

N6-methyladenosine (m6A) is the most prevalent internal modification that occurs in the messenger RNA (mRNA) of most eukaryotes. In this review, Yue et al. summarize recent progress in the study of the m6A mRNA methylation machineries across eukaryotes and discuss their newly uncovered roles in post-transcriptional gene expression regulation.
Keywords: N6-methyladenosine, m6A methyltransferase, RNA demethylase, METTL3–METTL14, mRNA methylation, post-transcriptional regulation
Abstract
N6-methyladenosine (m6A) is the most prevalent and internal modification that occurs in the messenger RNAs (mRNA) of most eukaryotes, although its functional relevance remained a mystery for decades. This modification is installed by the m6A methylation “writers” and can be reversed by demethylases that serve as “erasers.” In this review, we mainly summarize recent progress in the study of the m6A mRNA methylation machineries across eukaryotes and discuss their newly uncovered biological functions. The broad roles of m6A in regulating cell fates and embryonic development highlight the existence of another layer of epigenetic regulation at the RNA level, where mRNA is subjected to chemical modifications that affect protein expression.
Both DNA and histone proteins undergo dynamic and reversible chemical modifications to control gene expression (Strahl and Allis 2000; Bird 2001; Suzuki and Bird 2008; Bhutani et al. 2011; Jones 2012; Kohli and Zhang 2013). Although post-transcriptional modifications are known to occur to RNAs, the impact of these modifications on gene expression regulation has only recently begun to be explored (He 2010). To date, more than a hundred structurally distinct chemical modifications have been found in eukaryotic RNAs (Cantara et al. 2011; Machnicka et al. 2013); however, the enzymes responsible for each modification and the biological consequences of these modified RNAs are largely unknown. RNA modifications were once considered to be static, but a flurry of recent discoveries has demonstrated that some chemical modifications can be dynamic and participate in the regulation of diverse physiological processes (Motorin and Helm 2011; Yi and Pan 2011; Chan et al. 2012; Fu et al. 2014; Meyer and Jaffrey 2014; Kirchner and Ignatova 2015). The presence of N6-methyladenosine (m6A) in polyadenylated mRNA was first discovered in the 1970s (Desrosiers et al. 1974; Perry and Kelley 1974; Lavi and Shatkin 1975; Wei et al. 1975; Schibler et al. 1977; Wei and Moss 1977) by researchers who were characterizing the 5′ cap structure of messenger RNA (mRNA) in mammalian cells. Since then, m6A has been identified as the most prevalent internal modification in mRNA and long noncoding RNA (lncRNA) in higher eukaryotes. It is widely conserved among eukaryotic species that range from yeast, plants, and flies to mammals as well as among viral mRNAs that replicate inside host nuclei (Krug et al. 1976; Beemon and Keith 1977; Horowitz et al. 1984; Bokar 2005). In addition to its occurrence in mRNA, m6A also exists in various classes of RNA in eukaryotes, bacteria, and archaea, including ribosomal RNAs, small nuclear RNAs, and transfer RNAs (Bjork et al. 1987; Maden 1990; Shimba et al. 1995; Gu et al. 1996; Agris et al. 2007; Piekna-Przybylska et al. 2008). Despite its widespread distribution in the mammalian transcriptome (on average, approximately three m6A sites per mRNA), functional insight has been lacking, possibly due to the low abundance of m6A mRNA and technical difficulties in global detection.
Interest in the biological relevance of m6A in mRNA resurfaced after the discovery of two mammalian RNA demethylases, FTO (fat mass and obesity-associated protein) (Jia et al. 2011) and its homolog, ALKBH5 (Zheng et al. 2013), which selectively reverse m6A to adenosine in nuclear RNA. FTO is associated with human obesity (Dina et al. 2007; Frayling et al. 2007; Loos and Yeo 2014) and mental development (Hess et al. 2013), while ALKBH5 is shown to affect mouse spermatogenesis in a demethylation-dependent manner (Zheng et al. 2013), suggesting broad roles of m6A in various physiological processes. Shortly after these findings, YTHDF2 (YTH domain-containing family protein 2) was identified as the first m6A reader protein that preferentially recognizes m6A-containing mRNA (Dominissini et al. 2012; Wang et al. 2014a) and mediates mRNA decay (Wang et al. 2014a), thereby suggesting a role for m6A RNA as a negative regulator of gene expression. On the other hand, a transcriptome-wide m6A profiling method was developed to decipher the m6A RNA landscape (Dominissini et al. 2012; Meyer et al. 2012). Intriguingly, m6A sites in mammalian polyadenylated RNA are dominated by the conserved Pu[G > A]m6AC[A/C/U] motif that localizes near stop codons, in 3′ untranslated regions (UTRs), within long internal exons, and at 5′ UTRs (Dominissini et al. 2012; Meyer et al. 2012; Schwartz et al. 2013; Li et al. 2014; Luo et al. 2014), immediately raising the question of how this specificity is achieved. The m6A RNA landscape is initially sculptured by a methyltransferase complex, but for a long time, METTL3 (methyltransferase-like 3) was the only known SAM (S-adenosyl methionine)-binding subunit associated with mRNA methylation (Bokar et al. 1997). In 2014, a new mammalian methyltransferase, METTL14, was discovered to catalyze m6A methylation. Together with METTL3, these two proteins form a stable heterodimer complex that mediates cellular m6A deposition on mammalian mRNAs (Liu et al. 2014; Wang et al. 2014b). Recently, the mammalian splicing factor WTAP (Wilms’ tumor 1-associating protein) was identified as the third auxiliary factor of the core methyltransferase complex that affects cellular m6A methylation (Liu et al. 2014; Ping et al. 2014). The identification and characterization of the complete mammalian m6A methylation machinery are the first steps toward deciphering the selectivity and biological functions of m6A deposition in eukaryotic mRNAs.
In this review, we mainly summarize recent progress in the study of m6A methylation in mRNA across different eukaryotes and discuss their newly discovered roles in post-transcriptional gene expression regulation. We first describe the features of m6A on a global scale and briefly introduce the mammalian m6A writers, erasers, and readers that specifically install, remove, or bind to m6A at defined sequence motifs (Fig. 1). We then discuss the evolutional conservation of the m6A methylation machinery across eukaryotic species that range from yeast, plants, and flies to mammals, highlighting the broad roles of methyltransferases and m6A in regulating cell status and embryonic development. Finally, we discuss the emerging functions of m6A in several mechanisms of post-transcriptional gene expression regulation with a special focus on the effects of m6A on differentiation and reprograming of stem cells.
Figure 1.

Illustration of the cellular pathways of m6A in nuclear RNAs. The m6A methyltransferases and demethylases dynamically control the m6A methylation landscape within the nucleus. The m6A reader proteins preferentially bind to the methylated RNA and mediate specific functions. In the nucleus, m6A may affect alternative splicing of pre-mRNA and mature mRNA storage and export. After mature RNAs are exported to the cytoplasm, cytoplasmic m6A reader YTHDF2 can bind to the m6A-containing mRNAs to mediate mRNA decay. Other cytoplasmic readers could modulate mRNA translation and storage.
Features of m6A on a global scale
Studies in the 1970s revealed that m6A modification in mRNA mainly occurs at Pu[G > A]m6AC[U > A > C] (Pu represents purine) and is estimated to be present at an average level of approximately three m6A residues per mRNA (Rottman et al. 1974; Narayan and Rottman 1988; Csepany et al. 1990; Narayan et al. 1994). Transcriptome-wide mapping of m6A is hindered by the following two facts: (1) m6A, akin to A, reverse-transcribes to a thymine (T), and (2) m6A is not susceptible to chemical modifications that might promote its detection. In 2012, two groups independently developed an antibody-based high-throughput sequencing method (Dominissini et al. 2012; Meyer et al. 2012) and for the first time profiled the transcriptome-wide m6A distribution. In each method, mammalian mRNA is properly fragmented and immunoprecipitated by an m6A-specific antibody. Libraries are prepared from immunoprecipitated and input control fragments, respectively, and subjected to high-throughput sequencing. In general, ∼12,000 m6A sites in the transcripts of ∼7000 coding genes and ∼300 noncoding ones are identified in human cells. The resolution of the m6A peak site is ∼100 nucleotides (nt), which was further improved by later optimization (Schwartz et al. 2013, 2014b; Chen et al. 2015a). However, transcriptome-wide m6A detection at single-base resolution remains a challenge.
To date, m6A RNA methylomes across many eukaryotes, including human (Dominissini et al. 2012; Meyer et al. 2012; Batista et al. 2014; Schwartz et al. 2014b), mouse (Dominissini et al. 2012; Meyer et al. 2012; Batista et al. 2014; Schwartz et al. 2014b; Wang et al. 2014b; Geula et al. 2015), yeast (Schwartz et al. 2013), and plant (Li et al. 2014; Luo et al. 2014), have been profiled. In general, global mapping reveals a conserved, widespread, and dynamic mRNA methylation in eukaryotes. Three salient features of the m6A methylome are evident. First, m6A sites are mainly confined to the consensus motif Pu[G > A] m6AC[U > A > C], which is consistent with earlier studies. Second, m6A marks are not equally distributed across the transcriptome; they are preferentially enriched in a subset of consensus sequences near stop codons, in 3′ UTRs, and within long internal exons (Fig. 2). In particular, this topology is preserved upon endodermal differentiation of stem cells (Batista et al. 2014; Geula et al. 2015). Last, m6A-modified genes are well conserved between human and mouse embryonic stem cells (ESCs) and somatic cells (Batista et al. 2014). For instance, ∼70% of human ESC genes are also m6A-modified in the orthologous mouse gene, with ∼46% of the m6A peak sites in common. As expected, higher m6A peak intensities were detected in conserved sites compared with those that are not conserved. On the other hand, distinct m6A patterns can also be detected among different species or cells residing in different developmental stages (Meyer et al. 2012; Schwartz et al. 2013; Batista et al. 2014; Geula et al. 2015). Certain m6A modifications are tissue-specific and dynamically alter in response to different stimuli, indicating the potential role of m6A in regulating diverse cellular processes.
Figure 2.
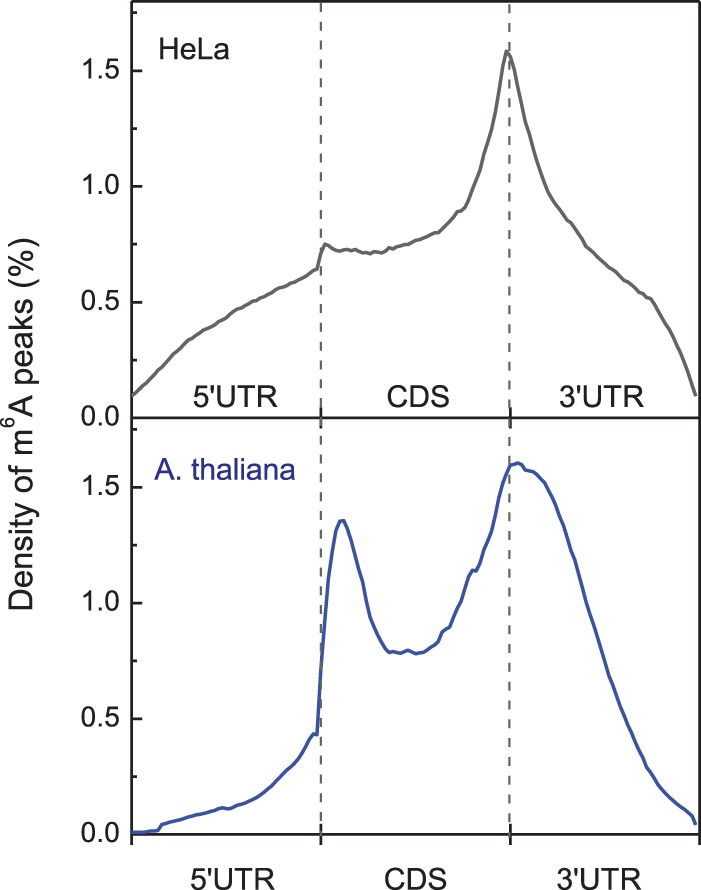
The normalized distribution (density) of m6A peaks along the mRNA transcripts in HeLa cells (top panel) and Arabidopsis thaliana (bottom panel), where each mRNA transcript is divided into the 5′ UTR, coding sequences (CDS), and the 3′ UTR.
m6A writers in mammals
The m6A modification is installed by a multicomponent methyltransferase complex (Fig. 1), which has not been fully characterized. In a pioneer work reported in 1997 (Bokar et al. 1997), a 200-kDa methyltransferase complex was isolated from the HeLa nuclear extract, which exhibits methyltransferase activity. Only a 70-kDa protein, termed MT-A70 or METTL3, was identified as one SAM-binding unit within this 200-kDa methyltransferase complex. The knockdown of METTL3 led to apoptosis of human HeLa cells and a concomitant reduction in cellular m6A level (Bokar 2005). METTL3 and m6A appear to be strongly associated with development and gametogenesis, since the depletion of the METTL3 homologs in yeast (Agarwala et al. 2012), flies (Hongay and Orr-Weaver 2011), and plants (Zhong et al. 2008; Bodi et al. 2012) readily lead to developmental arrest or defects in gametogenesis.
A phylogenetic analysis of the MT-A70 (METTL3) family methyltransferase has suggested METTL14, which shares 43% identity with METTL3 but belongs to a different lineage, as a homolog of METTL3 (Fig. 3; Bujnicki et al. 2002). The highly conserved nature of METTL14 in mammals together with the fact that the METTL14 protein can be pulled down by METTL3 has prompted researchers to consider METTL14 as a putative candidate for m6A deposition on mRNA (Liu et al. 2014). Intriguingly, knockdown of METTL14 results in a more pronounced decrease of m6A in polyadenylated RNA compared with knockdown of METTL3 in both HeLa and human 293 FT cell lines (Liu et al. 2014). The recombinant METTL3 and METTL14 proteins can form a stable METTL3–METTL14 complex in the gel filtration experiment, and subsequent two-dimensional native/SDS-PAGE analysis has further demonstrated the formation of a heterodimer between these two proteins, with a stoichiometry of 1/1 (Liu et al. 2014). While the METTL14 protein itself exhibits higher methylation activity compared with METTL3 in vitro, the combination of both methyltransferases substantially enhances methylation efficiency, demonstrating a synergistic effect that is further confirmed by in vivo studies (Liu et al. 2014; Wang et al. 2014b). The METTL3–METTL14 heterodimer preferentially methylates RNA substrates containing the previously identified consensus sequence GGACU and exhibits a modest preference for the less structured RNA substrate in vitro. Furthermore, the methyltransferase complex was isolated from the native HeLa cell nuclear extract. The nuclear extract fraction that exhibits the highest methylation activity was found to be mostly enriched with METTL3 and METTL14 (Liu et al. 2014), thus clearly indicating that the heterodimer of METTL3–METTL14 forms the catalytic core of the mammalian m6A methyltransferase complex.
Figure 3.
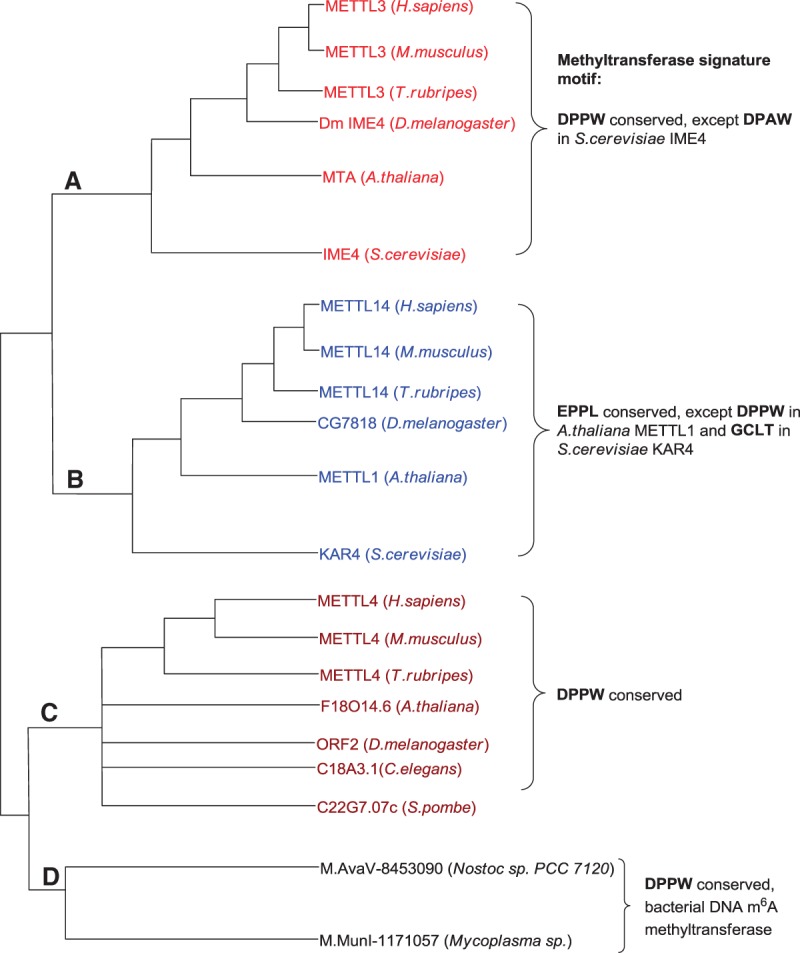
Simplified phylogenetic analysis of the MT-A70 (METTL3) superfamily. Each subfamily is marked with different colors; its corresponding conserved signature motif at the catalytic site is listed for comparison.
WTAP has been identified as the third crucial component of the mammalian m6A methyltransferase complex (Fig. 1; Liu et al. 2014; Ping et al. 2014). WTAP was initially shown to act as a splicing factor that binds to Wilms’ tumor 1 protein (Little et al. 2000) and plays a regulatory role in cell cycle progression and early embryo development (Horiuchi et al. 2006, 2013). The first evidence of WTAP as a third component of the methyltransferase complex came from the coimmunoprecipitation result, which showed that WTAP readily binds to the METTL3–METTL14 heterodimer inside cells, although the interactions between WTAP and the two methyltransferases are weaker compared with that between METTL3 and METTL14 (Liu et al. 2014). WTAP itself does not possess methylation activity, consistent with its lack of a conserved catalytic methylation domain, but interacts with the METTL3–METTL14 heterodimer to substantially affect cellular m6A deposition (Liu et al. 2014; Schwartz et al. 2014b). A subsequent study suggests that WTAP helps to coordinate the localization of the METTL3–METTL14 heterodimer into nuclear speckles, thereby facilitating m6A deposition (Ping et al. 2014).
Global analysis indicates that METTL3, METTL14, and WTAP share a large portion of common binding sites (∼36%) on their RNA substrates and exhibit a binding consensus motif similar, if not identical, to that of m6A (Liu et al. 2014). A PAR-CLIP (photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation) assay revealed that a large fraction of the binding sites fall into intergenic regions (∼46%) and introns (∼31%). This observation suggests that the core methyltransferase complex might work on precursor mRNAs (pre-mRNAs); however, whether and how m6A is installed is not yet known (Fig. 1). The m6A mark may play a regulatory role in alternative splicing pathways because alternative splicing can be directly affected by the presence of the m6A modification in the spliced region (Fig. 1; Geula et al. 2015). In addition, silencing of the methyltransferase complex leads to enhanced abundance of their m6A target transcripts, supporting the role of m6A as a negative regulator of gene expression (Batista et al. 2014; Liu et al. 2014; Schwartz et al. 2014b; Wang et al. 2014a,b; Geula et al. 2015).
The discovery of the core mammalian m6A methyltransferase complex comprised of METTL3, METTL14, and WTAP reveals several new insights. It is surprising and interesting that the core complex of the mRNA m6A methyltransferase contains two parallel active methyltransferases. Each is active and seems to impact different sets of transcripts. One potential explanation points to the selective regulation of different pathways and functions inside cells. Each component may be subjected to different post-translational modifications or binding of partner proteins for the tuning of specific pathways through m6A methylation. Meanwhile, how WTAP, a splicing factor with a noticeable mouse phenotype (Horiuchi et al. 2006), participates in and facilitates m6A methylation remains to be unveiled. Intriguingly, WTAP orthologs in yeast and plants are also shown to interact with the corresponding METTL3 orthologs (Zhong et al. 2008; Agarwala et al. 2012), while its presence in yeast is directly associated with m6A methylation activity (Agarwala et al. 2012). In principle, WTAP could recruit additional auxiliary proteins or RNAs to coordinate methylation of selective RNA substrates. Careful identification of its binding proteins or RNAs may provide a hint in the future. Recent work identified several mammalian WTAP-interacting protein candidates, many of which reside in the RNA processing machinery and have reported roles in alternative splicing (Horiuchi et al. 2013). Whether and how WTAP regulates alternative splicing in an m6A-dependent manner have yet to be systematically explored.
m6A erasers in mammals
FTO is the first identified demethylase that oxidatively reverses m6A to adenosine in mRNA (Jia et al. 2011). FTO is a member of the AlkB subfamily of FeII/α-ketoglutarate-dependent dioxygenases, which has eight other family members in humans (ALKBH1–ALKBH8) and catalyzes the oxidation of diverse biological substrates (Kurowski et al. 2003; Gerken et al. 2007; Fu et al. 2010; Zheng et al. 2014). FTO was initially thought to work on 3-methylthymidine (3mT) in ssDNA (Gerken et al. 2007) and 3-methyluracil (3mU) in ssRNA (Jia et al. 2008). In 2011, FTO was discovered to efficiently demethylate m6A in nuclear RNA (Jia et al. 2011). A subsequent study showed that FTO can oxidize m6A to two previously unknown intermediates—N6-hydroxymethyladenosine (hm6A) and N6-formyladenosine (f6A)—in a stepwise manner (Fu et al. 2013). Intriguingly, this process is similar to the oxidation of 5-methylcytosine (5mC) in genomic DNA to 5-hydroxymethylcytosine (5hmC) and then 5-formylcytosine (5fC) by the TET (ten eleven translocation) family proteins (Tahiliani et al. 2009; Ito et al. 2010, 2011), which also belong to the general family of FeII/α-ketoglutarate-dependent dioxygenases. TET proteins can further oxidize 5fC to 5-carboxylcytosine (5caC) (He et al. 2011; Ito et al. 2011; Zhang et al. 2012). While 5hmC, 5fC, and 5caC are stable cytosine derivatives, hm6A and f6A are short-lived intermediates with half-lives of ∼3 h in aqueous solution under physiological conditions (Fu et al. 2013). The continuous oxidation of 5hmC by the TET family proteins is a critical step in the active DNA demethylation pathway in mammals (He et al. 2011; Pastor et al. 2013; Shen et al. 2014). It is not yet clear whether hm6A and f6A have specific biological functions.
Immunostaining revealed that the FTO protein mainly resides in the nucleus and partially colocalizes with nuclear speckles (Jia et al. 2011), suggesting a dynamic model of m6A demethylation on mRNA coupled with m6A deposition and RNA processing. A recent study found that FTO can modulate alternative splicing of the important adipogenic factor RUNX1T1 by removing the m6A residues around the splice sites (Zhao et al. 2014). It is proposed that loss of m6A on RUNX1T1 transcripts prevents the binding of the splicing factor SRSF2 protein and promotes the production of a shorter isoform, which in turn acts to induce preadipocyte differentiation. FTO is also found in the cytoplasm in several cell types, suggesting a possible role of FTO in modulating cytosolic mRNA processing (Gulati et al. 2013; Vujovic et al. 2013).
Shortly after the discovery of FTO, ALKBH5 was identified and characterized as a second mammalian m6A demethylase that displays distinct biological functions (Zheng et al. 2013). Like FTO, ALKBH5 preferentially binds ssRNAs due to the presence of a unique loop in ALKBH5 that confers single-stranded substrate selectivity (Aik et al. 2014; Xu et al. 2014a). Distinct from FTO, though, ALKBH5 directly reverses m6A to adenosine with no detected intermediates. ALKBH5 is primarily colocalized with nuclear speckles and affects mRNA export and RNA metabolism in a demethylation-dependent manner (Zheng et al. 2013). ALKBH5 knockout mice exhibit impaired male fertility, consistent with the highest expression level of ALKBH5 being in the testis (Zheng et al. 2013). In contrast, FTO is most highly expressed in mouse brains, and FTO-deficient mice mainly suffer from early mortality and reduced body mass (Gerken et al. 2007; Fischer et al. 2009). Taken together, the diverse functions regulated by these two demethylases suggest broad physiological roles of m6A.
Further research is needed to delineate the mechanisms by which demethylases act on specific mRNAs and lncRNAs. Advanced sequencing methods coupled with global analysis approaches will help to define the demethylomes of FTO and ALKBH5.
m6A readers in mammals
While the transcriptome-wide RNA m6A landscape is sculpted by methyltransferases and demethylases in a dynamic and reversible manner, proteins that preferentially recognize m6A (termed m6A readers) bind to methylated RNA and confer downstream functions. Studies using methylated RNA probes to pull down binding proteins followed by mass spectrometry identification have identified several m6A reader candidates in mammalian cells (Dominissini et al. 2012). Among them, the YTH domain-containing family proteins (YTHDF1–3) were validated as m6A readers in cytoplasm, with binding affinities to methylated RNA ranging from ∼180 nM to ∼520 nM (Wang et al. 2014a). Subsequently, YTHDC1, another member of the YTH domain family, was identified as a mammalian m6A reader in the nucleus (Xu et al. 2014b). Mrb1 (methylated RNA-binding 1), a yeast protein with an YTH domain, was also shown to be an m6A reader (Schwartz et al. 2013). Crystal structure characterizations of the YTH domain containing a bound m6A further reveal a conserved hydrophobic pocket used for the binding of the methyl group of m6A as well as the preferential recognition of the GG(m6A)C motif by certain reader proteins (Xu et al. 2014b).
The binding sites and physiological targets of these m6A reader proteins can be readily profiled using transcriptome-wide methods, such as PAR-CLIP. In fact, changing the cellular level of the specific reader proteins could give functional insight into the roles of the reader proteins as well as the fate of the corresponding substrate mRNA. YTHDF2 was shown to mediate mRNA decay (Fig. 1) by selectively binding to its transcript targets at a defined G(m6A)C consequence motif (Wang et al. 2014a). YTHDF2 binds to m6A via its C-terminal YTH domain and facilitates the relocalization of the cognate mRNA from the actively translating pool to mRNA decay sites through its N-terminal domain. However, biological functions of YTHDF1, YTHDF3, and YTHDC1 remain to be unveiled. A recent study showed that YTHDF1 promotes translation of m6A-containing transcripts (Wang et al. 2015), presenting a novel mechanism of translation promotion by m6A in mRNA.
A recent study also suggests heterogeneous nuclear ribonucleoproteins (hnRNPs) as potential “indirect” nuclear m6A readers. When m6A is installed in a stem–loop of RNA, it can alter the local RNA structure by destabilizing the base-pairing between the m6A consensus motif and the uridine track and thus facilitate the binding of HNRNPC to the uridine track in the loop (Liu et al. 2015). Depletion of m6A impairs the binding of HNRNPC and thereby affects the abundance and alternative splicing of its target RNAs. This study reveals another function of m6A; namely, by altering the structure of RNA (termed m6A switch), m6A facilitates the binding of a regulatory protein and thereby modulates gene expression and RNA maturation. Indeed, structural mapping of mRNA inside mammalian cells has revealed that the methylation regions of mRNA tend to lack secondary structures, highlighting the potential role of m6A in shaping RNA structures (Schwartz et al. 2013; Wan et al. 2014; Spitale et al. 2015).
Conservation of m6A RNA methylation machinery and its related biological functions across eukaryotes
The identification and characterization of the m6A methylation machineries are the first steps toward elucidating the biological roles of m6A in mRNAs. Phylogenetic analysis revealed that the MT-A70 (METTL3) superfamily consists of four lineages of proteins with varied degrees of interrelatedness (Bujnicki et al. 2002). The simplified and updated version is shown in Figure 3. Lineages A, B, and C are unique to eukaryotes, while lineage D corresponds to a small group of bacterial DNA m6A methyltransferases associated with restriction/modification systems. Among eukaryotes, humans, mice, pufferfish, Drosophila melanogaster, and Arabidopsis thaliana each contain representatives of the A, B, and C lineages. For instance, humans have representative proteins METTL3, METTL14, and METTL4 that belong to the A–C subfamily, respectively. The budding yeast Saccharomyces cerevisiae specifies IME4 (inducer of meiosis 4) and KAR4 (karyogamy protein) in the A and B lineages, respectively, while the fission yeast Schizosaccharomyces pombe seems to have only one member in lineage C. Conservation of the methylation signature motifs such as DPPW and EPPL (Fig. 3) in the MT-A70 superfamily members suggests a common ancestry. Genetic studies of methyltransferases in different organisms have been performed in order to understand functional roles of m6A methylation on mRNA (Table 1). Below we focus on reviewing methyltransferases in different organisms and their associated biological functions.
Table 1.
Evolutionary conservation of nuclear RNA m6A methylation machinery
m6A methylation machinery in yeast: the MIS [MUM2 (muddled meiosis 2)–IME4–SLZ1 (sporulation-specific leucine zipper 1)] complex mediates m6A RNA deposition during yeast meiosis
Unlike mammals, m6A methylation in yeast S. cerevisiae is confined to meiosis; m6A starts to accumulate on mRNA at the onset of meiosis, peaks in premeiotic S and G2/prophase, and decreases as strains enter into the meiotic divisions. In fact, the modification is hardly detected in yeast undergoing mitotic growth (Clancy et al. 2002; Bodi et al. 2010; Agarwala et al. 2012). High-resolution mapping of m6A sites in meiotic yeast transcripts reveals that the methylation sites are primarily enriched in a consensus motif—RGAC (R = A/G), similar to the consensus motif in mammals—and are strongly biased toward the 3′ end of the transcripts (Schwartz et al. 2013). IME4 (yeast homolog of mammalian METTL3) is identified as an essential component for m6A deposition on yeast mRNA and regulates meiotic progression via RNA methylation. Depletion of IME4 in yeast is not lethal but delays cellular entry into meiosis divisions and hinders sporulation (Shah and Clancy 1992; Hongay et al. 2006; Agarwala et al. 2012). A two-hybrid screen in yeast has identified a core m6A RNA methyltransferase complex (termed MIS) composed of IME4, MUM2 (yeast homolog of mammalian WTAP), and a third crucial component, SLZ1 (not conserved in mammals) (Table 1; Agarwala et al. 2012). Intriguingly, each component of the MIS complex is expressed in a meiosis-specific manner, consistent with meiosis-confined methylation (Agarwala et al. 2012; Schwartz et al. 2013). At the onset of meiosis, SLZ1 expression is transcriptionally activated by IME1, a master regulator of yeast meiosis (Schwartz et al. 2013). Upon the induction of meiosis, SLZ1 shuttles IME4 and MUM2 from the cytoplasm into the nucleolus. Notably, nucleolar entry of the MIS complex is essential for m6A deposition on yeast mRNA, and the global m6A level subsequently reaches its maximum at meiotic prophase. After that, down-regulation of m6A deposition is induced by activation of NDT80, a transcription factor required for exit from meiotic G2/prophase (Chu and Herskowitz 1998). As a result, the MIS complex exits from the nucleolus, and m6A abundance returns to the basal level as cells enter into the meiotic divisions. Interestingly, researchers have found that IME4 also regulates IME1, which implies a putative positive feedback loop between m6A deposition and IME1 expression (Schwartz et al. 2013).
m6A methylation in D. melanogaster
D. melanogaster IME4 shows significant amino acid similarity to and a conserved catalytic domain with its eukaryotic homologs (Table 1; Fig. 3). Unlike in yeast, elimination of the full-length D. melanogaster IME4 in Drosophila is lethal (Hongay and Orr-Weaver 2011). Partial deletion of D. melanogaster IME4 is semilethal, with the rare viable adults showing significantly reduced fecundity. The catalytic domain of D. melanogaster IME4 is required for the rescue of this semilethality (Hongay and Orr-Weaver 2011), indicating a potential role for m6A RNA methylation in metazoan development. Further studies showed that D. melanogaster IME4 was primarily expressed in the gonads of adult flies. In females, D. melanogaster IME4 plays a crucial role in oogenesis; D. melanogaster ime4-deficient females exhibit compound egg chambers accompanied by significant defects in the Notch signaling pathway. The ancillary factor FL(2)D (female-lethal 2 D), the homolog of yeast MUM2 and mammalian WTAP, is conserved in Drosophila. This protein is required for the splicing regulation of Sxl (Sex lethal) and tra (transformer) pre-mRNAs, two critical gene transcripts associated with Drosophila sex determination and dosage compensation (Penalva et al. 2000; Ortega et al. 2003; Penn et al. 2008).
m6A methylation in plants
m6A is a ubiquitous modification found in the mRNAs of various plants, including monocot plants maize (Nichols 1979), wheat (Kennedy and Lane 1979), oat (Haugland and Cline 1980), A. thaliana (Zhong et al. 2008; Luo et al. 2014), and rice (Li et al. 2014). MTA (encoded by At4g10760), a METTL3 ortholog in Arabidopsis, has been identified as an active component of the m6A methyltransferase complex (Zhong et al. 2008). MTA interacts with FIP37 (encoded by At3g54170), an Arabidopsis homolog of mammalian WTAP and Drosophila FL(2)D, highlighting the highly conserved nature of the methyltransferase components across eukaryotes (Table 1). Intriguingly, MTA tends to be expressed in higher levels in dividing tissues, such as developing seeds, shoot meristems, and emerging lateral roots (Craigon et al. 2004; Zhong et al. 2008). Disruption of either MTA or FIP37 in Arabidopsis leads to developmental arrest of embryos at the globular stage (Vespa et al. 2004; Zhong et al. 2008), coupled to a loss of m6A from the mRNA in arrested seeds (Vespa et al. 2004; Zhong et al. 2008). Later in development, perturbation of MTA causes multiple growth defects, including reduced apical dominance, organ abnormality, and increased trichome branching (Bodi et al. 2012). Collectively, these results demonstrate that the methyltransferase and hence m6A methylation in mRNA play a crucial role in plant development. Very recently, transcriptome-wide m6A profiling was performed in two accessions of Arabidopsis (Luo et al. 2014)—Can-0 and Hen-16—as well as in the rice callus and leaf (Li et al. 2014). It is worth noting that Arabidopsis and rice are unique in their enrichment of m6A not only around the stop codon and within 3′ UTRs—as observed in yeast and mammals—but also around the start codon (Fig. 2). As genes possessing m6A sites around the start codon are associated with photosynthesis and appear to be highly expressed in Arabidopsis, this suggests a potential direct role of m6A at the 5′ UTR during translation (Luo et al. 2014). It will be interesting to determine whether this feature observed in plants is conserved in other organisms such as mammals.
m6A methylation machinery in vertebrates and mammals
We previously discussed the m6A methylation machinery of mammals in our description of writer proteins. The core m6A methyltransferase complex METTL3−METTL14−WTAP is highly conserved from zebrafish to mammals. In zebrafish, both METTL3 and WTAP proteins are ubiquitously expressed during embryogenesis and specifically enriched in the brain 36 h after fertilization (Ping et al. 2014). Embryos injected with either METTL3 or WTAP antisense morpholinos (MOs) suffer from various developmental defects, including smaller heads, eyes, and brain ventricles and curved notochord. In comparison with embryos injected with single-gene-targeted MOs, simultaneous knockdown of these two genes leads to a more pronounced phenotype in embryonic development as well as more severe decreases in the m6A level, indicating the in vivo synergistic effect of the methyltransferase complex. How METTL14 affects m6A deposition and zebrafish tissue differentiation remains to be studied.
Methyltransferases METTL3 and METTL14 are also shown to mediate the m6A formation in mouse ESCs (mESCs) (Batista et al. 2014; Wang et al. 2014b; Geula et al. 2015). Recent work has identified m6A as a crucial regulator in the differentiation and reprogramming of stem cells, which are discussed next.
Biological consequences of m6A methylation of mRNA and the underlying mechanisms
m6A RNA methylation determines stem cell fate by regulating pluripotency transition toward differentiation
ESCs are pluripotent stem cells derived from the inner cell mass (ICM) of a preimplantation embryo, exhibiting prolonged undifferentiated proliferation and stable developmental potential to form derivatives of all three embryonic germ layers (Thomson et al. 1998). The ESCs reside in a so-called “naïve” pluripotent state, while epiblast stem cells (EpiSC) that are derived from a post-implantation epiblast reside in a more differentiation-prepared, “primed” pluripotent state (Geula et al. 2015). The transition from naïve pluripotency to differentiation is tightly regulated by a plethora of pluripotency markers and developmental factors. Transcriptome-wide m6A profiling in mESCs and human ESCs showed that the majority of these core pluripotent genes (e.g., Nanog, Sox2, Klf4, Myc, Jarid2, and Smad3) and developmental regulators (e.g., Foxa2 and Sox17) have m6A modifications on their transcripts, with most of them being targets of Mettl3 (Batista et al. 2014; Wang et al. 2014b; Geula et al. 2015). Meanwhile, siRNA screening also identified Mettl3 as an epigenetic repressor that specifically destabilizes the primed EpiSCs (Geula et al. 2015). Importantly, both of the two methyltransferases, Mettl3 and Mettl14, are shown to catalyze m6A RNA deposition in mESCs (Batista et al. 2014; Wang et al. 2014b; Geula et al. 2015). Wang et al. (2014b) reported that the partial depletion of Mettl3 or Mettl14 by shRNAs leads to decreased m6A levels and reduced self-renewal of mESCs. However, in more recent studies (Batista et al. 2014; Geula et al. 2015) complete Mettl3 knockout mESCs and epiblasts were generated that actually displayed increased self-renewal but substantially impaired differentiation into mature cardiomyocytes and neurons (Batista et al. 2014). When subcutaneously injected into immunodeficient mice, Mettl3 knockout mESCs readily generate larger but poorly differentiated teratomas in vivo, further indicating that depletion of m6A in mESCs enhances self-renewal but hampers differentiation (Batista et al. 2014).
Recently, Geula et al. (2015) demonstrated that the m6A modification plays a key role in facilitating transition of mESCs from the naïve state to the primed state upon differentiation (Fig. 4). To resolve the role of m6A in the naïve pluripotent state, genetic ablation of Mettl3 was performed in mESCs, and mating the Mettl3+/− heterozygote mice yielded the Mettl3−/− knockout blastocysts. Consistent with previous results of Batista et al. (2014), Mettl3-depleted mESCs showed an almost complete loss of m6A and preserved naïve pluripotentcy but failed to proceed into the primed EpiSC-like state. Like Mettl3−/− mESCs, Mettl14−/− knockout mESCs resisted progression out of the naïve state. Taken together, this evidence suggests that m6A ablation in naïve mESCs impairs the transition of naïve mESCs into the primed state and hence blocks the subsequent differentiation. In contrast, mouse EpiSCs (mEpiSCs) at a primed pluripotency state showed a distinct response to m6A depletion; namely, Mettl3 knockdown in mEpiSCs resulted in attenuated stability and an enhanced tendency to lineage priming, which finally led to fast differentiation and/or cell death.
Figure 4.

Methyltransferases set m6A marks on mRNAs to balance the expression levels of pluripotency genes and lineage commitment genes in naïve and primed states of the ESCs. In the naïve state, the expression level of the pluripotency genes is dominant over that of lineage commitment genes, while in the primed state, the trend exhibits the opposite. The m6A methyltransferase depletion in naïve pluripotent cells further up-regulates already highly abundant naïve pluripotency genes, while the lineage commitment genes remain at very low residual levels. As a result, cells stay in a “hypernaïve” pluripotent state and fail to progress into the primed state. If the methyltransferase depletion occurs in the primed state, the expression level of the differentiation priming markers is further boosted, which pushes cells above the critical threshold toward differentiation, leading to fast differentiation and/or cell death.
The balance between naïve pluripotency and lineage priming is fine-tuned by the relative expression of naïve pluripotency markers and lineage commitment factors. Global analysis of methylomes of naïve ESCs and primed EBs showed that m6A modification was detected in 80% of the transcripts of naïve pluripotency genes (e.g., Nanog, Klf4, Sox2, and Esrrb) as well as multiple lineage commitment regulators (e.g., Foxa2 and Sox17). In general, m6A deposition in mESCs decreases the expression of methylated transcripts and directly reduces their stability. For both types of regulators, loss of m6A results in increased abundance of transcripts and longer mRNA lifetime (Batista et al. 2014; Wang et al. 2014b; Geula et al. 2015), reminiscent of the role of YTHDF2 in mediating the degradation of methylated mRNA (Wang et al. 2014a). Thus, depletion of m6A acts to boost the expression of the dominant regulators (pluripotent-promoting or lineage commitment genes) at a given pluripotency state, thereby driving stem cell differentiation. In the ground naïve state, where pluripotency-promoting transcripts prevail, Mettl3 depletion further amplifies the already highly expressed naïve pluripotency genes but leads to only a marginal increase in lineage commitment transcripts, resulting in a so-called “hypernaïve” pluripotency phenotype (Fig. 4; Batista et al. 2014; Geula et al. 2015). In the primed state, where lineage commitment transcripts dominate, Mettl3 depletion primarily up-regulates lineage commitment factors while leading to a minimal increase of naïve pluripotency markers, further tipping the balance toward lineage priming and differentiation (Fig. 4; Geula et al. 2015). Similar divergent effects were also found when m6A was depleted in different stages of cellular reprograming toward naïve pluripotency. During the reprogramming of primed mEpiSCs to naïve mESCs, early inactivation of Mettl3 compromises the pluripotency stability of primed cells and impairs their reversion, whereas late depletion of Mettl3 significantly enhances the reprogramming efficiency of mEpiSCs (Geula et al. 2015).
It should be noted that knockout of Mettl3 is embryonic-lethal (Geula et al. 2015). Post-implantation embryonic day 5.5 (E5.5)–E7.5 knockout embryos retained the widespread expression of pluripotent marker Nanog and failed to up-regulate early differentiation markers (e.g., Foxa2 and Brachyury), which recapitulated the in vitro resistance to differentiation and ultimately resulted in embryonic lethality (Batista et al. 2014; Geula et al. 2015).
Collectively, these studies showed that m6A modification precisely modulates the differentiation and reprograming of stem cells via regulation of the expression of dominant genes involved in corresponding processes. In addition to its role in RNA stability, m6A might regulate gene expression via other pathways, such as translation and alternative splicing (Geula et al. 2015). Interestingly, protein profiling showed that loss of m6A in mESCs enhances the overall protein production level; this trend is intensified for transcripts that bear more m6A peaks. Subsequent ribosomal profiling experiments revealed that the absence of m6A in mESCs and mouse EBs resulted in a modest yet significant increase in translation efficiency, which might also contribute to the maintenance of naïve pluripotency state in Mettl3 knockout mESCs (Geula et al. 2015). Alternative splicing is affected by the presence of m6A modification in the spliced region. Depletion of m6A significantly increases the frequency of two types of alternative splicing: skipped exons and retained introns (Geula et al. 2015). The underlying mechanism is not fully understood.
In general, dynamic mRNA modifications appear to be tightly correlated to the differentiation and reprograming of stem cells. In addition to m6A, recent studies have characterized the distribution of pseudouridine as another widespread and dynamic modification of mRNA (Carlile et al. 2014; Schwartz et al. 2014a). Intriguingly, mutations in dyskerin, an enzyme responsible for pseudouridine formation, lead to aberrant differentiation of hematopoietic stem cells, whereas the conditional expression of dyskerin with a catalytically active domain rescues the severe defects in differentiation (Bellodi et al. 2013). Recent work has also reported efficient generation of induced pluripotent stem cells (iPSCs) from human fibroblasts by using synthetics mRNA with certain modifications. Complete substitution of pseudouridine for uridine and 5mC for cytidine in synthetic mRNAs encoding reprogramming factors attenuated the interferon-mediated innate immune response and enhanced the protein expression yield, thereby remarkably increasing the reprogramming efficiency (Warren et al. 2010).
m6A RNA methylation controls the circadian clock
The mechanism of the mammalian circadian clock involves a negative transcription–translation feedback loop in which the transcription of the clock genes is suppressed by their own encoded proteins. The period of the circadian cycle is set according to this general principle. Around 10% of the transcriptome in livers is known to be rhythmic, but only about one-fifth is driven by de novo transcription, which indicates that mRNA processing could serve as a major circadian component. Recent work showed that many clock genes as well as clock output gene transcripts bear m6A modifications (Fustin et al. 2013). Inhibition of m6A formation by silencing METTL3 causes an mRNA processing delay and circadian period elongation. It appears that m6A depletion prolongs nuclear retention of mature mRNAs of the clock genes Per2 and Arntl. This result reveals an important physiological function of m6A methylation in setting the pace of the circadian cycle and determining clock speed and stability.
Perspectives
The last few years have witnessed breakthrough discoveries on biological functions of m6A in mRNA, but the field is still in its infancy. Methylation specificity stands out as one of several challenging questions that remain to be addressed. In mammals, m6A occurs in only ∼15% of all methylation consensus Pu[G > A]m6AC[A/C/U] motifs, and these methylated sites are primarily enriched near the stop codon, at the 3′ UTR, within long exons, and at the 5′ UTR. How the methylation machinery selectively targets a subset of consensus motifs in the transcriptome remains to be understood. This specificity likely has functional implications on the methylated RNAs. The METTL3–METTL14 heterodimer exhibits higher activity to the GGACU sequence located in a random structure region compared with that residing in the stem or loop (Liu et al. 2014). In agreement with the biochemistry results, global analysis also shows that methylated sites are significantly less structured when compared with randomly selected counterparts from the same genes, possibly because these sites are more exposed and accessible to the methylation machinery (Schwartz et al. 2013). However, more complicated pathways/mechanisms must be involved to achieve target selectivity. A recent study indicated that microRNAs (miRNAs) could partially regulate m6A modification via a sequence-pairing mechanism (Chen et al. 2015b), whereby miRNA expression may modulate the binding of METTL3 to mRNA substrates. Further biochemical and cellular validations are required to confirm this model. Interestingly, another recent study revealed that the m6A mark on primary miRNA (pri-miRNA) plays critical roles in miRNA maturation (Alarcon et al. 2015). METTL3 methylates pri-miRNAs, which facilitates their recognition and processing by the RNA-binding protein DGCR8 in the initiation of miRNA biogenesis. Collectively, these studies suggest a potential regulatory network between the miRNA-based regulation and the m6A-dependent regulation as two main pathways that post-transcriptionally control gene expression (Alarcon et al. 2015; Berulava et al. 2015; Chen et al. 2015b).
The multicomponent mammalian methyltransferase complex still needs to be completely resolved because auxiliary components in the complex may play roles in recruiting the catalytic core to the particular locations of the cognate pre-mRNAs and/or tuning activities of the methyltransferases. Thus, careful characterizations of proteins that interact with METTL3/METTL14/WTAP within the nuclear speckles will shed further insights on the origination of the m6A specificity.
Transcriptome-wide mapping of m6A at single-base resolution will greatly facilitate our understanding of selective m6A installation by the methyltransferase complex. With a base-resolution m6A map, single and clustered m6A sites can be differentiated from each other; m6A fractions on particular transcripts and nearby cis elements can be derived. Additionally, one can study the knockout cell lines to determine whether METTL3 and METTL14 control individual groups of transcripts or share the same targets. Most m6A-seq studies to date have profiled the steady-state polyadenylated RNA inside cells, with the majority of them being mature mRNA rather than highly labile pre-mRNA. Therefore, it is necessary to carry out m6A sequencing on pre-mRNA in order to thoroughly examine the prevalence and distribution of m6A within the intronic regions and estimate the percentage of mRNAs that could be methylated either cotranscriptionally or, potentially, post-transcriptionally.
Emerging results suggest that m6A serves as a dynamic mark on a large number of mRNAs and lncRNAs, which help cells rapidly respond and/or adapt to external signaling and stimuli. By virtue of the reversible nature of the m6A modification, the stability, localization, and translatability of a large group of mRNA transcripts and lncRNAs can be regulated by m6A reader proteins and thereby participate in a timely manner in various biological pathways. The methyltransferases, demethylases, and reader proteins can all direct the methylation-based signaling process. Development of small molecule inhibitors or gene therapy tools for targeting these proteins could lead to new ways of controlling gene expression and potential new therapies for human diseases.
Last, m6A in eukaryotic mRNA exhibits substantial contributions to post-transcriptional gene expression regulation. This same modification, N6-methyladenie (6mA or m6dA), in DNA has been known to play important roles in bacterial genomes. Very recently, three independent studies reported the presence and characterizations of 6mA/m6dA in three different eukaryotic genomes (green alga, worm, and fly) with proposed transcription regulation functions (Fu et al. 2015; Greer et al. 2015; Zhang et al. 2015). Indeed, the adenine methylation appears to be a common mechanism to control gene expression.
Acknowledgments
This work was supported by the National Institutes of Health (GM071440 to C.H.). C.H. is an investigator of the Howard Hughes Medical Institute. S.F. Reichard contributed editing.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.262766.115.
References
- Agarwala SD, Blitzblau HG, Hochwagen A, Fink GR. 2012. RNA methylation by the MIS complex regulates a cell fate decision in yeast. PLoS Genet 8: e1002732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agris PF, Vendeix FAP, Graham WD. 2007. tRNA's wobble decoding of the genome: 40 years of modification. J Mol Biol 366: 1–13. [DOI] [PubMed] [Google Scholar]
- Aik W, Scotti JS, Choi H, Gong L, Demetriades M, Schofield CJ, McDonough MA. 2014. Structure of human RNA N6-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res 42: 4741–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. 2015. N6-methyladenosine marks primary microRNAs for processing. Nature 519: 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista Pedro J, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley Donna M, Lujan E, Haddad B, Daneshvar K, et al. 2014. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15: 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beemon K, Keith J. 1977. Localization of N6-methyladenosine in the Rous sarcoma virus genome. J Mol Biol 113: 165–179. [DOI] [PubMed] [Google Scholar]
- Bellodi C, McMahon M, Contreras A, Juliano D, Kopmar N, Nakamura T, Maltby D, Burlingame A, Savage SA, Shimamura A, et al. 2013. H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell Rep 3: 1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berulava T, Rahmann S, Rademacher K, Klein-Hitpass L, Horsthemke B. 2015. N6-adenosine methylation in miRNAs. PLoS One 10: e0118438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutani N, Burns DM, Blau HM. 2011. DNA demethylation dynamics. Cell 146: 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. 2001. Methylation talk between histones and DNA. Science 294: 2113–2115. [DOI] [PubMed] [Google Scholar]
- Bjork GR, Ericson JU, Gustafsson CED, Hagervall TG, Jonsson YH, Wikstrom PM. 1987. Transfer RNA Modification. Annu Rev Biochem 56: 263–285. [DOI] [PubMed] [Google Scholar]
- Bodi Z, Button JD, Grierson D, Fray RG. 2010. Yeast targets for mRNA methylation. Nucleic Acids Res 38: 5327–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodi Z, Zhong S, Mehra S, Song J, Graham N, Li H, May S, Fray RG. 2012. Adenosine methylation in Arabidopsis mRNA is associated with the 3′ end and reduced levels cause developmental defects. Front Plant Sci 3: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokar JA. 2005. The biosynthesis and functional roles of methylated nucleosides in eukaryotic mRNA. In Topics in Current Genetics Vol. 12, Fine-tuning of RNA functions by modification and editing (ed. Grosjean H), pp. 141–177. Springer, Heidelberg. [Google Scholar]
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. 1997. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N-6-adenosine)-methyltransferase. RNA 3: 1233–1247. [PMC free article] [PubMed] [Google Scholar]
- Bujnicki JM, Feder M, Radlinska M, Blumenthal RM. 2002. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m6A methyltransferase. J Mol Evol 55: 431–444. [DOI] [PubMed] [Google Scholar]
- Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FAP, Fabris D, Agris PF. 2011. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res 39: D195–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. 2014. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515: 143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CTY, Pang YLJ, Deng W, Babu IR, Dyavaiah M, Begley TJ, Dedon PC. 2012. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun 3: 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, Han D, Dominissini D, Dai Q, Pan T, et al. 2015a. High-resolution N6-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angew Chem Int Ed Engl 54: 1587–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Hao YJ, Zhang Y, Li MM, Wang M, Han W, Wu Y, Lv Y, Hao J, Wang L, et al. 2015b. m6A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 16: 289–301. [DOI] [PubMed] [Google Scholar]
- Chu S, Herskowitz I. 1998. Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol Cell 1: 685–696. [DOI] [PubMed] [Google Scholar]
- Clancy MJ, Shambaugh ME, Timpte CS, Bokar JA. 2002. Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res 30: 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craigon DJ, James N, Okyere J, Higgins J, Jotham J, May S. 2004. NASCArrays: a repository for microarray data generated by NASC's transcriptomics service. Nucleic Acids Res 32: D575–D577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csepany T, Lin A, Baldick CJ, Beemon K. 1990. Sequence specificity of mRNA N6-adenosine methyltransferase. J Biol Chem 265: 20117–20122. [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, Rottman F. 1974. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci 71: 3971–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, Carlsson LMS, Kiess W, Vatin V, Lecoeur C, et al. 2007. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet 39: 724–726. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201–206. [DOI] [PubMed] [Google Scholar]
- Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, Ruther U. 2009. Inactivation of the FTO gene protects from obesity. Nature 458: 894–898. [DOI] [PubMed] [Google Scholar]
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JRB, Elliott KS, Lango H, Rayner NW, et al. 2007. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316: 889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Dai Q, Zhang W, Ren J, Pan T, He C. 2010. The AlkB domain of mammalian ABH8 catalyzes hydroxylation of 5-methoxycarbonylmethyluridine at the wobble position of tRNA. Angew Chem Int Ed Engl 49: 8885–8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, Smemo S, Dai Q, Bailey KA, Nobrega MA, et al. 2013. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun 4: 1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, He C. 2014. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet 15: 293–306. [DOI] [PubMed] [Google Scholar]
- Fu Y, Luo G-Z, Chen K, Deng X, Yu M, Han D, Hao Z, Liu J, Lu X, Doré Louis C, et al. 2015. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 161: 879–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, et al. 2013. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155: 793–806. [DOI] [PubMed] [Google Scholar]
- Gerken T, Girard CA, Tung Y-CL, Webby CJ, Saudek V, Hewitson KS, Yeo GSH, McDonough MA, Cunliffe S, McNeill LA, et al. 2007. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318: 1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. 2015. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347: 1002–1006. [DOI] [PubMed] [Google Scholar]
- Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizábal-Corrales D, Hsu C-H, Aravind L, He C, Shi Y. 2015. DNA methylation on N6-adenine in C. elegans. Cell 161: 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Patton JR, Shimba S, Reddy R. 1996. Localization of modified nucleotides in Schizosaccharomyces pombe spliceosomal small nuclear RNAs: modified nucleotides are clustered in functionally important regions. RNA 2: 909–918. [PMC free article] [PubMed] [Google Scholar]
- Gulati P, Cheung MK, Antrobus R, Church CD, Harding HP, Tung YC, Rimmington D, Ma M, Ron D, Lehner PJ, et al. 2013. Role for the obesity-related FTO gene in the cellular sensing of amino acids. Proc Natl Acad Sci 110: 2557–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugland RA, Cline MG. 1980. Post-transcriptional modifications of oat coleoptile ribonucleic acids. Eur J Biochem 104: 271–277. [DOI] [PubMed] [Google Scholar]
- He C. 2010. RNA epigenetics? Nat Chem Biol 6: 863–865. [DOI] [PubMed] [Google Scholar]
- He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. 2011. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333: 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess ME, Hess S, Meyer KD, Verhagen LAW, Koch L, Bronneke HS, Dietrich MO, Jordan SD, Saletore Y, Elemento O, et al. 2013. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci 16: 1042–1048. [DOI] [PubMed] [Google Scholar]
- Hongay CF, Orr-Weaver TL. 2011. Drosophila inducer of meiosis 4 (IME4) is required for Notch signaling during oogenesis. Proc Natl Acad Sci 108: 14855–14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongay CF, Grisafi PL, Galitski T, Fink GR. 2006. Antisense transcription controls cell fate in Saccharomyces cerevisiae. Cell 127: 735–745. [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Umetani M, Minami T, Okayama H, Takada S, Yamamoto M, Aburatani H, Reid PC, Housman DE, Hamakubo T, et al. 2006. Wilms’ tumor 1-associating protein regulates G(2)/M transition through stabilization of cyclin A2 mRNA. Proc Natl Acad Sci 103: 17278–17283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T, Hamakubo T. 2013. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem 288: 33292–33302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz S, Horowitz A, Nilsen TW, Munns TW, Rottman FM. 1984. Mapping of N6-methyladenosine residues in bovine prolactin mRNA. Proc Natl Acad Sci 81: 5667–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. 2010. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466: 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333: 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Yang C-G, Yang S, Jian X, Yi C, Zhou Z, He C. 2008. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett 582: 3313–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang Y-G, et al. 2011. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7: 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492. [DOI] [PubMed] [Google Scholar]
- Kennedy TD, Lane BG. 1979. Wheat embryo ribonucleates. XIII. Methyl-substituted nucleoside constituents and 5′-terminal dinucleotide sequences in bulk poly(A)-rich RNA from imbibing wheat embryos. Can J Biochem 57: 927–931. [DOI] [PubMed] [Google Scholar]
- Kirchner S, Ignatova Z. 2015. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat Rev Genet 16: 98–112. [DOI] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y. 2013. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502: 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug RM, Morgan MA, Shatkin AJ. 1976. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J Virol 20: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurowski M, Bhagwat A, Papaj G, Bujnicki J. 2003. Phylogenomic identification of five new human homologs of the DNA repair enzyme AlkB. BMC Genomics 4: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi S, Shatkin AJ. 1975. Methylated simian virus 40-specific RNA from nuclei and cytoplasm of infected BSC-1 cells. Proc Natl Acad Sci 72: 2012–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang X, Li C, Hu S, Yu J, Song S. 2014. Transcriptome-wide N6-methyladenosine profiling of rice callus and leaf reveals the presence of tissue-specific competitors involved in selective mRNA modification. RNA Biol 11: 1180–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little NA, Hastie ND, Davies RC. 2000. Identification of WTAP, a novel Wilms’ tumour 1-associating protein. Hum Mol Genet 9: 2231–2239. [DOI] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. 2014. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10: 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. 2015. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 518: 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos RJ, Yeo GS. 2014. The bigger picture of FTO—the first GWAS-identified obesity gene. Nat Rev Endocrinol 10: 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G-Z, MacQueen A, Zheng G, Duan H, Dore LC, Lu Z, Liu J, Chen K, Jia G, Bergelson J, et al. 2014. Unique features of the m6A methylome in Arabidopsis thaliana. Nat Commun 5: 5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. 2013. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res 41: D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maden BE. 1990. The numerous modified nucleotides in eukaryotic ribosomal RNA. Prog Nucleic Acid Res Mol Biol 39: 241–303. [DOI] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. 2014. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol 15: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, Helm M. 2011. RNA nucleotide methylation. Wiley Interdiscip Rev RNA 2: 611–631. [DOI] [PubMed] [Google Scholar]
- Narayan P, Rottman FM. 1988. An invitro system for accurate methylation of internal adenosine residues in messenger RNA. Science 242: 1159–1162. [DOI] [PubMed] [Google Scholar]
- Narayan P, Ludwiczak RL, Goodwin EC, Rottman FM. 1994. Context effects on N6-adenosine methylation sites in prolactin mRNA. Nucleic Acids Res 22: 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols JL. 1979. N6-methyladenosine in maize poly(A)-containing RNA. Plant Sci Lett 15: 357–361. [Google Scholar]
- Ortega A, Niksic M, Bachi A, Wilm M, Sánchez L, Hastie N, Valcárcel J. 2003. Biochemical function of female-lethal (2)D/Wilms’ tumor suppressor-1-associated proteins in alternative pre-mRNA splicing. J Biol Chem 278: 3040–3047. [DOI] [PubMed] [Google Scholar]
- Pastor WA, Aravind L, Rao A. 2013. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 14: 341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penalva LO, Ruiz MF, Ortega A, Granadino B, Vicente L, Segarra C, Valcárcel J, Sánchez L. 2000. The Drosophila fl(2)d gene, required for female-specific splicing of Sxl and tra pre-mRNAs, encodes a novel nuclear protein with a HQ-rich domain. Genetics 155: 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn JK, Graham P, Deshpande G, Calhoun G, Chaouki AS, Salz HK, Schedl P. 2008. Functioning of the Drosophila Wilms’-tumor-1-associated protein homolog, Fl(2)d, in sex-lethal-dependent alternative splicing. Genetics 178: 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RP, Kelley DE. 1974. Existence of methylated messenger RNA in mouse L cells. Cell 1: 37–42. [Google Scholar]
- Piekna-Przybylska D, Decatur WA, Fournier MJ. 2008. The 3D rRNA modification maps database: with interactive tools for ribosome analysis. Nucleic Acids Res 36: D178–D183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang W-J, Adhikari S, Shi Y, Lv Y, Chen Y-S, et al. 2014. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottman F, Shatkin AJ, Perry RP. 1974. Sequences containing methylated nucleotides at the 5′ termini of messenger RNAs: possible implications for processing. Cell 3: 197–199. [DOI] [PubMed] [Google Scholar]
- Schibler U, Kelley DE, Perry RP. 1977. Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J Mol Biol 115: 695–714. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, Tabach Y, Mikkelsen TS, Satija R, Ruvkun G, et al. 2013. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155: 1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, et al. 2014a. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159: 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. 2014b. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8: 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah JC, Clancy MJ. 1992. IME4, a gene that mediates MAT and nutritional control of meiosis in Saccharomyces cerevisiae. Mol Cell Biol 12: 1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Song C-X, He C, Zhang Y. 2014. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem 83: 585–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimba S, Bokar JA, Rottman F, Reddy R. 1995. Accurate and efficient N-6-adenosine methylation in spliceosomal U6 small nuclear RNA by HeLa cell extract in vitro. Nucleic Acids Res 23: 2421–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung J-W, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, et al. 2015. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519: 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403: 41–45. [DOI] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. 1998. Embryonic stem cell lines derived from human blastocysts. Science 282: 1145–1147. [DOI] [PubMed] [Google Scholar]
- Vespa L, Vachon G, Berger F, Perazza D, Faure J-D, Herzog M. 2004. The immunophilin-interacting protein AtFIP37 from Arabidopsis is essential for plant development and is involved in trichome endoreduplication. Plant Physiol 134: 1283–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujovic P, Stamenkovic S, Jasnic N, Lakic I, Djurasevic SF, Cvijic G, Djordjevic J. 2013. Fasting induced cytoplasmic Fto expression in some neurons of rat hypothalamus. PLoS One 8: e63694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, et al. 2014. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505: 706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. 2014a. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505: 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. 2014b. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16: 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. 2015. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161: 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren L, Manos PD, Ahfeldt T, Loh Y-H, Li H, Lau F, Ebina W, Mandal P, Smith ZD, Meissner A, et al. 2010. Highly efficient reprogramming to pluripotency and directed differentiation of human cells using synthetic modified mRNA. Cell Stem Cell 7: 618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, Moss B. 1977. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry 16: 1672–1676. [DOI] [PubMed] [Google Scholar]
- Wei CM, Gershowitz A, Moss B. 1975. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 4: 379–386. [DOI] [PubMed] [Google Scholar]
- Xu C, Liu K, Tempel W, Demetriades M, Aik W, Schofield CJ, Min J. 2014a. Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single stranded m6A RNA demethylation. J Biol Chem 289: 17299–17311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, Lu Z, He C, Min J. 2014b. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol 10: 927–929. [DOI] [PubMed] [Google Scholar]
- Yi C, Pan T. 2011. Cellular dynamics of RNA modification. Acc Chem Res 44: 1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu G-L, Luo C, Jiang H, He C. 2012. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol 8: 328–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, et al. 2015. N6-methyladenine DNA modification in Drosophila. Cell 161: 893–906. [DOI] [PubMed] [Google Scholar]
- Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, Hao YJ, Ping X-L, Chen YS, Wang WJ, et al. 2014. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res 24: 1403–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, Vågbø CB, Shi Y, Wang W-L, Song S-H, et al. 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Fu Y, He C. 2014. Nucleic acid oxidation in DNA damage repair and epigenetics. Chem Rev 114: 4602–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, Fray RG. 2008. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 20: 1278–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]