis by our study is a novel finding with important implications for understanding the pathogenesis of these disorders. A particularly important result of our investigation was the rescue of liver inflammation and fibrosis by Wnt3a treatment. This finding established a causal link between altered Wnt signaling and NAFLD. This effect of Wnt3a was consistent with earlier reports on its function as an inhibitor of inflammatory processes (27). Thus, pathways involving inhibition of noncanonical Wnt by Wnt3a may be important targets for treatment of NAFLD.
TGF-β signaling has been implicated in liver fibrosis (24). Thus, liver fibrosis of LRP6mut/mut mice may be partially accounted for by increased activity of TGF-β pathways. TGF-β has been shown to be activated by noncanonical Wnt in smooth muscle cells (33). TGF-β1 synthesis is triggered by activated PKC (23), which was one of the activated noncanonical Wnt proteins in LRP6mut/mut mouse liver. One mechanism for induction of liver fibrosis by TGF-β1 is via increased expression of α-SMA and generation of myofibroblasts (21, 22). Consistent with these findings, there was intense staining of α-SMA in LRP6mut/mut mouse liver compared to WT mice. Overall, the noncanonical Wnt pathway appears to play the major role in liver fibrosis and inflammation in these mice. This is also evident by the fact that Wnt3a normalized the noncanonical Wnt and has almost no effect in enhancing canonical Wnt.
Strikingly, α-SMA colocalized with albumin in LRP6mut/mut mouse liver. This intriguing finding raises the possibility of hepatocyte transdifferentiation into myofibroblasts, a process known as EMT. Several studies have rebutted the role of EMT in formation of myofibroblasts in the liver. Epithelial cell differentiation into myofibroblasts was ruled out as the cause of liver fibrosis by lineage tracing in α-fetoprotein Cre mice (34). Similar results were obtained in an albumin Cre mouse model of liver fibrosis (35). There are, however, significant differences between these models and LRP6mut/mut mice. Most importantly, these are experimental models of fibrosis, whereas fibrosis of LRP6mut/mut mice occurs spontaneously as a consequence of a human disease variant. The time frame stimuli in these experimental models may also be critical because the short duration of insult may not be sufficient to reproduce the cellular pathophysiology that occurs in chronic human liver disease or in our mouse model. These experimental models also provide no alternative explanation for the coexpression of epithelial and mesenchymal markers in biopsies of patients with primary biliary cirrhosis or primary sclerosing cholangitis. Furthermore, they do not provide any explanation for the fibrotic gene expression profile in injured hepatocytes (36) and cholangiocytes (37). Thus, despite controversies about the role of EMT as injury-induced changes in epithelial cells, phenotypic changes observed in human hepatocytes and in animal models such as LRP6mut/mut mice are important findings that demand further investigations.
In summary, our study establishes a link between noncanonical Wnt signaling and NAFLD and identifies these pathways as potential targets for drug development against this emerging disease.
Supplementary Material
Acknowledgments
This study was supported by U.S. National Institutes of Health (NIH) National Heart, Lung, and Blood Institute Grants R01HL122830-01 and R01HL122822-01 (to A.M.) and by NIH National Institute of Diabetes and Digestive and Kidney Diseases Grant R01DK082600 (to Y.I.).
Glossary
- α-SMA
smooth muscle α-actin
- CAD
coronary artery disease
- CAMK
calcium/calmodulin-dependent protein kinase
- EMT
epithelial-to-mesenchymal transition
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HCD
high-cholesterol diet
- H&E
hematoxylin and eosin
- HRP
horseradish peroxidase
- LRP6
LDL receptor-related protein 6
- LRP6mut/mut
homozygote LRP6R611C
- MetS
metabolic syndrome
- MPO
myeloperoxidase
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OCT
optimum cutting temperature
- PNPLA3
patatin-like phospholipase domain-containing protein 3
- Rac1
Ras-related C3 botulinum toxin substrate 1
- RhoA
Ras homolog family member A
- rmWnt3a
recombinant mouse Wnt3a
- ROCK
Rho-associated protein kinase
- shRNA
small hairpin RNA
- SREBP
sterol regulatory element-binding protein
- TCF7L2
transcription factor 7-like 2
- TG
triglyceride
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Cohen J. C., Horton J. D., Hobbs H. H. (2011) Human fatty liver disease: old questions and new insights. Science 332, 1519–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yki-Järvinen H. (2014) Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2, 901–910 [DOI] [PubMed] [Google Scholar]
- 3.Hebbard L., George J. (2011) Animal models of nonalcoholic fatty liver disease. Nat.Rev. Gastroenterol. Hepatol. 8, 35–44 [DOI] [PubMed] [Google Scholar]
- 4.Zeb I., Katz R., Nasir K., Ding J., Rezaeian P., Budoff M. J. (2013) Relation of nonalcoholic fatty liver disease to the metabolic syndrome: the Multi-Ethnic Study of Atherosclerosis. J. Cardiovasc. Comput. Tomogr. 7, 311–318 [DOI] [PubMed] [Google Scholar]
- 5.Kim N. H., Park J., Kim S. H., Kim Y. H., Kim D. H., Cho G. Y., Baik I., Lim H. E., Kim E. J., Na J. O., Lee J. B., Lee S. K., Shin C. (2014) Non-alcoholic fatty liver disease, metabolic syndrome and subclinical cardiovascular changes in the general population. Heart 100, 938–943 [DOI] [PubMed] [Google Scholar]
- 6.Idilman I. S., Akata D., Hazirolan T., Doganay Erdogan B., Aytemir K., Karcaaltincaba M. (2015) Nonalcoholic fatty liver disease is associated with significant coronary artery disease in type 2 diabetic patients: a computed tomography angiography study. J. Diabetes 7, 279–286 [DOI] [PubMed] [Google Scholar]
- 7.Mani A., Radhakrishnan J., Wang H., Mani A., Mani M. A., Nelson-Williams C., Carew K. S., Mane S., Najmabadi H., Wu D., Lifton R. P. (2007) LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 315, 1278–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh R., Smith E., Fathzadeh M., Liu W., Go G. W., Subrahmanyan L., Faramarzi S., McKenna W., Mani A. (2013) Rare nonconservative LRP6 mutations are associated with metabolic syndrome. Hum. Mutat. 34, 1221–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Go G. W., Srivastava R., Hernandez-Ono A., Gang G., Smith S. B., Booth C. J., Ginsberg H. N., Mani A. (2014) The combined hyperlipidemia caused by impaired Wnt-LRP6 signaling is reversed by Wnt3a rescue. Cell Metab. 19, 209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y., Cohen J. C., Hobbs H. H. (2011) Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 286, 37085–37093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rotman Y., Koh C., Zmuda J. M., Kleiner D. E., Liang T. J.; NASH CRN (2010) The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 52, 894–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryja V., Andersson E. R., Schambony A., Esner M., Bryjová L., Biris K. K., Hall A. C., Kraft B., Cajanek L., Yamaguchi T. P., Buckingham M., Arenas E. (2009) The extracellular domain of Lrp5/6 inhibits noncanonical Wnt signaling in vivo. Mol. Biol. Cell 20, 924–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulte D. M., Müller N., Neumann K., Oberhäuser F., Faust M., Güdelhöfer H., Brandt B., Krone W., Laudes M. (2012) Pro-inflammatory wnt5a and anti-inflammatory sFRP5 are differentially regulated by nutritional factors in obese human subjects. PLoS One 7, e32437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu H., Wu J. Y., Kudo T., Ohno T., Graham D. Y., Yamaoka Y. (2005) Regulation of interleukin-6 promoter activation in gastric epithelial cells infected with Helicobacter pylori. Mol. Biol. Cell 16, 4954–4966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LoGrasso P. V., Feng Y. (2009) Rho kinase (ROCK) inhibitors and their application to inflammatory disorders. Curr. Top. Med. Chem. 9, 704–723 [DOI] [PubMed] [Google Scholar]
- 16.Ouchi N., Higuchi A., Ohashi K., Oshima Y., Gokce N., Shibata R., Akasaki Y., Shimono A., Walsh K. (2010) Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 329, 454–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh R., De Aguiar R. B., Naik S., Mani S., Ostadsharif K., Wencker D., Sotoudeh M., Malekzadeh R., Sherwin R. S., Mani A. (2013) LRP6 enhances glucose metabolism by promoting TCF7L2-dependent insulin receptor expression and IGF receptor stabilization in humans. Cell Metab. 17, 197–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J., Kim J., Kim D. W., Ha Y., Ihm M. H., Kim H., Song K., Lee I. (2010) Wnt5a induces endothelial inflammation via beta-catenin-independent signaling. J. Immunol. 185, 1274–1282 [DOI] [PubMed] [Google Scholar]
- 19.Watsky M. A., Weber K. T., Sun Y., Postlethwaite A. (2010) New insights into the mechanism of fibroblast to myofibroblast transformation and associated pathologies. Int. Rev. Cell Mol. Biol. 282, 165–192 [DOI] [PubMed] [Google Scholar]
- 20.Eyden B. (2008) The myofibroblast: phenotypic characterization as a prerequisite to understanding its functions in translational medicine. J. Cell. Mol. Med. 12, 22–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morishima Y., Nomura A., Uchida Y., Noguchi Y., Sakamoto T., Ishii Y., Goto Y., Masuyama K., Zhang M. J., Hirano K., Mochizuki M., Ohtsuka M., Sekizawa K. (2001) Triggering the induction of myofibroblast and fibrogenesis by airway epithelial shedding. Am. J. Respir. Cell Mol. Biol. 24, 1–11 [DOI] [PubMed] [Google Scholar]
- 22.Lemoinne S., Cadoret A., El Mourabit H., Thabut D., Housset C. (2013) Origins and functions of liver myofibroblasts. Biochim. Biophys. Acta 1832, 948–954 [DOI] [PubMed] [Google Scholar]
- 23.Ha H., Yu M. R., Lee H. B. (2001) High glucose-induced PKC activation mediates TGF-beta 1 and fibronectin synthesis by peritoneal mesothelial cells. Kidney Int. 59, 463–470 [DOI] [PubMed] [Google Scholar]
- 24.Gressner A. M., Weiskirchen R., Breitkopf K., Dooley S. (2002) Roles of TGF-beta in hepatic fibrosis. Front. Biosci. 7, d793–d807 [DOI] [PubMed] [Google Scholar]
- 25.Taura K., Miura K., Iwaisako K., Osterreicher C. H., Kodama Y., Penz-Osterreicher M., Brenner D. A. (2010) Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 51, 1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rygiel K. A., Robertson H., Marshall H. L., Pekalski M., Zhao L., Booth T. A., Jones D. E., Burt A. D., Kirby J. A. (2008) Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab. Invest. 88, 112–123 [DOI] [PubMed] [Google Scholar]
- 27.Schaale K., Neumann J., Schneider D., Ehlers S., Reiling N. (2011) Wnt signaling in macrophages: augmenting and inhibiting mycobacteria-induced inflammatory responses. Eur. J. Cell Biol. 90, 553–559 [DOI] [PubMed] [Google Scholar]
- 28.Tickenbrock L., Schwäble J., Strey A., Sargin B., Hehn S., Baas M., Choudhary C., Gerke V., Berdel W. E., Müller-Tidow C., Serve H. (2006) Wnt signaling regulates transendothelial migration of monocytes. J. Leukoc. Biol. 79, 1306–1313 [DOI] [PubMed] [Google Scholar]
- 29.Argo C. K., Northup P. G., Al-Osaimi A. M., Caldwell S. H. (2009) Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J. Hepatol. 51, 371–379 [DOI] [PubMed] [Google Scholar]
- 30.Ross S. E., Hemati N., Longo K. A., Bennett C. N., Lucas P. C., Erickson R. L., MacDougald O. A. (2000) Inhibition of adipogenesis by Wnt signaling. Science 289, 950–953 [DOI] [PubMed] [Google Scholar]
- 31.Gunaratnam K., Vidal C., Gimble J. M., Duque G. (2014) Mechanisms of palmitate-induced lipotoxicity in human osteoblasts. Endocrinology 155, 108–116 [DOI] [PubMed] [Google Scholar]
- 32.Neuschwander-Tetri B. A. (2010) Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 52, 774–788 [DOI] [PubMed] [Google Scholar]
- 33.Kumawat K., Menzen M. H., Bos I. S., Baarsma H. A., Borger P., Roth M., Tamm M., Halayko A. J., Simoons M., Prins A., Postma D. S., Schmidt M., Gosens R. (2013) Noncanonical WNT-5A signaling regulates TGF-β-induced extracellular matrix production by airway smooth muscle cells. FASEB J. 27, 1631–1643 [DOI] [PubMed] [Google Scholar]
- 34.Chu A. S., Diaz R., Hui J. J., Yanger K., Zong Y., Alpini G., Stanger B. Z., Wells R. G. (2011) Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology 53, 1685–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Österreicher C. H., Penz-Österreicher M., Grivennikov S. I., Guma M., Koltsova E. K., Datz C., Sasik R., Hardiman G., Karin M., Brenner D. A. (2011) Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. USA 108, 308–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jung Y., Witek R. P., Syn W. K., Choi S. S., Omenetti A., Premont R., Guy C. D., Diehl A. M. (2010) Signals from dying hepatocytes trigger growth of liver progenitors. Gut 59, 655–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patsenker E., Popov Y., Stickel F., Jonczyk A., Goodman S. L., Schuppan D. (2008) Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology 135, 660–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.