

Abstract
Elevated levels of serum retinol-binding protein 4 (RBP4) contribute to insulin resistance and correlate with increased prevalence of hypertension and myocardial infarction. We sought to determine whether lowering RBP4 would improve blood pressure (BP) and protect against obesity- or angiotensin (Ang)-II-induced hypertension. Systolic and diastolic BP were lower in the RBP4-knockout (RBP4-KO) mice and higher in the RBP4-overexpressing (RBP4-Tg) mice compared with BP in the wild-type (WT) littermates. Carbachol-induced vasodilatation was increased in arteries from the RBP4-KO compared with the WT mice and was impaired in the RBP4-Tg mice. Aortic eNOSSer1177 phosphorylation was enhanced ∼50% in the RBP4-KO mice, with no change in total eNOS protein. Feeding a high-fat diet increased BP in the RBP4-KO mice only to the level in the WT mice fed chow and had no effect on aortic eNOSSer1177 phosphorylation. Ang-II infusion resulted in 22 mmHg lower systolic BP in the RBP4-KO than in the WT mice, although the relative BP increase over saline infusion was ∼30% in both. Ang-II treatment decreased aortic eNOSSer1177 phosphorylation in the WT and RBP4-KO mice, but phosphorylation remained higher in the RBP4-KO mice. Cardiac hypertrophy with Ang-II treatment was diminished by 56% in the RBP4-KO mice. Thus, elevated serum RBP4 raises BP and lack of RBP4 reduces it, with commensurate changes in aortic eNOSSer1177 phosphorylation. Lowering RBP4 may reduce BP through enhanced eNOS-mediated vasodilatation and may be a novel therapeutic approach for hypertension.—Kraus, B. J., Sartoretto, J. L., Polak, P., Hosooka, T., Shiroto, T., Eskurza, I., Lee, S.-A., Jiang, H., Michel, T., Kahn, B. B. Novel role for retinol-binding protein 4 in the regulation of blood pressure.
Keywords: angiotensin II, hypertension, obesity, endothelial NO-synthase
Retinol-binding protein 4 (RBP4) was first described in 1968 as the major transport protein for vitamin A from the liver to the peripheral tissues (1). RBP4 serum levels are elevated in obesity and diabetes, and RBP4 mediates insulin resistance (2, 3), at least partially, by inducing proinflammatory cytokine production from macrophages (4). Large epidemiologic studies have shown that RBP4 levels correlate closely with insulin resistance and cardiovascular risk factors, such as dyslipidemia and hypertension and with cardiovascular disease (CVD) (5–7). Genetic studies suggest a causative role for elevated RBP4 in metabolic disease (8). In a smaller study, correlation of RBP4 with BP was independent of metabolic syndrome (9). Data from the Nurses’ Health Study show a high predictive value of elevated serum RBP4 for occurrence of myocardial infarction (10). In studies of endothelial function, RBP4 correlated negatively with flow-mediated vasodilatation (11, 12). Atherosclerosis parameters, such as intima media thickness and plaque echogenicity, are strongly and independently associated with the serum retinol:RBP4 ratio, even after adjustment for metabolic syndrome indicators (7, 13, 14). Moreover, in patients with hypertension without metabolic syndrome, serum RBP4 levels are elevated (9). Thus, RBP4 may play a direct role in blood pressure (BP) regulation.
Decreased NO production due to defective insulin signaling is an important mediator of endothelial dysfunction in insulin resistance and obesity-induced hypertension (15, 16). In addition, flow-mediated vasodilatation and angiotensin (Ang-II)-induced vasoconstriction depend on NO-modulated signaling (17). Therefore, we hypothesized that RBP4 causes hypertension through impaired vascular reactivity via attenuated eNOS-signaling. Our aim was to investigate whether lowering RBP4 would improve BP and vascular eNOS signaling and protect against obesity- and Ang-II induced hypertension.
MATERIALS AND METHODS
Animals
C57Bl/SF129 mice lacking RBP4 systemically [RBP4-knockout (KO)] and C57Bl mice with a muscle creatine kinase-driven overexpression of RBP4 (RBP4-Tg) were bred in house. Study groups of RBP4-KO mice consisted of males generated from HetxHet breeding, compared with their respective WT littermates at the indicated ages. For the generation of RBP4-Tg mice, male RBP4-overexpressing mice were bred with female C57BL mice. Genotype was determined by detecting RBP4 in a 20× dilution of serum in lysis buffer (20 mM Tris-Cl, pH 7.4, 5 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, 1% Igepal, 6.7 µg/ml aprotinin and leupeptin, 1 mM PMSF, and 2 mM Na3VO4), and proteins were separated by 15% SDS-PAGE and transferred to nitrocellulose membranes. RBP4 protein was detected with anti-human RBP4 polyclonal antisera (Dako, Carpinteria, CA, USA). The anti-human antibody also recognizes mouse RBP4, but with lower affinity.
Mice were housed in a barrier facility in individually ventilated autoclaved cages, water was also autoclaved, and diet was irradiated. They were maintained in a 14/10 h light-dark cycle, no daylight savings was observed, and the air temperature in the room was controlled between 72 and 74°F.
RBP4-KO mice and their WT littermates were fed Teklad 7904 (Harlan Laboratories, Indianapolis, IN, USA) with 11% fat (29% calories from fat) and 37 IU vitamin A per gram of food. RBP4-Tg mice and their WT littermates were fed chow Formulab 5008 (LabDiet; Nestle Purina Petcare Company, St. Louis, MO, USA) containing 6.5% fat (17% of calories from fat) and 15 IU vitamin A per gram of food.
Experiments were initiated 4 h after food was removed at 7 am (euthanasia started at 11 am) except for insulin-injection studies, which were performed after withholding food overnight. For endothelial insulin signaling, mice on chow or a high-fat diet (HFD) were injected with saline or 6 IU insulin i.p. and killed by cervical dislocation 30 minutes later. All animal experimentation was performed according to protocols approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Hypertension models
Hypertension was induced by infusion of Ang-II (1 mg/kg/d) (A9525; Sigma-Aldrich, St. Louis, MO, USA) via osmotic minipump (Model 1002; Alzet Osmotic Pumps, Cupertino, CA, USA) in 10- to 12-wk-old male WT and RBP4-KO mice for 2 wk.
Diet-induced obesity and hypertension were induced by feeding the WT and RBP4-KO mice an HFD ad libitum for 12 wk, starting at 6 wk of age. The HFD (Teklad 93075; Harlan Laboratories) contained 29.3% fat (55% of calories derived from fat) and 27 IU vitamin A per gram of food.
Echocardiography
Two-dimensional guided M-mode echocardiography was performed on conscious mice. Chest hair was removed with Nair cream (Church & Dwight Co., Ewing, NJ, USA), and ultrasound transmission gel was applied. Echocardiography was conducted with a 15-Mhz linear-array probe and a Vivid FiVe ultrasonograph (GE Healthcare Life Sciences, Marlborough, MA, USA) with a heart rate (HR) of 500–600 bpm. M-mode imaging was obtained with a parasternal short-axis view. Three consecutive cardiac cycles were measured and averaged according to the Recommendations for a Standard Report for Adult Transthoracic Echocardiography of the American Society of Echocardiography (18). Care was used to obtain exactly symmetrical short-axis images at the level of the papillary muscles. Ventricular septum thickness was used to quantify hypertrophy, and systolic and diastolic left ventricular diameters were measured to calculate fractional shortening, as a measure of systolic function. At least 5 measurements were obtained and averaged for every data point from each mouse.
BP measurement
BP and HR were assessed with a noninvasive Volume Pressure Recording tail-cuff system (CODA; Kent Scientific Corp., Torrington, CT, USA) in RBP4-KO or -Tg mice and their respective WT littermates. We used a rigorous protocol for noninvasive BP measurements, as suggested in tail-cuff validation studies (19).
All measurements were performed on a warming platform in a controlled environment of 24–26°C, constant humidity and noise-free at the same time between 8 am and noon. A maximum of 4 animals were measured simultaneously, to allow monitoring of behavior and reading quality during the measurements. Each animal was trained for 3 consecutive days, to minimize intraindividual variance between measurements. On the fourth and fifth days, 20 readings were collected from every animal. To avoid observer bias, the measurements were performed with the postdoctoral fellow or technician not knowing the genotype or treatment group. For analysis, only accurate readings with full sets of systolic and diastolic pressure, as well as HR and clean pressure curves, where used. For each mouse, all valid readings were used to calculate the average that was used for group analysis.
This method yielded strikingly consistent and reproducible values in all cohorts studied. Therefore, considering the disadvantages of invasive BP measurements, such as additional stress and increased surgical mortality in obese animals fed an HFD and with additional surgery for implantation of Ang-II pumps, we decided that telemetry would not be the optimal technique for this study.
Arterial preparation and vascular reactivity studies
Vascular reactivity was tested on carotid arteries, as described previously (20). After the mice were killed with pentobarbital, the carotid arteries were removed and mounted onto glass cannulae in a pressure myograph (Living Systems Inc., St. Albans, VT, USA) and maintained with perfusion (80 mmHg pressure, 37°C) in physiologic saline solution (PSS), containing 130 mM NaCl, 4.7 mM KCl, 1.17 mM MgSO4, 14.9 mM NaHCO3, 1.6 mM CaCl2, 1.18 mM KH2PO4, 0.026 mM EDTA, and 5.5 mM glucose. Continuous buffer aeration (95% O2, 5% CO2) maintained pH at 7.2 to 7.4. Vessel diameter was continuously recorded by video (IonWizard 4.4; IonOptix Corp., Milton, MA, USA). Vessels were equilibrated in PSS alone for 60 minutes before phenylephrine (PE) constriction (10−5 M, 60 minutes). Vessel-relaxation dose-response curves were generated with increasing carbachol concentrations (10−8–10−5 M) added to the organ bath, expressing relaxation as the percentage change in diameter after PE preconstriction compared with the diameter before PE constriction, using the following equation: percentage dilation = 100% × [(Dx – Di)/(B – Di)], where D is the measured arterial diameter, subscripts x and i denote arterial diameters at each dose of agonist (x) and initial diameter following PE constriction (i), and B is the basal arterial diameter before PE constriction. For each experiment, carotid arteries from WT and KO mice were studied simultaneously in a 2-vessel myograph. Experiments were repeated at least 4 times in independent preparations.
In vitro protein synthesis
Protein synthesis in cultured H9C2 cardiomyocytes (ATCC, Manassas, VA, USA) treated with Ang-II, with or without RBP4, was quantified as incorporation of the radioactive-labeled amino acid [3H]leucine as described previously (21). The cardiomyocytes were grown in 6-well plates with Dulbecco’s modified Eagle’s medium (Cellgro; Mediatech, Inc., Manassas, VA, USA) supplemented with 10% (v/v) fetal calf serum (not heat-inactivated; Invitrogen, Carlsbad, CA, USA) and 1% penicillin-streptomycin (Invitrogen). The cells were serum starved overnight, incubated with the indicated treatment (1 μM Ang-II A9525; Sigma-Aldrich), with or without 50 μg/ml recombinant mRBP4 (courtesy of Pratik Aryal, Beth Israel Deaconess Medical Center, Boston, MA, USA), and 1 µCi/ml l-4,5-[3H]leucine (Perkin Elmer; Wellesley, MA, USA) was added for 24 h at 37°C. The cells were washed 3 times with PBS (Gibco; Life Technologies, Waltham, MA, USA) and harvested in ice-cold lysis buffer (20 mM Tris-Cl, pH 7.4, 5 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, 1% Igepal, 6.7 µg/ml aprotinin and leupeptin, 1 mM PMSF, and 2 mM Na3VO4). The incorporated [3H]leucine was determined by liquid scintillation counting.
Western blot analysis
Western blot analysis of total and phosphorylated eNOS was performed on protein lysates of whole aorta. Snap-frozen tissue was homogenized in lysis buffer (20 mM Tris-Cl, pH 7.4, 5 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, 1% Igepal, 6.7 µg/ml aprotinin and leupeptin, 1 mM PMSF, and 2 mM Na3VO4). Proteins (50 μg) were separated by electrophoresis on a 10% polyacrylamide gel and transferred to a nitrocellulose membrane. Nonspecific binding sites were blocked with 5% skim milk in Tris-buffered solution for 1 h at room temperature. The membranes were then incubated at 4°C overnight (1:1000) with the following primary antibodies: RBP4 (DAKO), phospho-eNOSSer1177 (Cell Signaling; Beverly, MA, USA), total eNOS (Cell Signaling), phospho-AktSer473 (Cell Signaling), and total Akt (EMD Millipore, Billerica, MA, USA). The membranes where washed with Tris-buffered solution and incubated for 1 h at room temperature with secondary horseradish peroxidase-conjugated goat anti-rabbit antibody (1:5000; Pierce Scientific, Rockford, IL, USA). Thereafter, the membranes where washed again, and the signals revealed with chemiluminescence, visualized by autoradiography film, and quantified by densitometry. The same membrane was used to determine phosphorylated isoforms, total protein content after stripping (Restore Plus Western Blot Stripping Buffer; Thermo Fisher Scientific, Inc., Waltham, MA, USA), and p85-PI3K (Millipore). The p85 subunit of PI3K was used to normalize protein expression in each sample.
Retinoid serum and tissue measurements
Tissue and serum retinol and retinyl ester (RE) levels were determined by HPLC, as described previously (22). In brief, serum and hearts of 5-month-old male WT and RBP4-KO mice were collected under red light to prevent retinoid degradation, snap-frozen in liquid nitrogen, and stored immediately at −80°C for analysis. The tissues were homogenized in 10 vol PBS (10 mM sodium phosphate, pH 7.2, and 150 mM sodium chloride) on a Polytron homogenizer (Brinkmann Instruments, Westbury, NY, USA) at half-maximum speed for 10 seconds. An aliquot of serum or tissue was treated with an equal volume of absolute ethanol containing a known amount of retinyl acetate as an internal standard. The retinoids in the homogenates were extracted into hexane. After 1 backwash against double-distilled water, the hexane extract was evaporated to dryness under a gentle stream of N2. Immediately upon reaching dryness, the retinoid-containing film was redissolved in 40 µl of benzene for injection onto the HPLC column. The extracted retinoids were separated on a 4.6 × 250 mm Ultrasphere C-18 column (Beckman Coulter, Brea, CA, USA) preceded by a C-18 guard column (Supelco, Bellefonte, PA, USA), with 70% acetonitrile, 15%–methanol, and 15% methylene chloride used as the running solvent flowing at 1.8 ml/min. Retinol and REs (retinyl palmitate, oleate, linoleate, and stearate) were detected at 325 nm and identified by comparing the retention times and spectral data of experimental compounds with those of authentic standards. Concentrations of retinol and REs in the tissues were quantitated by comparing integrated peak areas of each retinoid against those of known amounts of purified standards. Loss during extraction was accounted for by adjusting for the recovery of internal standard added immediately after homogenization of the samples.
Statistical analysis
Data are expressed as means ± sem with the number (n) of experiments or animals indicated. Statistical analysis using GraphPad Prism (GraphPad Software, San Diego, CA, USA) and Daniel’s XL toolbox software (opensource software) was performed by Student t test when comparing 2 groups and by 2-way ANOVA followed by the Bonferroni post hoc analysis when more than 2 groups were compared. P < 0.05 indicated statistical significance.
RESULTS
RBP4-KO mice exhibit lower BP and RBP4-overexpressing mice have higher BP
Systolic and diastolic (Fig. 1A) and mean BP (Supplemental Table S1) were lower in the RBP4-KO mice than in their WT littermates at 6 and 14 wk of age. BP of the RBP4-Tg mice at 12 to 14 wk was higher than in their WT littermates (Fig. 1B). HR was not different between genotypes (Supplemental Table S1).
Figure 1.
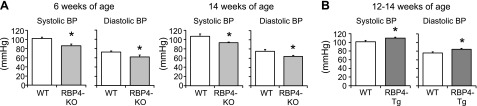
RBP4-KO mice have lower and RBP4-overexpressing mice have higher BP than WT mice. Systolic and diastolic BP in WT and RBP4-KO (A) and RBP4-Tg (B) mice (n = 8–14/genotype). *P < 0.05 vs. WT.
Endothelium-dependent vasodilatation is increased in RBP4-KO and reduced in RBP4-Tg mice
To determine whether these differences in BP were caused by vascular effects, we tested endothelium-dependent and -independent vasodilatation of isolated carotid arteries of the RBP4-KO and RBP4-Tg mice. There was no difference in maximum PE-induced preconstriction between the RBP4-KO (Fig. 2A) or RBP4-Tg mice (Fig. 2B) and their respective WT littermates. Carbachol-induced vasodilatation was more pronounced in the RBP4-KO than in the WT mice (Fig. 2C) and was impaired in the RBP4-Tg mice (P < 0.05 at 60 nM and for EC50; Fig. 2D). Endothelium-independent vasodilatation was not different among genotypes (data not shown).
Figure 2.

RBP4-KO and RBP4-Tg mice show reciprocally altered endothelium-dependent vasodilatation and eNOS phosphorylation. Maximum preconstriction response to 10−5 M PE of carotid arteries ex vivo from WT and RBP4-KO, P = 0.41 (A) and RBP4-Tg mice, P = 0.91 (B). n = 7–10/group. Vasorelaxation of carotid arteries ex vivo from WT and RBP4-KO (C) and RBP4-Tg (D) mice in response to carbachol (n = 7–10/group). Aortic eNOS protein and eNOSSer1177 phosphorylation in WT and RBP4-KO (E) and RBP4-Tg (F) mice at 12–14 wk of age (n = 4–6/group). *P < 0.05 vs. WT.
These differences in endothelium-dependent effects on vasodilatation could be eNOS dependent. Therefore we studied arterial eNOS phosphorylation. In RBP4-KO aorta, basal eNOSSer1177 phosphorylation, reflecting eNOS activation, was enhanced to ∼150% of WT (P = 0.01; Fig. 2E) with no change in total eNOS protein. eNOS phosphorylation in aorta of the RBP4-Tg mice was reduced to ∼80% of that in the WT mice (Fig. 2F), with no change in total eNOS.
RBP4-KO protects against Ang-II-induced hypertension
To assess whether hypertension mediated by impaired eNOS function would be influenced by RBP4-KO, we infused Ang-II (1 mg/kg/d for 2 wk), resulting in 22 mmHg lower systolic BP in the RBP4-KO mice than in the WT (Fig. 3A), although the relative increase in BP over saline infusion was ∼30% in both genotypes. Serine1177 phosphorylation of aortic eNOS was markedly decreased with Ang-II treatment in both the WT and RBP4-KO mice. eNOS phosphorylation tended to remain higher in the RBP4-KO mice than in the WT (P = 0.18; Fig. 3B). Total eNOS expression was not different between the groups (Supplemental Fig. S1).
Figure 3.
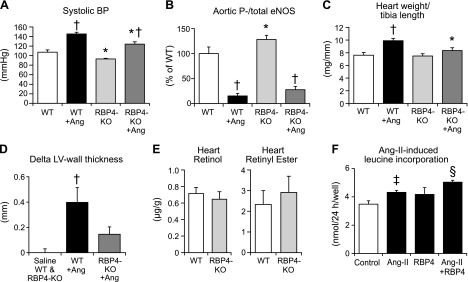
RBP4-KO mice are partially protected from Ang-II-induced hypertension. RBP4 promotes Ang-II-induced cardiac hypertrophy. Effect of Ang-II treatment on BP (A) and aortic eNOSSer1177 phosphorylation (B) and myocardial hypertrophy (C, D) in male mice. Ang-II (1 mg/kg/d) or saline was infused for 2 wk via subcutaneous minipumps starting at 10–12 wk of age. E) Cardiac retinol and RE contents per gram of tissue were not different between WT and RBP4-KO 5-month-old male mice (n = 7 per group). P = 0.56 for cardiac retinol, P = 0.58 for cardiac RE. F) Effect of RBP4 (50 µg/ml) on Ang-II (1 µM)-induced hypertrophy in H9C2-cardiomyocytes (n = 5–6/group). *P < 0.05 vs. WT, same treatment; †P < 0.05 vs. saline, same genotype; ‡P < 0.05 vs. control; §P < 0.05 vs. Ang-II.
Hearts of RBP4-KO mice are partially protected from Ang-II-induced hypertrophy
Ang-II treatment increased the heart weight and heart weight/tibia length ratio by 32% in the WT mice but had no significant effect in the RBP4-KO mice (Fig. 3C, and Supplemental Fig. S2). Likewise, Ang-II-treatment increased echocardiographic left ventricular wall thickness in WT mice but not in RBP4-KO mice (Fig. 3D). The RBP4-KO mice showed normal systolic left ventricular function (Supplemental Fig. S3). Histologically, the Ang-II-induced increase in cardiomyocyte diameter tended to be reduced in the RBP4-KO mice, although the difference was not statistically significant (Supplemental Fig. S4). Retinol and RE levels were not different in hearts of the RBP4-KO mice, compared with those in the hearts of the WT mice (Fig. 3E). To investigate whether the effect of RBP4 on hypertrophy is BP independent, we tested direct effects of RBP4 on cardiomyocyte hypertrophy in vitro. Ang-II increased cardiomyocyte leucine incorporation by 25%. Physiologic concentrations of RBP4 alone had no effect, but nearly doubled the Ang-II response (Fig. 3F).
RBP4-KO mice fed an HFD tend to have lower BP
Because RBP4-KO mice are more insulin sensitive and lowering RBP4 protects from HFD-induced insulin resistance (23, 24), we tested the BP response in the insulin-resistant state caused by HFD-induced obesity. The RBP4-KO mice showed similar weight gain while consuming an HFD for 3 months (Fig. 4A).
Figure 4.
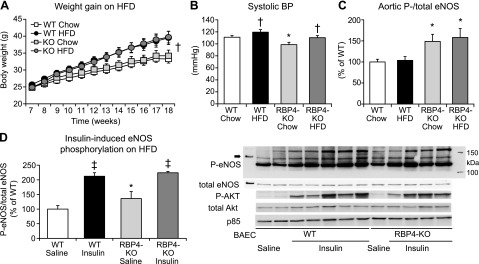
RBP4-KO does not protect from HFD-induced obesity or hypertension. Effect of chow vs. HFD on weight gain (A), BP (B), and ambient (C) and insulin-induced (D) aortic eNOSSer1177 phosphorylation in 18-wk-old WT and RBP4-KO mice. The mice were killed after withholding food overnight (C) or 15 minutes after intraperitoneal injection of saline or 6 mIU/g insulin in the unfed state (D). Bovine aortic endothelial cell (BAEC) lysates were run on the same gel as positive controls for eNOS. Twelve wk on an HFD [(A–C); n = 9–10/group; (D) n = 4–6/group]. *P < 0.05 vs. other genotypes, same diet; †P < 0.05 vs. same genotype, chow diet; ‡P < 0.05 vs. same genotype, saline injection.
It is noteworthy that both WT and RPB4-KO mice had an ∼10% increase in BP (Fig. 4B). In WT mice, the systolic BP increased from 111 ± 3 mmHg on the chow diet to 119 ± 4 mmHg on the HFD. In RBP4-KO mice, BP was lower on a chow diet (99 ± 4 mmHg) and increased to 110 ± 3 mmHg on the HFD, a reading that tended to be lower than that in WT mice on the HFD (P = 0.09) and was similar to that in WT mice on chow.
eNOS phosphorylation was increased by ∼50% in aorta of RBP4-KO mice on either chow or HFD, compared with that in WT mice (Fig. 4C), consistent with increased eNOS activation in the younger RBP4-KO mice (Fig. 2E). HFD did not alter aortic eNOS protein or eNOSSer1177 phosphorylation in either genotype (Fig. 4C). eNOS phosphorylation was increased 50% in saline-injected KO mice, compared with that in WT mice on HFD (Fig. 4D). Insulin-induced eNOS phosphorylation was not different between genotypes (Fig. 4D).
DISCUSSION
Our study provides the first evidence of a role of RBP4 in BP regulation. BP was lower in mice lacking RBP4 and higher in mice overexpressing it. This result supports the possibility of a direct effect of elevated serum RBP4-levels on BP (9) and endothelial function in humans (11, 12). Furthermore, large epidemiologic studies show a high predictive value of RBP4 for the incidence of myocardial infarction and association with coronary artery disease (7, 10, 25). It has been unclear whether increased serum RBP4 levels solely reflect metabolic abnormalities leading to hypertension and CVD, or whether RBP4 impairs vascular reactivity independent of its effects on glucose-insulin homeostasis. This uncertainty is due both to the lack of mechanistic data and to the contradictory findings in clinical studies, which may result from differences in patient populations or methodological deficiencies in RBP4 assays (26). Therefore, our results advance the understanding of the impact of RBP4 on CVD by demonstrating a causal role for chronic absence or elevation of RBP4 on vascular endothelial function and BP.
In our experiments, RBP4-KO mice were protected from Ang-II-induced hypertension. Although Ang-II infusion increased BP in both RBP4-KO and WT mice, the increased systolic BP was 22 mmHg lower in RBP4-KO mice and remained in the physiological range. This difference could have a significant impact on clinical end points if comparable responses are achieved in humans. Similar conclusions derive from our observation that an HFD increased BP in both the WT and RBP4-KO mice, but BP in the RBP4-KO mice fed an HFD did not exceed levels in WT mice fed chow. Because we did not perform invasive BP measurement by radiotelemetry, we may have missed differences in circadian BP variations between the models. However, we decided that telemetry would not be the optimal technique for this work, as discussed in Materials and Methods.
The greater effect of Ang-II treatment than an HFD on BP elevation in both WT and RBP4-KO mice is consistent with findings in many studies that showed a greater increase in BP with Ang-II (27) than with an HFD (28). The greater beneficial effect of RBP4-KO on BP elevation resulting from Ang-II treatment compared to an HFD may lie in the finding that RBP4-KO maintains eNOS phosphorylation, even with Ang-II treatment. eNOS phosphorylation was dramatically reduced in our studies in WT mice with Ang-II treatment but not with an HFD. The different responses may also reflect the different pathophysiologies of high BP in these models. HFD-induced hypertension is a multifactorial disease based on complex hormonal changes and a proinflammatory state, whereas Ang-II increases BP mainly through increased renal sodium reabsorption and direct vasoconstriction.
Nevertheless, the effects of both Ang-II treatment and obesity on BP appear to be RBP4 independent, given that they occurred in RBP4-KO mice. It is noteworthy that the absence of RBP4 reduced BP and RBP4 elevation alone increased BP. Therefore, RBP4 is a potential novel target for treatment of hypertension. Our data do not exclude the possibility that these effects are retinol dependent, as retinol serum levels are lower in the RBP4-KO mice (Supplemental Fig. S5) and higher in RBP4-Tg mice [57 vs. 18.1 µg/dl in the WT mice; (29)]. In human hypertension, there are no data regarding whether excess or deficient vitamin A directly affects BP. Vitamin A supplementation has been associated with an increased risk for CVD in randomized trials (30, 31), but data also show an inverse correlation of vitamin A levels with cardiovascular mortality (32). Correlation with BP was not reported in those studies. Experimental data on retinoid’s effects on endothelial function and atherosclerosis are also inconsistent [for review, see Rhee et al. (33)].
The fat and vitamin A content of the diets fed to the RBP4-KO mice and their WT littermates was relatively high, to prevent vitamin A deficiency (34). These high levels are necessary because RBP4-KO mice cannot mobilize hepatic retinol stores and rely fully on recently absorbed dietary vitamin A to maintain normal tissue levels. On a vitamin A-sufficient diet (22 IU vitamin A per gram of food) the retinol levels in liver, kidney, spleen, testis, and ovary in a prior study were not different between WT and RBP4-KO mice of the same age as those in our study (35). As myocardial retinol levels in RBP4-KO mice have not been reported previously, we assessed myocardial retinol and REs in our animals and found no differences between the WT and RBP4-KO mice (Fig. 3E).
Our data suggest that RBP4 also exerts direct effects, independent of BP, on myocardial hypertrophy, because it enhanced Ang-II-induced hypertrophy in cardiomyocytes in vitro. This role for RBP4 is supported by the reduced cardiac hypertrophy in our RBP4-KO mice. Retinoids have been shown to protect against cardiac hypertrophy (36), but retinol and RE levels were similar in the hearts of the WT and RBP4-KO mice in our study and therefore do not explain the differences in hypertrophy in our model. Likewise, the differences cannot be explained by alterations in local RBP4 expression, as we did not detect any cardiac RBP4 protein in lean or obese mice (data not shown). Therefore, enhanced cardiac hypertrophy appears to be a retinol-independent effect of serum RBP4, but the clinical implication is currently unknown. Results in one small study showed elevated serum RBP4 in patients with heart failure and systolic dysfunction (37), but to date, there are no clinical data correlating RBP4 levels with cardiac hypertrophy or diastolic dysfunction. As these are important clinical outcomes in patients with hypertension, evaluation of the relationship of RBP4 to cardiac function warrants further study. It may also be interesting to address the effect of RBP4-KO or lowering RBP4 on vascular remodeling and plaque morphology, because RBP4 induces inflammation in human endothelial cells (38) and in macrophages (4), which can promote foam cell formation.
To better understand the role of RBP4 in BP regulation, we investigated endothelial function and eNOSSer1177 phosphorylation in the RBP4-KO and RBP4-Tg mice. eNOS phosphorylation on the activating residue Serine1177 was higher in aorta of the RBP-KO mice and lower in aorta of the RBP4-Tg mice. Accordingly, endothelium-dependent vasodilatation was increased in the RBP4-KO and decreased in the RBP4-Tg carotid arteries compared to WT mice, whereas endothelial-independent vasodilatation was unchanged in either genotype compared to WT mice. Our findings may seem contradictory to those in a publication that showed that RBP4 stimulates the eNOS pathway in endothelial cells and in aortic rings in vitro (39). These differences may be attributable to the fact that those were acute studies in vitro, whereas we analyzed the chronic effects of in vivo treatments. The observed changes in eNOS signaling in our study seem to be independent of the RBP4 receptor Stra6, as we did not detect Stra6 mRNA expression in aorta of the WT or RBP4-KO mice (data not shown). This result is in line with results in a previous report demonstrating the absence of Stra6 expression in HUVECs and retinal endothelial cells (38) and with the fact that other actions of RBP4 (e.g., adipose tissue inflammation) are Stra6-independent (4).
Because we did not find a difference in maximum PE-stimulated vasoconstriction (Fig, 2A and B), we limited our studies to vasodilatation. For these studies, we used maximum PE preconstriction, which may have diminished the differences in vasodilation between genotypes. Although we measured eNOS phosphorylation and vasodilatation in different vessels, we think the alterations in eNOS phosphorylation may explain the effects on vasodilatation, because both the aorta and carotids are central arteries with similar endothelial properties. Furthermore, they are both affected in patients with diabetes or hypertension (40). The findings in eNOS phosphorylation and vasodilatation are complementary and consistent with the differences in systemic BP in the RBP4-KO and RBP4-Tg mice. Together, these observations provide evidence for alterations in endothelial function in these mice.
RBP4 is elevated in many people with metabolic syndrome (5–7) or hypertension (9). Our data show that RBP4 can play a direct role in elevating BP and increased serum RBP4 may lead to endothelial dysfunction, at least partly through attenuation of the NO-mediated vascular responses. We speculate that pharmacological strategies targeting RBP4 would be an effective therapeutic approach to increase vasodilatation, improve BP, and protect against adverse myocardial remodeling.
Supplementary Material
Acknowledgments
The authors thank William Blaner (Columbia Medical School, New York, NY, USA) for helpful discussions; Odile Peroni, Anna Lee, and Ed Hadro (Beth Israel Deaconess Medical Center, Boston, MA, USA) for technical assistance with mouse breeding, phenotyping, and euthanasia; Anthony Rosenzweig (Beth Israel Deaconess Medical Center) for use of his echo machine; and Pratik Aryal (Beth Israel Deaconess Medical Center) for kindly providing the recombinant RBP4 that was used for cell culture studies. This work was supported by U.S. National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases Grants R37DK43051, R24DK83938, and P30DK57521 (to B.B.K.); NIH National Heart, Lung, and Blood Institute Grants HL46457 and HL48743 (to T.M.); a postdoctoral Bayer fellowship from the German Cardiac Society and a research grant from MSD Sharp & Dohme, GmbH (to B.J.K.); an EMBO long-term fellowship (to P.P.); a research grant from the Manpei Suzuki Diabetes Foundation (to T.H.); and the Japan Heart Foundation and Uehara Medical Foundation (to T.S.). B.B. Kahn is an inventor on a patent related to RBP4 and insulin resistance.
Glossary
- Akt
protein kinase B
- Ang-II
angiotensin-II
- BP
blood pressure
- CVD
cardiovascular disease
- HFD
high-fat diet
- HR
heart rate
- KO
knockout
- PE
phenylephrine
- PSS
physiologic saline solution
- RBP4
retinol-binding protein 4
- RE
retinyl ester
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Blaner W. S. (1989) Retinol-binding protein: the serum transport protein for vitamin A. Endocr. Rev. 10, 308–316 [DOI] [PubMed] [Google Scholar]
- 2.Graham T. E., Yang Q., Blüher M., Hammarstedt A., Ciaraldi T. P., Henry R. R., Wason C. J., Oberbach A., Jansson P.-A., Smith U., Kahn B. B. (2006) Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med. 354, 2552–2563 [DOI] [PubMed] [Google Scholar]
- 3.Yang Q., Graham T. E., Mody N., Preitner F., Peroni O. D., Zabolotny J. M., Kotani K., Quadro L., Kahn B. B. (2005) Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 436, 356–362 [DOI] [PubMed] [Google Scholar]
- 4.Norseen J., Hosooka T., Hammarstedt A., Yore M. M., Kant S., Aryal P., Kiernan U. A., Phillips D. A., Maruyama H., Kraus B. J., Usheva A., Davis R. J., Smith U., Kahn B. B. (2012) Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol. Cell. Biol. 32, 2010–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meisinger C., Rückert I. M., Rathmann W., Döring A., Thorand B., Huth C., Kowall B., Koenig W. (2011) Retinol-binding protein 4 is associated with prediabetes in adults from the general population: the Cooperative Health Research in the Region of Augsburg (KORA) F4 Study. Diabetes Care 34, 1648–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qi Q., Yu Z., Ye X., Zhao F., Huang P., Hu F. B., Franco O. H., Wang J., Li H., Liu Y., Lin X. (2007) Elevated retinol-binding protein 4 levels are associated with metabolic syndrome in Chinese people. J. Clin. Endocrinol. Metab. 92, 4827–4834 [DOI] [PubMed] [Google Scholar]
- 7.Lambadiari V., Kadoglou N. P. E., Stasinos V., Maratou E., Antoniadis A., Kolokathis F., Parissis J., Hatziagelaki E., Iliodromitis E. K., Dimitriadis G. (2014) Serum levels of retinol-binding protein-4 are associated with the presence and severity of coronary artery disease. Cardiovasc. Diabetol. 13, 121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Hoek M., Dehghan A., Zillikens M. C., Hofman A., Witteman J. C., Sijbrands E. J. G. (2008) An RBP4 promoter polymorphism increases risk of type 2 diabetes. Diabetologia 51, 1423–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solini A., Santini E., Madec S., Rossi C., Muscelli E. (2009) Retinol-binding protein-4 in women with untreated essential hypertension. Am. J. Hypertens. 22, 1001–1006 [DOI] [PubMed] [Google Scholar]
- 10.Sun Q., Kiernan U. A., Shi L., Phillips D. A., Kahn B. B., Hu F. B., Manson J. E., Albert C. M., Rexrode K. M. (2013) Plasma retinol-binding protein 4 (RBP4) levels and risk of coronary heart disease: a prospective analysis among women in the nurses’ health study. Circulation 127, 1938–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park S. E., Kim D. H., Lee J. H., Park J. S., Kang E. S., Ahn C. W., Lee H. C., Cha B. S. (2009) Retinol-binding protein-4 is associated with endothelial dysfunction in adults with newly diagnosed type 2 diabetes mellitus. Atherosclerosis 204, 23–25 [DOI] [PubMed] [Google Scholar]
- 12.Solini A., Stea F., Santini E., Bruno R. M., Duranti E., Taddei S., Ghiadoni L. (2012) Adipocytokine levels mark endothelial function in normotensive individuals. Cardiovasc. Diabetol. 11, 103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bobbert T., Raila J., Schwarz F., Mai K., Henze A., Pfeiffer A. F. H., Schweigert F. J., Spranger J. (2010) Relation between retinol, retinol-binding protein 4, transthyretin and carotid intima media thickness. Atherosclerosis 213, 549–551 [DOI] [PubMed] [Google Scholar]
- 14.Ingelsson E., Lind L. (2009) Circulating retinol-binding protein 4 and subclinical cardiovascular disease in the elderly. Diabetes Care 32, 733–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muniyappa R., Montagnani M., Koh K. K., Quon M. J. (2007) Cardiovascular actions of insulin. Endocr. Rev. 28, 463–491 [DOI] [PubMed] [Google Scholar]
- 16.Michel T., Vanhoutte P. M. (2010) Cellular signaling and NO production. Pflugers Arch. 459, 807–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Förstermann U., Sessa W. C. (2012) Nitric oxide synthases: regulation and function. Eur Heart J 33, 829–837, 837a–837d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardin JM, Adams DB, Douglas PS, Feigenbaum H, Forst DH, Fraser AG, Grayburn PA, Katz AS, Keller AM, Kerber RE, Khandheria BK, Klein AL, Lang RM, Pierard LA, Quinones MA, Schnittger I; American Society of Echocardiography. (2002) Recommendations for a standardized report for adult transthoracic echocardiography: a report from the American Society of Echocardiography's Nomenclature and Standards Committee and Task Force for a Standardized Echocardiography Report. J. Am. Soc. Echocardiogr. 15, 275–290 [DOI] [PubMed] [Google Scholar]
- 19.Krege J. H., Hodgin J. B., Hagaman J. R., Smithies O. (1995) A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension 25, 1111–1115 [DOI] [PubMed] [Google Scholar]
- 20.Dantas A. P. V., Igarashi J., Michel T. (2003) Sphingosine 1-phosphate and control of vascular tone. Am. J. Physiol. Heart Circ. Physiol. 284, H2045–H2052 [DOI] [PubMed] [Google Scholar]
- 21.Stuck B. J., Lenski M., Böhm M., Laufs U. (2008) Metabolic switch and hypertrophy of cardiomyocytes following treatment with angiotensin II are prevented by AMP-activated protein kinase. J. Biol. Chem. 283, 32562–32569 [DOI] [PubMed] [Google Scholar]
- 22.Wongsiriroj N., Jiang H., Piantedosi R., Yang K. J. Z., Kluwe J., Schwabe R. F., Ginsberg H., Goldberg I. J., Blaner W. S. (2014) Genetic dissection of retinoid esterification and accumulation in the liver and adipose tissue. J. Lipid Res. 55, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan Y., Sun L.-Q., Kamal M. A., Wang X., Seale J. P., Qu X. (2011) Suppression of retinol-binding protein 4 with RNA oligonucleotide prevents high-fat diet-induced metabolic syndrome and non-alcoholic fatty liver disease in mice. Biochim. Biophys. Acta 1811, 1045–1053 [DOI] [PubMed] [Google Scholar]
- 24.Yu X., Watts L., Manchem P., Monia B., Bhanot S. (2008) Antisense reduction of retinol-binding protein 4 expression in liver and adipose tissues causes robust improvements in insulin sensitivity in diabetic and obese mice. 68th Scientific Sessions Meeting, Abstract 59-LB, http://professional.diabetes.org/Abstracts_Display.aspx?TYP=1&CID=71035 [Google Scholar]
- 25.Mallat Z., Simon T., Benessiano J., Clément K., Taleb S., Wareham N. J., Luben R., Khaw K.-T., Tedgui A., Boekholdt S. M. (2009) Retinol-binding protein 4 and prediction of incident coronary events in healthy men and women. J. Clin. Endocrinol. Metab. 94, 255–260 [DOI] [PubMed] [Google Scholar]
- 26.Graham T. E., Wason C. J., Blüher M., Kahn B. B. (2007) Shortcomings in methodology complicate measurements of serum retinol binding protein (RBP4) in insulin-resistant human subjects. Diabetologia 50, 814–823 [DOI] [PubMed] [Google Scholar]
- 27.Romero J. C., Reckelhoff J. F. (1999) State-of-the-Art lecture: role of angiotensin and oxidative stress in essential hypertension. Hypertension 34, 943–949 [DOI] [PubMed] [Google Scholar]
- 28.Kennedy A. J., Ellacott K. L. J., King V. L., Hasty A. H. (2010) Mouse models of the metabolic syndrome. Dis. Model. Mech. 3, 156–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quadro L., Blaner W. S., Hamberger L., Van Gelder R. N., Vogel S., Piantedosi R., Gouras P., Colantuoni V., Gottesman M. E. (2002) Muscle expression of human retinol-binding protein (RBP): suppression of the visual defect of RBP knockout mice. J. Biol. Chem. 277, 30191–30197 [DOI] [PubMed] [Google Scholar]
- 30.Törnwall M. E., Virtamo J., Korhonen P. A., Virtanen M. J., Taylor P. R., Albanes D., Huttunen J. K. (2004) Effect of alpha-tocopherol and beta-carotene supplementation on coronary heart disease during the 6-year post-trial follow-up in the ATBC study. Eur. Heart J. 25, 1171–1178 [DOI] [PubMed] [Google Scholar]
- 31.Cook N. R., Albert C. M., Gaziano J. M., Zaharris E., MacFadyen J., Danielson E., Buring J. E., Manson J. E. (2007) A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: results from the Women’s Antioxidant Cardiovascular Study. Arch. Intern. Med. 167, 1610–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brazionis L., Walker K. Z., Itsiopoulos C., O’Dea K. (2012) Plasma retinol: a novel marker for cardiovascular disease mortality in Australian adults. Nutr. Metab. Cardiovasc. Dis. 22, 914–920 [DOI] [PubMed] [Google Scholar]
- 33.Rhee E.-J., Nallamshetty S., Plutzky J. (2012) Retinoid metabolism and its effects on the vasculature. Biochim. Biophys. Acta 1821, 230–240 [DOI] [PubMed] [Google Scholar]
- 34.Quadro L., Blaner W. S., Hamberger L., Novikoff P. M., Vogel S., Piantedosi R., Gottesman M. E., Colantuoni V. (2004) The role of extrahepatic retinol binding protein in the mobilization of retinoid stores. J. Lipid Res. 45, 1975–1982 [DOI] [PubMed] [Google Scholar]
- 35.Quadro L., Blaner W. S., Salchow D. J., Vogel S., Piantedosi R., Gouras P., Freeman S., Cosma M. P., Colantuoni V., Gottesman M. E. (1999) Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 18, 4633–4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maier L. S. (2008) Vitamin A for the heart: progress for cardiac hypertrophy regression? Am. J. Physiol. Heart Circ. Physiol. 294, H588–H589 [DOI] [PubMed] [Google Scholar]
- 37.Chavarria N., Kato T. S., Khan R., Chokshi A., Collado E., Akashi H., Takayama H., Naka Y., Farr M., Mancini D., Schulze P. C. (2012) Increased levels of retinol binding protein 4 in patients with advanced heart failure correct after hemodynamic improvement through ventricular assist device placement. Circ. J. 76, 2148–2152 [DOI] [PubMed] [Google Scholar]
- 38.Farjo K. M., Farjo R. A., Halsey S., Moiseyev G., Ma J.-X. (2012) Retinol-binding protein 4 induces inflammation in human endothelial cells by an NADPH oxidase- and nuclear factor kappa B-dependent and retinol-independent mechanism. Mol. Cell. Biol. 32, 5103–5115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takebayashi K., Sohma R., Aso Y., Inukai T. (2011) Effects of retinol binding protein-4 on vascular endothelial cells. Biochem. Biophys. Res. Commun. 408, 58–64 [DOI] [PubMed] [Google Scholar]
- 40.Vlachopoulos C., Aznaouridis K., Stefanadis C. (2010) Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J. Am. Coll. Cardiol. 55, 1318–1327 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.