

Abstract
Dietary influences may affect microbiome composition and host immune responses, thereby modulating propensity toward inflammatory bowel diseases (IBDs): Crohn disease (CD) and ulcerative colitis (UC). Dietary n-6 fatty acids have been associated with UC in prospective studies. However, the critical developmental period when (n-6) consumption may induce UC is not known. We examined the effects of transiently increased n-6 consumption during pediatric development on subsequent dextran-sulfate-sodium (DSS)-induced acute murine colitis. The animals transiently became obese then rapidly lost this phenotype. Interestingly, mice were protected against DSS colitis 40 days after n-6 consumption. The transient high n-6-induced protection against colitis was fat type- and dietary reversal-dependent and could be transferred to germ-free mice by fecal microbiota transplantation. We also detected decreased numbers of chemokine receptor (Cxcr)5+ CD4+ T cells in the mesenteric lymph nodes (MLNs) of transiently n-6-fed mice. Further experiments revealed that anti-chemokine ligand (Cxcl)13 (the ligand of Cxcr5) antibody treatment decreased DSS colitis severity, implicating the importance of the Cxcr5-Cxcl13 pathway in mammalian colitis. Consecutively, we found elevated CXCL13 concentrations (CD: 1.8-fold, P = 0.0077; UC: 1.9-fold, P = 0.056) in the serum of untreated pediatric IBD patients. The human serologic observations supported the translational relevance of our findings.—Nagy-Szakal, D., Mir, S. A. V., Harris, R. A., Dowd, S. E., Yamada, T., Lacorazza, H. D., Tatevian, N., Smith, C. W., de Zoeten, E. F., Klein, J., Kellermayer, R. Loss of n-6 fatty acid induced pediatric obesity protects against acute murine colitis.
Keywords: diet, inflammatory bowel disease, microbiome, CXCL13
IBDs (CD and UC) affect over 1.5 million people in the United States alone with significant morbidity (1). There has been an emergence of IBDs over the past 5–6 decades associated with spread of industrialization around the world (2, 3). The incidence of the disease group peaks in young adulthood and prevalence continues to rise in both children (4–7) and adults (8). Although IBD is thought to be influenced by genetics, host immune dysfunction, mucosal barrier defects, and the gut microbiome (9, 10), its exact etiology remains unknown. However, nongenetic factors may be as or even more significant in the etiology of the disease group (11–13). The “developmental origins of disease” (DOD) hypothesis postulates that transient environmental exposures can induce critical changes in biologic structures during finite periods of development thereby modifying susceptibility to disorders later in life. IBDs are examples of human disorders to which the concept of DOD appears to strongly pertain (14, 15), although this notion has been rarely examined directly in the scientific literature (16).
Changes in dietary habits and nutrition have been implicated as potential causes for the rising incidence of IBD (17). A large-scale prospective nutritional study linked n-6 (or w-6) fatty acid consumption to the development of UC (18). However, clear results from human epidemiologic observations (even in prospective and well-controlled studies) are very difficult to obtain for obvious ethical and technical considerations (19). Therefore, the timing and nature of environmental factors critical to IBD development have remained largely unknown, as have the molecular mechanisms, which may uncover the environmental contribution to disease induction (16).
Environmental influences may trigger critical pathogenic changes at any time within an individual prior to the onset of disease, which can occur during a broad age range from fertilization to adulthood in case of IBD. Here, we examined the complex effects of transient high n-6 fat diet consumption during pediatric development on the young adult metagenome and immune system in a murine model of IBD.
MATERIALS AND METHODS
Animals, diets, and experimental design
Postnatal day (P) 21 male C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME, USA) received standard rodent diet (2920X; Harlan-Teklad, Madison, WI, USA) until P30. Then, mice were randomly assigned to artificial diets: low 12% caloric content corn oil/linoleic acid control diet (#102460; Dyets Inc., Bethlehem, PA, USA) or high n-6 polyunsaturated fatty acids (40% caloric content corn oil/linoleic acid, n-6 diet (#102459; Dyets Inc.) from P30 to P80 (Supplemental Table 1). Following this 50-day period, the animals were reversed to control diet for 10 (n6-R10) or 40 (n6-R40) days. High milk fat (MF), high cholesterol diet (#112734; Dyets Inc.) was used with the same feeding protocol to examine fat-dependent effects. Fat-to-body weight ratio was interrogated on P90 and P120 by QMRI. Six-week-old Swiss-Webster germ-free (GF) mice were derived by cesarean section under GF conditions at Taconic Farms Inc. (Hudson, New York, USA) and delivered with specific GF shipping for the microbiota transfer experiments.
All mice were housed in our specific pathogen-free animal facility during the experiment.
All applicable institutional and governmental regulations concerning the ethical use of animals were followed. The protocol was approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Dextran sulfate sodium exposure
Susceptibility to colitis was tested by administering 2 or 3% (wt/vol) DSS (MW = 36,000–50,000, MP Biomedicals, LLC, Solon, OH, USA) in the drinking water at P90 or P120 ad libitum for 5 days followed by regular water for an additional 1–3 days. DSS of this molecular weight induces diffuse colitis from cecum to distal large bowel (20). The animals were weighed daily and colonic length measurements were performed at the end of the experiments following CO2 asphyxiation. Weight loss during DSS administration in mice has been shown to correlate well with molecular and histologic outcome measures of colitis severity (21–23). Therefore, we decided to follow weight loss and colon length as the primary outcomes to determine colitis severity in our DSS experiments. Mice with more than 25% weight loss during DSS challenge had to be euthanized according to our protocol (i.e., mortality group).
Colons were longitudinally transected (Swiss roll technique, which provides whole length assessment of the colon) and processed for standard hematoxylin-eosin staining after fixation in 10% formaldehyde. Histological severity of inflammation was determined based upon an established colitis recording system scored blindly by a pathologist (17).
Tissue collection
At the end of the feeding periods, mice were euthanized by CO2 inhalation between 11:00 am and 2:00 pm without any previous food restriction. The colons were placed on ice, transected longitudinally, cleansed from feces, washed with ice cold normal saline, followed by the collection of colonic mucosa with a microscope slide (24) (excluding the cecum). The mucosal scrapings were flash frozen on dry ice and stored at −80°C as described earlier (23).
Microbiota transplantation
Cecal contents were pooled from n6-R40 or C120 littermates. Cecal extracts were suspended in PBS (2.5 ml per cecum) and were administered orally by gavage (0.1 ml per mouse) immediately to sterilely packed 6-week-old Swiss-Webster GF male GF mice (Taconic) as described by Vijay-Kumar et al. (25). Transplanted mice were fed with control diet for 5 days, and then 3% DSS was given to induce acute colitis. Body weight was measured daily. The animals were killed and tissues were collected as above.
DNA extraction for microbial studies
Mucosal scrapings were centrifuged at 14,000 rpm for 30 seconds and resuspended in 500 μl RLT buffer (Qiagen, Valencia, CA, USA) (with β-mercaptoethanol). Sterile 5 mm steel beads (Qiagen) and 500 μl sterile 0.1 mm glass beads (Scientific Industries, Incorporated, New York, NY, USA) were added for complete bacterial lyses in a Qiagen TissueLyser (Qiagen), run at 30 Hz for 5 minutes. Samples were centrifuged briefly and 100 μl of 100% ethanol was added to a 100 μl aliquot of the sample supernatant. This mixture was added to a DNA spin column, and DNA recovery protocols were followed as instructed in the QIAamp DNA Mini Kit (Qiagen) starting at step 5 of the tissue protocol. DNA was eluted and diluted to a final concentration of 20 ng/μl.
Massively parallel bacterial tag-encoded FLX-titanium amplicon pyrosequencing
Bacterial tag-encoded FLX-titanium amplicon pyrosequencing (bTEFAP) was performed as described previously (26). bTEFAP utilizes titanium reagents and procedures, and a 1-step PCR, mixture of Hot Start and HotStar high-fidelity taq polymerases, and amplicons originating from the 27 F region numbered in relation to Escherichia coli rRNA. The bTEFAP procedures were performed at the Research and Testing Laboratory (www.researchandtesting.com; Lubbock, TX, USA).
Bacterial diversity data analysis
All failed sequence reads, low-quality sequence ends and tags, and sequences shorter than 300 bp were removed, and any nonbacterial ribosome sequences and chimeras were depleted using custom software described previously (26) and the Black Box Chimera Check software B2C2 (described and freely available at http://www.researchandtesting.com/B2C2.html). This process provided 4303 to 8871 filtered sequences in the individual mucosal DNA samples, which were queried using a distributed BLASTn .NET algorithm (27) against a database of high quality 16S rRNA bacterial sequences derived from NCBI. Database sequences were characterized as high-quality based upon similar criteria as described for RDP version 9 (http://rdp.cme.msu.edu/) (28). Using a .NET and C# analysis pipeline the resulting BLASTn outputs were validated using taxonomic distance methods and compiled as described previously (26). Sequences with identity scores greater than 97% (<3% divergence) were resolved at the species level, between 95 and 97% at the genus level, between 90 and 95% at the family level, between 85 and 90% at the order level, between 80 and 85% at the class level, and below this to the phylum level (77 and 80%). Statistical tests including Principle Component Analysis using Unifrac-based distances, ANOVA with Tukey-Kramer post hoc, and hierarchal clustering (Wards minimum variance clustering with Manhattan distances) to evaluate microbiome results were evaluated using NCSS 2007 (Kaysville, UT, USA).
Flow cytometry
To conduct lymphocyte population analysis, MLNs and spleens underwent mechanical disruption, erythrocyte lysis, and preparation for flow cytometry as described previously (29). Lymphocytes were labeled for the markers CD4 and CXCR5 (BD Biosciences, San Diego, CA, USA). Flow cytometry was performed using a FACSCanto instrument (BD Biosciences), and the data was analyzed using FlowJo software (Treestar, Ashland, OR, USA).
In vivo chemokine blocking
C57BL/6 mice were treated with 0.2 mg of neutralizing CXCL13 antibody) (#MAB470; R&D Systems, Minneapolis, MN, USA) or isotype control antibody (#MAB006; R&D Systems). Both mouse antibodies were delivered by intraperitoneal injection in 1 mg/ml concentration dissolved in PBS. To examine the effect of CXCL13 antibody on DSS-induced colitis model, antibodies were administered on the first, third, and fifth days (3 times) after initiating 3% DSS.
Human samples and ELISA
Thirty-three pediatric patients were recruited prior to endoscopy following informed consent though the institutional review board-approved tissue bank of the Pediatric Inflammatory Bowel Disease Consortium Registry at the Baylor College of Medicine (H-17654). Sera of 12 UC, 11 CD, and 10 healthy controls were used for quantification of circulating CXCL13 levels. Pediatric controls included children who underwent colonoscopic evaluation for diagnoses of hematochezia, diarrhea, or abdominal pain, but whose endoscopy was grossly and histologically normal. BD Vacutainer CPT Cell Preparation Tube with Sodium Citrate (BD Biosciences, San Jose, CA, USA) was used to isolate peripheral blood mononuclear cells from peripheral blood leucocytes. Peripheral blood leucocytes were centrifuged at 1000 g for 10 minutes and stored at −80°C. QIAamp DNA mini kit (Qiagen) was utilized on the isolated peripheral blood mononuclear cells to retract DNA. Plasma concentrations of CXCL13 were quantified by Quantikine ELISA methodology according to the manufacturer’s instructions (#DCX130; R&D Systems). All samples were measured in duplicate. Color intensity of the assay was measured by a standard ELISA microplate reader. Color intensity correlates with the amount of bound CXCL13. Quantikine kit standards were used for generation of standard curves.
Additional statistical and bioinformatic analysis
Unpaired 2-tailed Student’s t tests, correlation and odds ratio calculations were utilized in the group comparisons where statistical significance was declared at P < 0.05. Error bars represent sem. Fatigo functional enrichment analysis (http://babelomics3.bioinfo.cipf.es/) was used to identify gene ontology classifications significantly over- or underrepresented adjusted (P < 0.05 in this case) among the gene lists.
RESULTS
Reversal from n-6 fat diet protected against colitis
Mice fed for 50 days with high n-6 fat diet exhibited significantly higher body fat compared with controls. This body composition difference was lost following 40 days of reversal (n6-R40; Fig. 1A). In the meantime, dietary reversal from high n-6 fat diet induced prolonged protection against DSS-induced colitis. The n6-R40 animals displayed lower body weight loss (Fig. 1B) and lower histologic severity of colitis than control (Fig. 1C–E). These findings were replicated in independent experiments.
Figure 1.

n-6 fat diet-dependent protection against colitis. A) qMRI measurements showed significantly increased fat composition following n-6 fat dietary intake in mice (n6-P80) compared with controls (C-P80), which diminished after 40 days of reversal (C-P120, compared with n6-R40) (n = 14, 8, 15, 11, respectively). ns, not significant. B) However, 5 days following 5-day DSS exposure (2%), the n6-R40 group lost less weight compared with control (C-P120) (n = 5-5). C–E) n6-R40 group had milder tissue damage upon DSS challenge compared with controls. C) Histologic severity of colitis was higher in control compared with n6-R40. D, E) A greater degree of tissue damage and inflammatory cell infiltration was observed in control colons compared with n6-R40 (magnification ×200). Error bars represent sem. *P < 0.05, **P < 0.01, *** P < 0.001, unpaired Student’s t test.
Protection against colitis was n-6 and reversal dependent
We were interested to investigate whether the colitis protective effect was specific to n-6 or whether the transient pediatric consumption of any type of fat would induce a similar phenotype. Using a high MF diet (Supplemental Table 1) instead of n-6 fat, we observed the opposite response. The transiently MF fed mice lost more weight upon DSS challenge than controls (Supplemental Fig. 1A). Thereafter, we sought to establish whether exposure to high n-6 is sufficient to protect against colitis, or if dietary reversal to control diet is necessary to establish the phenotype. Without dietary reversal, mice had a similar degree of weight loss as controls upon DSS challenge at both P90 (60 days of n-6 feeding) and P120 (90 days of n-6 feeding) (Supplemental Fig. 1B, C).
The velocity and extent of weight loss was similar in both the n-6 and milk-fat reversal groups (Supplemental Fig. 1D).
n6-reversal did not affect other models of colitis
We examined the effects of the transient high n-6 feeding in both IL-10−/− mice (30), and in the adoptive transfer model of CD4+, CD45RBhi+ T cells, into Rag1−/− mice (31). The nutritional intervention did not have any significant effect on these chronic models of colitis (data not shown).
The gut microbiome of n6-reversed mice transmitted the colitis suppression
We were interested to elucidate whether the gut microbiome of n6-R40 mice may be functionally modified to transfer protection against chemically induced colitis. We collected fecal material from transiently high n-6 fat-fed and control mice and transferred it into GF mice (fecal microbiota transplantation). The transplanted animals were then exposed to DSS to examine the effects of the dietary intervention induced microbiome changes on acute colitis. The recipients of the n6-R40 microbiome had a 100% survival rate compared with a 60% survival in controls (Fig. 2A). Colons retained their lengths and exhibited less severe colitis even in the survived n6-R40 mice compared with controls (Fig. 2B, C).
Figure 2.
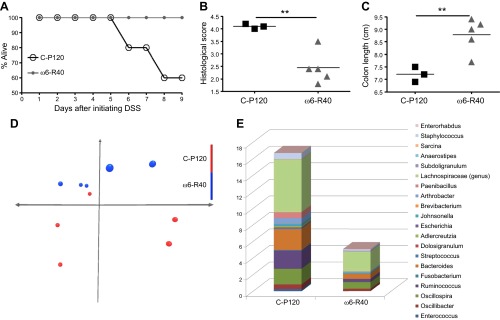
Microbial changes following transient n-6 fat diet. A–C) GF murine recipients of feces from the reversed high-fat dietary group (n6-R40) were protected against colitis. After initiation of DSS treatment, (A) increased mortality in the control group was observed (C-P120) compared with the n6-R40 group. B) Histologic colitis severity was less and (C) colons length remained longer in n6-R40 compared with C-P120. D) Unweighted principal component analysis of the colonic mucosal microbiomes showed separation between the experimental groups, which persisted after 40 days of reversal from high n-6 fat diet (n = 5, 5). E) Genera-level (most abundant 20) microbiome separation between the n6-R40 and control groups. Only Enterococcus abundance was statistically significantly lower in the n6-R40 group (Supplemental Table 2). **P < 0.01, unpaired Student’s t test.
The microbiome of n6-reversed mice segregated from control
Analysis of the gut microbiome revealed separation between the dietary groups. Unweighted principal component analysis of colonic mucosal microbiomes showed a segregation of mice transiently fed with high n-6 fat diet from controls even after 40 days of dietary reversal (Fig. 2D). Upon examination of the 20 most abundant genera in the microbiomes (Fig. 2E), only the Enterococcus genus showed a significant decrease in n6-R40 group compared with control (P = 0.0079) (Supplemental Table 2). At the species level, the abundance of Hespellia porcina, Enterococcus faecalis, Clostridium innocuum, and Oscillibacter species decreased in the reversal group (n6-R40) compared with control (P = 0.0317, 0.0159, 0.0159, and 0.0159, respectively) (Supplemental Table 2).
Cxcr5+ CD4+ T cells were suppressed in n6-reversed mice
We next explored how the colonic epithelium/mucosa was influenced by a transient consumption of high n-6 fat diet. DNA methylation specific amplification microarrays and gene expression microarrays on isolated colonic mucosa were employed to interrogate persistent diet-induced epigenetic changes (32). Even though these high-throughput approaches did not yield statistically significant results (data not shown), we observed a small reduction of Cxcr5 expression in the colonic mucosa of n6-reversed mice. We hypothesized that Cxcr5 transcripts may have originated from mucosa associated leukocytes derived from MLNs rather than from epithelial cells (which generally comprise >90% of the cell population in our colonic mucosal scrapings). Flow cytometric analysis of different T-cell subsets confirmed a significant reduction in the numbers of Cxcr5+ CD4+ T cells in the MLNs of n6-R40 animals in independent experiments (Fig. 3A, B).
Figure 3.

Cxcr5-Cxcl13 immunologic pathway revealed to play an important role in DSS-induced colitis upon transient n-6 fat diet. A, B) Flow cytometry showed significantly decreased number of Cxcr5+ CD4+ T cells in the MLNs, but not in the spleens (SPL) of n6-R40 compared with C-P120 (n = 3, 3 animals, 2 MLNs per mice, altogether 6 lymph nodes per group). C–E) Cxcl13 antibody (ab) treatment protected mice against DSS-induced colitis (n = 5, 5). Arrows indicate intraperitoneal ab treatment. Cxcl13 ab-treated mice had a tendency for decreased weight loss during DSS challenge. This finding was associated with longer colons and decreased histologic severity of colitis in the Cxcl13 ab-treated group compared with controls. F) Significantly increased CXCL13 concentration was measured in serum of treatment naïve human pediatric patients with CD, with a similar trend in UC. Error bars represent sem. *P < 0.05, **P < 0.01, unpaired Student’s t test.
Cxcr5 ligand B-lymphocyte chemoattractant (or Cxcl13) modulated colitis susceptibility
The observed decrease in the number of Cxcr5+ CD4+ T cells implicated the importance of Cxcr5 and its ligand B-lymphocyte chemoattractant (or Cxcl13) in murine acute colitis. Therefore, we examined the effect of Cxcl13 antibody treatment in the severity of DSS-induced colitis. Cxcl13 antibody-treated mice had a tendency to lose less weight during DSS challenge (Fig. 3C). Additionally, we observed increased colonic length and decreased colitis severity in the Cxcl13 antibody-treated mice compared with isotype controls (Fig. 3D, E), indicating that the inhibition of Cxcr5 signaling is protective against murine acute colitis.
CXCL13 levels in the plasma of patients with IBD
Finally, we sought to establish whether the CXCR5-CXCL13 pathway may be activated in human IBD based on our murine experiments. CXCL13 was measured in the blood of untreated pediatric patients with UC, CD, and controls. Plasma samples were collected at the time of diagnostic endoscopy. Plasma concentration of CXCL13 was significantly elevated in children with treatment naive CD with a similar trend in UC, compared with controls (Fig. 3F).
DISCUSSION
Western diets (usually containing high fat, especially n-6 fatty acid) have been proposed to increase the risk of IBDs (15). Dietary n-6 and its metabolites have been shown to have proinflammatory properties and to exaggerate immunologic responses in animal models (33). The increased intake of essential n-6 fatty acid, such as linoleic acid, has been associated with UC in a prospective human trial (18). These findings are supported in studies on rodent models of colitis (34). In the meantime, the effects of transient pediatric exposure to high n-6 fatty acid on successive colitis susceptibility have not been examined. Early adulthood is the peak for IBD presentation, which indicates the potential importance of pediatric dietary exposures (including increased n-6 consumption) in the developmental origins of the diseases. In this study, we examined how n-6-induced transient obesity during pediatric development may impact colitis in young adult mice. On the contrary, to our expectations, we found that reversal from n-6 induced pediatric obesity protected mice from acute colitis in young adulthood. The colitis protection was exclusive to the acute/chemical injury model, because chronic colitis severity was not modulated by the same dietary intervention in both IL-10−/− mice (30), and in the adoptive transfer model of CD4+, CD45RBhi+ T cells into Rag1−/− mice (31).
The acute colitis protection was specific to n-6 (corn oil), at least in comparison with saturated fat (MF). This finding is consistent with recent work showing that saturated fatty acids (such as palm oil, which is similar to MF in fatty acid composition) augmented small intestinal inflammation in a CD relevant murine model independently of obesity (35). Additionally, Devkota et al. found that saturated fat, but not polyunsaturated fat, triggers colitis in genetically susceptible mice in association with the bloom of Bilophila wadsworthia (36). Indeed, clinical studies indicate that the type of dietary fat consumed rather than obesity itself is risk factor for the development of IBD (15, 37, 38). However, with the experiments in this work, it cannot be ruled out that other immunomodulator substances both in corn oil and MF may have played a role in the observed phenotype modifications. It is important to highlight that the phenotype observed in our work was also dependent on the cessation of increased n-6 consumption, because obese mice during high n-6 feeding were not protected from DDS-induced colonic inflammation. Importantly, the velocity and extent of weight loss was similar in both the n-6 and MF reversal groups. Therefore, the colitis protection by transient n-6 supplementation cannot be explained simply on the basis of weight loss. Rather, n-6 supplementation during mammalian pediatric development appears to specifically induce a less colitis prone microbiome and host immunophenotype in a prolonged fashion.
Genetic work indicates that the development of IBD can be influenced by diverse (over 163 susceptibility loci) (39), potentially interactive genetic polymorphisms (40), each of which contribute to the diseases with low penetrance. Monozygotic twin and other epidemiologic studies underscore the involvement of primarily nongenetically modulated, environmentally responsive, intercalating biologic systems (16). The disease-specific disruption of a single biologic system, such as the microbiome, appears to be relatively limited in IBD (41, 42), in agreement with the genetic observations. Rather, the individually small, but interactive dysregulation of multiple biologic systems within the host-microbiome network (43) may lead to the onset of IBD. Therefore, if one wishes to explore the consequences of a potentially critical environmental change in respect to IBD development, then a parallel multi-omic approach is warranted (10). We set out to examine metagenomic, colonic mucosal, and immunologic associations of transient n-6 feeding-induced acute colitis protection. Importantly, the colitis protection could be transmitted by the fecal microbiome into GF mice, underscoring the participation of the microbiome in the dietary intervention-induced phenotype. The different genetic background of GF recipients in comparison with the donor mice (used in all the other experiments) highlights the translational relevance of the microbiome findings by overarching the boundaries of single mouse strains. Enterococcus was specifically decreased in the colonic mucosal microbiome of the n6-R40 group (Fig. 2E). Recently Enterococcus has been shown to trigger colonic inflammation in mouse model experiments (44, 45). Additionally, increased abundance of Enterococcus was found in patients with UC, and it correlated with disease activity and severity (46). These observations support the potential importance of the n-6 fat reversal-induced decrease of mucosal Enterococcus to participate in colitis protection.
With respect to the colonic mucosa, we did not detect significant epigenetic (DNA methylation) and transcriptional changes associated with the examined dietary intervention. However, a trend for decreased mucosal Cxcr5 expression was found in the reversed animals. Count to threshold values for Cxcr5 were very low, suggesting that the signal may have been originated from mucosa-associated leukocytes derived from MLNs rather than from epithelial cells (which generally comprise >90% of the cell population in the colonic mucosal scrapings) (23). Multiparameter flow cytometric analysis of T-cell subsets revealed a significant reduction of Cxcr5+ CD4+ T cells in the MLN of the high n-6-reversed animals. Cxcr5 and its ligand B-lymphocyte chemoattractant (or Cxcl13) has been shown to orchestrate lymphoid organogenesis and the migration of CD4+ T cells from the mantle zone to the more central regions in lymphoid follicles. The expression of Cxcr5 on CD4+ T cells is induced by the interaction of OX40 (or CD134) and its ligand OX40L, thereby serving as an upstream event for the Cxcr5-dependent T-cell migration (49, 50). Constitutive OX40/OX40L interaction stimulates autoimmune-like diseases, such as interstitial pneumonia and spontaneous colitis (51). Inhibition of the OX40/OX40L interaction and secondary Cxcr5 expression in CD4+ T cells decreased the severity of DSS-induced colitis in mice (52). CXCR5+, IL-21 producing CD4+ T lymphocytes have been shown to play an important role in IBD (53). All these publications indicated that CXCR5-CXCL13 pathway may play in an important role in controlling mammalian intestinal inflammation. Therefore, we examined the effects of anti-Cxcl13 (the ligand of Cxcr5) immunotherapy in modulating DSS colitis. Anti-Cxcl13 treatment was protective. Consequently, we examined CXCL13 levels in treatment naïve IBD patients. The plasma concentration of CXCL13 was significantly elevated in children at the diagnosis of CD with a similar trend in UC compared with controls. This result was consistent with our prediction that the CXCL13-CXCR5 pathway may be important in the pathology of IBD. The observed elevation of serum chemokines was not mirrored by gene expression changes at the intestinal mucosal level. Our recent work with treatment naïve pediatric UC colon biopsy samples (54), and the observations of others in untreated ileal mucosal samples of pediatric IBD patients (43) has not detected differences in CXCR5 or CXCL13 expression compared with controls. These results coincide with our findings in the murine model system of the present work, indicating that the CXCL13-CXCR5 pathway is more relevant for leukocyte than epithelial signaling with respect to IBD development. The lack of colitis protection in the chronic murine models suggests that this specific chemokine signaling is more important in modulating the onset and flares of disease rather than the maintenance of chronic inflammation in IBD.
Chemokines have been recognized as novel therapeutic targets for IBD, but CXCL13 specifically was not implicated (55). Our results may set the stage for developing CXCL13 directed biologic treatments for the disease group in the future. Interestingly, tofacitinib is an emerging Janus kinase inhibitor utilized in IBD therapy (56), which has been observed to suppress CXCL13 concentration in synovia of rheumatoid arthritis patients. This finding supports our predicament that the CXCR5-CXCL13 pathway may be a target for future biologic treatments in IBD (57).
In summary, transient high n-6 diet during pediatric development in mice induced prolonged protection against murine colitis, which associated with persistent colonic mucosal microbiome and lymphoid organ composition changes. Our study underscores that transient dietary changes during critical periods of mammalian development can lead to the prolonged and concomitant modulation of the host-microbiome network relevant for intestinal inflammation. These findings may bare important implications for the nutritional developmental origins of IBD and promote the future development of novel preventive and therapeutic solutions for this disease group.
Supplementary Material
Acknowledgments
The authors thank the Gutsy Kids Fund, including philanthropic donation from the Karen and Brock Wagner family, and the support of the Houston Men of Distinction. The authors also acknowledge support from the U.S. National Institutes of Health for the Texas Medical Center Digestive Diseases Center (P30 DK56338). Measurements of body composition were performed in the Mouse Metabolic Research Unit at the U.S. Department of Agriculture/Agricultural Research Service (USDA/ARS) Children’s Nutrition Research Center, which is supported by funds from the USDA ARS (www.bcm.edu/cnrc/mmru). The authors acknowledge the assistance of the Mouse Metabolic Research Unit (Baylor College of Medicine) staff and faculty in performing these measurements. D.N.S. and R.K. developed the study concept and design; D.N.S., S.A.V.M., R.A.H., S.E.D., T.Y., H.D.L., N.T., E.Z., J.K., and R.K. acquired the data; D.N.S., R.A.H., S.E.D., and R.K. analyzed and interpreted the data; D.N.S. and R.K. drafted the manuscript; H.D.L., N.T., C.W.S., and R.K. critically revised the manuscript for important intellectual content; R.A.H., S.E.D., and R.K. performed statistical analysis; R.K. obtained funding; C.W.S. and R.K. provided material support; and T.Y., H.D.L., and N.T. provided technical support. The authors declare no conflicts of interest.
Glossary
- bTEFAP
bacterial tag-encoded FLX-titanium amplicon pyrosequencing
- CD
Crohn disease
- CXCL
chemokine ligand
- CXCR
chemokine receptor
- DOD
developmental origins of disease
- DSS
dextran-sulfate-sodium
- GF
germ-free
- IBD
inflammatory bowel disease
- MF
milk fat
- MLN
mesenteric lymph node
- P
postnatal day
- UC
ulcerative colitis
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Gunnarsson C., Chen J., Rizzo J. A., Ladapo J. A., Lofland J. H. (2012) Direct health care insurer and out-of-pocket expenditures of inflammatory bowel disease: evidence from a US national survey. Dig. Dis. Sci. 57, 3080–3091 [DOI] [PubMed] [Google Scholar]
- 2.Molodecky N. A., Soon I. S., Rabi D. M., Ghali W. A., Ferris M., Chernoff G., Benchimol E. I., Panaccione R., Ghosh S., Barkema H. W., Kaplan G. G. (2012) Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142, 46–54.e42; quiz e30 [DOI] [PubMed] [Google Scholar]
- 3.Cosnes J., Gower-Rousseau C., Seksik P., Cortot A. (2011) Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 140, 1785–1794 [DOI] [PubMed] [Google Scholar]
- 4.Benchimol E. I., Guttmann A., Griffiths A. M., Rabeneck L., Mack D. R., Brill H., Howard J., Guan J., To T. (2009) Increasing incidence of paediatric inflammatory bowel disease in Ontario, Canada: evidence from health administrative data. Gut 58, 1490–1497 [DOI] [PubMed] [Google Scholar]
- 5.Abramson O., Durant M., Mow W., Finley A., Kodali P., Wong A., Tavares V., McCroskey E., Liu L., Lewis J. D., Allison J. E., Flowers N., Hutfless S., Velayos F. S., Perry G. S., Cannon R., Herrinton L. J.(2010) Incidence, Prevalence, and Time Trends of Pediatric Inflammatory Bowel Disease in Northern California, 1996 to 2006. J. Pediatr. 157, 233.- [DOI] [PubMed] [Google Scholar]
- 6.Gupta N., Bostrom A. G., Kirschner B. S., Ferry G. D., Gold B. D., Cohen S. A., Winter H. S., Baldassano R. N., Abramson O., Smith T., Heyman M. B. (2010) Incidence of stricturing and penetrating complications of Crohn’s disease diagnosed in pediatric patients. Inflamm. Bowel Dis. 16, 638–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malaty H. M., Fan X., Opekun A. R., Thibodeaux C., Ferry G. D. (2010) Rising incidence of inflammatory bowel disease among children: a 12-year study. J. Pediatr. Gastroenterol. Nutr. 50, 27–31 [DOI] [PubMed] [Google Scholar]
- 8.Jason, K., Hou, J. R. K., Richardson, P., Mei, M., and El-Serag, H. (2011) The incidence and prevalence of inflammatory bowel disease among veterans: a national cohort study. Texas Medical Center Digestive Diseases Center 3rd Annual “Frontiers in Digestive Diseases” Symposium, Houston, Texas, USA [Google Scholar]
- 9.Sartor R. B. (2011) Key questions to guide a better understanding of host-commensal microbiota interactions in intestinal inflammation. Mucosal Immunol. 4, 127–132 [DOI] [PubMed] [Google Scholar]
- 10.Kellermayer R. (2010) Genetic drift. “Omics” as the filtering gateway between environment and phenotype: The inflammatory bowel diseases example. Am. J. Med. Genet. A. 152A, 3022–3025 [DOI] [PubMed] [Google Scholar]
- 11.Halfvarson J. (2011) Genetics in twins with Crohn’s disease: less pronounced than previously believed? Inflamm. Bowel Dis. 17, 6–12 [DOI] [PubMed] [Google Scholar]
- 12.Franke A., McGovern D. P., Barrett J. C., Wang K., Radford-Smith G. L., Ahmad T., Lees C. W., Balschun T., Lee J., Roberts R., Anderson C. A., Bis J. C., Bumpstead S., Ellinghaus D., Festen E. M., Georges M., Green T., Haritunians T., Jostins L., Latiano A., Mathew C. G., Montgomery G. W., Prescott N. J., Raychaudhuri S., Rotter J. I., Schumm P., Sharma Y., Simms L. A., Taylor K. D., Whiteman D., Wijmenga C., Baldassano R. N., Barclay M., Bayless T. M., Brand S., Büning C., Cohen A., Colombel J. F., Cottone M., Stronati L., Denson T., De Vos M., D’Inca R., Dubinsky M., Edwards C., Florin T., Franchimont D., Gearry R., Glas J., Van Gossum A., Guthery S. L., Halfvarson J., Verspaget H. W., Hugot J. P., Karban A., Laukens D., Lawrance I., Lemann M., Levine A., Libioulle C., Louis E., Mowat C., Newman W., Panés J., Phillips A., Proctor D. D., Regueiro M., Russell R., Rutgeerts P., Sanderson J., Sans M., Seibold F., Steinhart A. H., Stokkers P. C., Torkvist L., Kullak-Ublick G., Wilson D., Walters T., Targan S. R., Brant S. R., Rioux J. D., D’Amato M., Weersma R. K., Kugathasan S., Griffiths A. M., Mansfield J. C., Vermeire S., Duerr R. H., Silverberg M. S., Satsangi J., Schreiber S., Cho J. H., Annese V., Hakonarson H., Daly M. J., Parkes M. (2010) Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 42, 1118–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson C. A., Boucher G., Lees C. W., Franke A., D’Amato M., Taylor K. D., Lee J. C., Goyette P., Imielinski M., Latiano A., Lagacé C., Scott R., Amininejad L., Bumpstead S., Baidoo L., Baldassano R. N., Barclay M., Bayless T. M., Brand S., Büning C., Colombel J. F., Denson L. A., De Vos M., Dubinsky M., Edwards C., Ellinghaus D., Fehrmann R. S., Floyd J. A., Florin T., Franchimont D., Franke L., Georges M., Glas J., Glazer N. L., Guthery S. L., Haritunians T., Hayward N. K., Hugot J. P., Jobin G., Laukens D., Lawrance I., Lémann M., Levine A., Libioulle C., Louis E., McGovern D. P., Milla M., Montgomery G. W., Morley K. I., Mowat C., Ng A., Newman W., Ophoff R. A., Papi L., Palmieri O., Peyrin-Biroulet L., Panés J., Phillips A., Prescott N. J., Proctor D. D., Roberts R., Russell R., Rutgeerts P., Sanderson J., Sans M., Schumm P., Seibold F., Sharma Y., Simms L. A., Seielstad M., Steinhart A. H., Targan S. R., van den Berg L. H., Vatn M., Verspaget H., Walters T., Wijmenga C., Wilson D. C., Westra H. J., Xavier R. J., Zhao Z. Z., Ponsioen C. Y., Andersen V., Torkvist L., Gazouli M., Anagnou N. P., Karlsen T. H., Kupcinskas L., Sventoraityte J., Mansfield J. C., Kugathasan S., Silverberg M. S., Halfvarson J., Rotter J. I., Mathew C. G., Griffiths A. M., Gearry R., Ahmad T., Brant S. R., Chamaillard M., Satsangi J., Cho J. H., Schreiber S., Daly M. J., Barrett J. C., Parkes M., Annese V., Hakonarson H., Radford-Smith G., Duerr R. H., Vermeire S., Weersma R. K., Rioux J. D. (2011) Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 43, 246–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnett M., Bermingham E., McNabb W., Bassett S., Armstrong K., Rounce J., Roy N. (2010) Investigating micronutrients and epigenetic mechanisms in relation to inflammatory bowel disease. Mutat. Res. 690, 71–80 [DOI] [PubMed] [Google Scholar]
- 15.Hou J. K., Abraham B., El-Serag H. (2011) Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature. Am. J. Gastroenterol. 106, 563–573 [DOI] [PubMed] [Google Scholar]
- 16.Kellermayer R. (2012) Epigenetics and the developmental origins of inflammatory bowel diseases. Can. J. Gastroenterol. 26, 909–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagy-Szakal D., Mir S. A., Ross M. C., Tatevian N., Petrosino J. F., Kellermayer R. (2013) Monotonous diets protect against acute colitis in mice: epidemiologic and therapeutic implications. J. Pediatr. Gastroenterol. Nutr. 56, 544–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tjonneland A., Overvad K., Bergmann M. M., Nagel G., Linseisen J., Hallmans G., Palmqvist R., Sjodin H., Hagglund G., Berglund G., Lindgren S., Grip O., Palli D., Day N. E., Khaw K. T., Bingham S., Riboli E., Kennedy H., Hart A.; IBD in EPIC Study Investigators (2009) Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: a nested case-control study within a European prospective cohort study. Gut 58, 1606–1611 [DOI] [PubMed] [Google Scholar]
- 19.Petronis A. (2010) Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 465, 721–727 [DOI] [PubMed] [Google Scholar]
- 20.Kitajima S., Takuma S., Morimoto M. (2000) Histological analysis of murine colitis induced by dextran sulfate sodium of different molecular weights. Exp. Anim. 49, 9–15 [DOI] [PubMed] [Google Scholar]
- 21.Maslowski K. M., Vieira A. T., Ng A., Kranich J., Sierro F., Yu D., Schilter H. C., Rolph M. S., Mackay F., Artis D., Xavier R. J., Teixeira M. M., Mackay C. R. (2009) Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tao R., de Zoeten E. F., Ozkaynak E., Chen C., Wang L., Porrett P. M., Li B., Turka L. A., Olson E. N., Greene M. I., Wells A. D., Hancock W. W. (2007) Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 13, 1299–1307 [DOI] [PubMed] [Google Scholar]
- 23.Kellermayer R., Balasa A., Zhang W., Lee S., Mirza S., Chakravarty A., Szigeti R., Laritsky E., Tatevian N., Smith C. W., Shen L., Waterland R. A. (2010) Epigenetic maturation in colonic mucosa continues beyond infancy in mice. Hum. Mol. Genet. 19, 2168–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perret V., Lev R., Pigman W. (1977) Simple method for the preparation of single cell suspensions from normal and tumorous rat colonic mucosa. Gut 18, 382–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vijay-Kumar M., Aitken J. D., Carvalho F. A., Cullender T. C., Mwangi S., Srinivasan S., Sitaraman S. V., Knight R., Ley R. E., Gewirtz A. T. (2010) Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328, 228–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bailey M. T., Walton J. C., Dowd S. E., Weil Z. M., Nelson R. J. (2010) Photoperiod modulates gut bacteria composition in male Siberian hamsters (Phodopus sungorus). Brain Behav. Immun. 24, 577–584 [DOI] [PubMed] [Google Scholar]
- 27.Dowd S. E., Zaragoza J., Rodriguez J. R., Oliver M. J., Payton P. R. (2005) Windows. NET Network Distributed Basic Local Alignment Search Toolkit (W.ND-BLAST). BMC Bioinformatics 6, 93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cole J. R., Wang Q., Cardenas E., Fish J., Chai B., Farris R. J., Kulam-Syed-Mohideen A. S., McGarrell D. M., Marsh T., Garrity G. M., Tiedje J. M. (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamada T., Park C. S., Mamonkin M., Lacorazza H. D. (2009) Transcription factor ELF4 controls the proliferation and homing of CD8+ T cells via the Krüppel-like factors KLF4 and KLF2. Nat. Immunol. 10, 618–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaefer J. S., Montufar-Solis D., Vigneswaran N., Klein J. R. (2010) ICOS promotes IL-17 synthesis in colonic intraepithelial lymphocytes in IL-10-/- mice. J. Leukoc. Biol. 87, 301–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Zoeten E. F., Wang L., Butler K., Beier U. H., Akimova T., Sai H., Bradner J. E., Mazitschek R., Kozikowski A. P., Matthias P., Hancock W. W. (2011) Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol. Cell. Biol. 31, 2066–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaible T. D., Harris R. A., Dowd S. E., Smith C. W., Kellermayer R. (2011) Maternal methyl-donor supplementation induces prolonged murine offspring colitis susceptibility in association with mucosal epigenetic and microbiomic changes. Hum. Mol. Genet. 20, 1687–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmitz G., Ecker J. (2008) The opposing effects of n-3 and n-6 fatty acids. Prog. Lipid Res. 47, 147–155 [DOI] [PubMed] [Google Scholar]
- 34.Marion-Letellier R., Savoye G., Beck P. L., Panaccione R., Ghosh S. (2013) Polyunsaturated fatty acids in inflammatory bowel diseases: a reappraisal of effects and therapeutic approaches. Inflamm. Bowel Dis. 19, 650–661 [DOI] [PubMed] [Google Scholar]
- 35.Gruber L., Kisling S., Lichti P., Martin F. P., May S., Klingenspor M., Lichtenegger M., Rychlik M., Haller D. (2013) High fat diet accelerates pathogenesis of murine Crohn’s disease-like ileitis independently of obesity. PLoS ONE 8, e71661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Devkota S., Wang Y., Musch M. W., Leone V., Fehlner-Peach H., Nadimpalli A., Antonopoulos D. A., Jabri B., Chang E. B. (2012) Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 487, 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan S. S., Luben R., Olsen A., Tjonneland A., Kaaks R., Teucher B., Lindgren S., Grip O., Key T., Crowe F. L., Bergmann M. M., Boeing H., Hallmans G., Karling P., Overvad K., Palli D., Masala G., Kennedy H., vanSchaik F., Bueno-de-Mesquita B., Oldenburg B., Khaw K. T., Riboli E., Hart A. R. (2013) Body mass index and the risk for Crohn’s disease and ulcerative colitis: data from a European Prospective Cohort Study (The IBD in EPIC Study). Am. J. Gastroenterol. 108, 575–582 [DOI] [PubMed] [Google Scholar]
- 38.Walters W. A., Xu Z., Knight R. (2014) Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 588, 4223–4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jostins L., Ripke S., Weersma R. K., Duerr R. H., McGovern D. P., Hui K. Y., Lee J. C., Schumm L. P., Sharma Y., Anderson C. A., Essers J., Mitrovic M., Ning K., Cleynen I., Theatre E., Spain S. L., Raychaudhuri S., Goyette P., Wei Z., Abraham C., Achkar J. P., Ahmad T., Amininejad L., Ananthakrishnan A. N., Andersen V., Andrews J. M., Baidoo L., Balschun T., Bampton P. A., Bitton A., Boucher G., Brand S., Büning C., Cohain A., Cichon S., D’Amato M., De Jong D., Devaney K. L., Dubinsky M., Edwards C., Ellinghaus D., Ferguson L. R., Franchimont D., Fransen K., Gearry R., Georges M., Gieger C., Glas J., Haritunians T., Hart A., Hawkey C., Hedl M., Hu X., Karlsen T. H., Kupcinskas L., Kugathasan S., Latiano A., Laukens D., Lawrance I. C., Lees C. W., Louis E., Mahy G., Mansfield J., Morgan A. R., Mowat C., Newman W., Palmieri O., Ponsioen C. Y., Potocnik U., Prescott N. J., Regueiro M., Rotter J. I., Russell R. K., Sanderson J. D., Sans M., Satsangi J., Schreiber S., Simms L. A., Sventoraityte J., Targan S. R., Taylor K. D., Tremelling M., Verspaget H. W., De Vos M., Wijmenga C., Wilson D. C., Winkelmann J., Xavier R. J., Zeissig S., Zhang B., Zhang C. K., Zhao H., Silverberg M. S., Annese V., Hakonarson H., Brant S. R., Radford-Smith G., Mathew C. G., Rioux J. D., Schadt E. E., Daly M. J., Franke A., Parkes M., Vermeire S., Barrett J. C., Cho J. H.; International IBD Genetics Consortium (IIBDGC) (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magyari L., Kovesdi E., Sarlos P., Javorhazy A., Sumegi K., Melegh B. (2014) Interleukin and interleukin receptor gene polymorphisms in inflammatory bowel diseases susceptibility. World J. Gastroenterol. 20, 3208–3222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lepage P., Häsler R., Spehlmann M. E., Rehman A., Zvirbliene A., Begun A., Ott S., Kupcinskas L., Doré J., Raedler A., Schreiber S. (2011) Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 141, 227–236 [DOI] [PubMed] [Google Scholar]
- 42.Gevers D., Kugathasan S., Denson L. A., Vázquez-Baeza Y., Van Treuren W., Ren B., Schwager E., Knights D., Song S. J., Yassour M., Morgan X. C., Kostic A. D., Luo C., González A., McDonald D., Haberman Y., Walters T., Baker S., Rosh J., Stephens M., Heyman M., Markowitz J., Baldassano R., Griffiths A., Sylvester F., Mack D., Kim S., Crandall W., Hyams J., Huttenhower C., Knight R., Xavier R. J. (2014) The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haberman Y., Tickle T. L., Dexheimer P. J., Kim M. O., Tang D., Karns R., Baldassano R. N., Noe J. D., Rosh J., Markowitz J., Heyman M. B., Griffiths A. M., Crandall W. V., Mack D. R., Baker S. S., Huttenhower C., Keljo D. J., Hyams J. S., Kugathasan S., Walters T. D., Aronow B., Xavier R. J., Gevers D., Denson L. A. (2014) Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest. 124, 3617–3633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steck N., Hoffmann M., Sava I. G., Kim S. C., Hahne H., Tonkonogy S. L., Mair K., Krueger D., Pruteanu M., Shanahan F., Vogelmann R., Schemann M., Kuster B., Sartor R. B., Haller D. (2011) Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 141, 959–971 [DOI] [PubMed] [Google Scholar]
- 45.Szigeti R., Pangas S. A., Nagy-Szakal D., Dowd S. E., Shulman R. J., Olive A. P., Popek E. J., Finegold M. J., Kellermayer R. (2012) SMAD4 haploinsufficiency associates with augmented colonic inflammation in select humans and mice. Ann. Clin. Lab. Sci. 42, 401–408 [PMC free article] [PubMed] [Google Scholar]
- 46.Fite A., Macfarlane S., Furrie E., Bahrami B., Cummings J. H., Steinke D. T., Macfarlane G. T. (2013) Longitudinal analyses of gut mucosal microbiotas in ulcerative colitis in relation to patient age and disease severity and duration. J. Clin. Microbiol. 51, 849–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker L. S., Gulbranson-Judge A., Flynn S., Brocker T., Raykundalia C., Goodall M., Förster R., Lipp M., Lane P. (1999) Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J. Exp. Med. 190, 1115–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brocker T., Gulbranson-Judge A., Flynn S., Riedinger M., Raykundalia C., Lane P. (1999) CD4 T cell traffic control: in vivo evidence that ligation of OX40 on CD4 T cells by OX40-ligand expressed on dendritic cells leads to the accumulation of CD4 T cells in B follicles. Eur. J. Immunol. 29, 1610–1616 [DOI] [PubMed] [Google Scholar]
- 49.Murata K., Nose M., Ndhlovu L. C., Sato T., Sugamura K., Ishii N. (2002) Constitutive OX40/OX40 ligand interaction induces autoimmune-like diseases. J. Immunol. 169, 4628–4636 [DOI] [PubMed] [Google Scholar]
- 50.Obermeier F., Schwarz H., Dunger N., Strauch U. G., Grunwald N., Schölmerich J., Falk W. (2003) OX40/OX40L interaction induces the expression of CXCR5 and contributes to chronic colitis induced by dextran sulfate sodium in mice. Eur. J. Immunol. 33, 3265–3274 [DOI] [PubMed] [Google Scholar]
- 51.Sarra M., Monteleone I., Stolfi C., Fantini M. C., Sileri P., Sica G., Tersigni R., Macdonald T. T., Pallone F., Monteleone G. (2010) Interferon-gamma-expressing cells are a major source of interleukin-21 in inflammatory bowel diseases. Inflamm. Bowel Dis. 16, 1332–1339 [DOI] [PubMed] [Google Scholar]
- 52.Harris R. A., Nagy-Szakal D., Mir S. A., Frank E., Szigeti R., Kaplan J. L., Bronsky J., Opekun A., Ferry G. D., Winter H., Kellermayer R. (2014) DNA methylation-associated colonic mucosal immune and defense responses in treatment-naïve pediatric ulcerative colitis. Epigenetics 9, 1131–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishimura M., Kuboi Y., Muramoto K., Kawano T., Imai T. (2009) Chemokines as novel therapeutic targets for inflammatory bowel disease. Ann. N. Y. Acad. Sci. 1173, 350–356 [DOI] [PubMed] [Google Scholar]
- 54.Boland B. S., Sandborn W. J., Chang J. T. (2014) Update on Janus kinase antagonists in inflammatory bowel disease. Gastroenterol. Clin. North Am. 43, 603–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boyle D. L., Soma K., Hodge J., Kavanaugh A., Mandel D., Mease P., Shurmur R., Singhal A. K., Wei N., Rosengren S., Kaplan I., Krishnaswami S., Luo Z., Bradley J., Firestein G. S. (2014) The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. [E-pub ahead of print] Ann. Rheum. Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.