

In this study, Sallee et al. demonstrate that E-protein dimer formation can promote C. elegans and human bHLH protein instability. By investigating HLH-2, the sole C. elegans E protein, the authors show that HLH-2 functions as a homodimer for sequential roles in AC specification and differentiation and that the functional dimer is targeted for degradation in VUs, the “opposite” fate. The findings indicate that dimerization-driven regulation of bHLH protein stability may be a conserved mechanism for differential regulation in specific cell contexts.
Keywords: TCF3, E2A, HLH-2, bHLH, C. elegans
Abstract
E proteins are conserved regulators of growth and development. We show that the Caenorhabditis elegans E-protein helix–loop–helix-2 (HLH-2) functions as a homodimer in directing development and function of the anchor cell (AC) of the gonad, the critical organizer of uterine and vulval development. Our structure–function analysis of HLH-2 indicates that dimerization drives its degradation in other uterine cells (ventral uterine precursor cells [VUs]) that initially have potential to be the AC. We also provide evidence that this mode of dimerization-driven down-regulation can target other basic HLH (bHLH) dimers as well. Remarkably, human E proteins can functionally substitute for C. elegans HLH-2 in regulating AC development and also display dimerization-dependent degradation in VUs. Our results suggest that dimerization-driven regulation of bHLH protein stability may be a conserved mechanism for differential regulation in specific cell contexts.
The E subfamily of basic helix–loop–helix (bHLH) proteins encompasses essential regulators of cell growth and differentiation in mammalian development and tissue homeostasis. Dysregulated expression or activity of E proteins has been associated with leukemias, lymphomas, and autoimmune diseases (for review, see Belle and Zhuang 2014). Thus, understanding the mechanisms that govern E-protein activity and stability are highly relevant to human development and disease.
E proteins bind DNA as obligate dimers, forming either homodimers or heterodimers with “class II” bHLH proteins (Murre et al. 1994; Massari and Murre 2000). Dimer composition provides DNA-binding specificity for the transcriptional activation of target genes (Chien et al. 1996; Grove et al. 2009). Drosophila and mammalian E proteins are widely expressed, and, in most developmental contexts that have been studied, E proteins function in heterodimers with class II bHLH partners (Murre et al. 1994; Grove et al. 2009). Expression of these partners is precisely controlled spatially and temporally to achieve tissue-appropriate E-protein activity and target gene expression.
There are also dimerization-based mechanisms that regulate E-protein activity and bHLH stability. The best characterized is the negative regulatory mechanism involving expression of Drosophila Emc or mammalian Id, which contain an HLH domain but no DNA-binding domain: Emc/Id forms a heterodimer with E that cannot bind DNA, thereby acting as dominant-negative to reduce E activity (Benezra et al. 1990; Ellis et al. 1990). Id regulates E in diverse developmental and physiological processes, and how it regulates E continues to be investigated (Ling et al. 2014; Miyazaki et al. 2015). In addition, in some contexts, E proteins may stabilize their dimerization partners (Viñals et al. 2004; Roark et al. 2012). In this study, we made the novel observation that dimer formation can instead promote bHLH protein instability. We started with studies of HLH-2, the sole Caenorhabditis elegans E protein, and then explored the behavior of other C. elegans bHLH proteins and human E2A isoforms.
HLH-2 is central to the proper specification and differentiation of the anchor cell (AC) of the hermaphrodite gonad. The AC is a unique, specialized cell in the ventral uterus that serves as a signaling nexus for uterine and vulval patterning. Four cells initially have the potential to be the AC; only one AC is specified, and the other three become ventral uterine precursor cells (VUs) (Kimble 1981; Seydoux and Greenwald 1989; Seydoux et al. 1990). hlh-2 plays multiple and sequential roles in the specification and differentiation of the AC (Karp and Greenwald 2003, 2004; Hwang and Sternberg 2004). hlh-2 endows prospective uterine cells with the potential to be the AC, is essential to resolve which cell becomes the AC in a LIN-12/Notch-mediated interaction, and promotes differentiation and function of the AC by directly activating transcription of target genes that organize uterine and vulval development.
Here we show that HLH-2 functions as a homodimer for all of these roles. Surprisingly, we found that while the HLH-2 homodimer is active and required for HLH-2 function in the AC, HLH-2 homodimerization also drives its post-translational degradation in VUs, thereby spatially restricting its activity to the AC. By analyzing ectopically expressed class II proteins in the VUs, we show that stability of some class II proteins may also be regulated in a dimerization-dependent manner. Finally, we show that human E proteins can functionally substitute for C. elegans HLH-2 and that their stability is also regulated in a dimerization-dependent manner, suggesting that a similar mechanism may be conserved in humans.
Results and Discussion
HLH-2 is down-regulated post-translationally in the presumptive VUs
Specification of the AC is a critical event early in gonadogenesis (Fig. 1A). Four cells of the ventral uterine region of the somatic gonad primordium, which we term “α” and “β” cells, initially have the potential to be the AC. The two β cells lose the potential to be the AC quickly and always become VUs; the two α cells interact with each other via LIN-12/Notch, engaging feedback mechanisms that amplify initial differences between them; the α cell with relatively lower activity becomes the AC, and the other becomes a VU (Seydoux and Greenwald 1989; Seydoux et al. 1990; Greenwald 2012).
Figure 1.
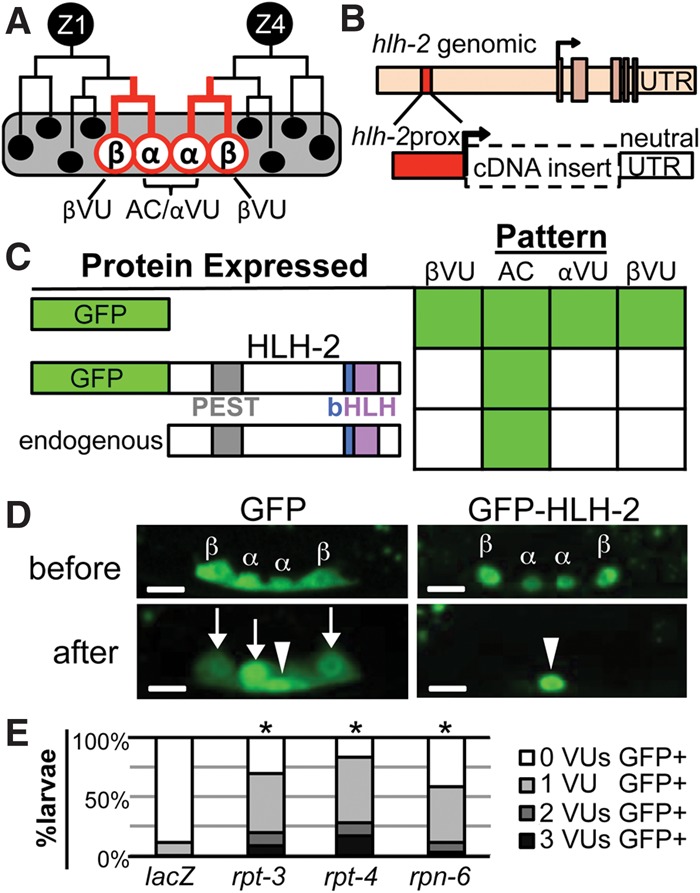
HLH-2 is post-translationally degraded in VUs. (A) Two cells, Z1 and Z4, generate the cells of the somatic gonad primordium (Kimble and Hirsh 1979). The proximal region contains two α cells (Z1.ppp and Z4.aaa) and their sisters, the β cells. lin-12/Notch-mediated signaling between the α cells specifies one as the AC and the other as a VU. The β cells always become VUs; the βVU fate does not require lin-12 activity and is specified earlier than the αVU fate. (B) hlh-2prox (red box) is a 327-base-pair (bp) element that drives transcription of GFP exclusively in α and β cells and their parents (red lines in A) with a “neutral” unc-54 untranslated region (UTR). hlh-2prox continues to drive transcription after the AC and VUs have been specified, although, after primordium formation occurs, the αVU is brighter than the βVUs, which were specified earlier. (C) hlh-2prox::GFP-HLH-2 displays the same pattern of accumulation as Karp and Greenwald (2003) observed for endogenous HLH-2, indicating post-translational regulation of HLH-2 stability. As described in the Supplemental Material, we set a 400-msec exposure time as a stringent threshold for analysis to facilitate scoring large numbers of individuals; two to four independent transgenic lines were scored per construct (see Supplemental Fig. S9 for details). To present a large amount of data for individual animals scored in summary form, in all figures, solid green indicates that ≥80% of cells are GFP+, green stripes indicate that 20%–80% of cells are GFP+, and solid white indicates that ≤20% of cells are GFP+. (D) Representative photomicrographs of hlh-2prox::GFP (left) and hlh-2prox::GFP-HLH-2 (right) before (top) and after (bottom) primordium formation (see the Supplemental Material for staging details). After primordium formation, GFP-HLH-2 has been reliably turned over in all three VUs, but GFP alone is still readily detectable in all three VUs. In all photomicrographs, maximum intensity projection allows visualization of all four cells; an arrowhead marks the AC, and arrows mark the VUs. Bar, 5 μm. (E) RNAi targeting proteasome components rpt-3, rpt-4, and rpn-6 stabilizes GFP-HLH-2 in VUs (see the Supplemental Material for details). (*) P < 0.005 by two-tailed Fisher's exact test compared with lacZ(RNAi).
Although hlh-2(0) mutants are embryonic-lethal, reducing hlh-2 activity post-embryonically by RNAi revealed its essential roles in AC specification and function (Karp and Greenwald 2003, 2004; Hwang and Sternberg 2004; Schindler and Sherwood 2011). Previous work showed that hlh-2 is transcribed in the four α and β cells and continues to be transcribed in the specified AC and VUs but that endogenous HLH-2 protein is detected by antibody staining only in the AC (Karp and Greenwald 2003). These observations suggested that a post-transcriptional mechanism results in differential HLH-2 accumulation in early ventral uterine cells.
Since hlh-2 is an essential gene, manipulations that alter its expression level or activity might have deleterious pleiotropic effects. To facilitate analysis of HLH-2 regulation, we identified “hlh-2prox,” an element from the hlh-2 5′ flanking region (Supplemental Fig. S1) that is necessary and sufficient to promote transcription only in the four α and β cells of the developing ventral uterus (Fig. 1B–D) with no detectable expression elsewhere. When hlh-2prox drives expression of GFP alone, the level of fluorescence is initially similar in the α and β cells; as the primordium forms and the cells adopt their fates, GFP remains evident in the presumptive AC and the three VUs, although the βVUs sometimes appear dimmer than the AC and αVU (Fig. 1D). In contrast, when hlh-2prox drives expression of GFP fused to HLH-2, GFP-HLH-2 fluorescence is initially observed in all four α and β cells, but as the primordium forms and the cell fates are specified, fluorescence is evident only in the presumptive AC and not in the VUs (Fig. 1C,D). Thus, GFP-HLH-2(+) has the same pattern of accumulation as endogenous HLH-2 (or HLH-2 marked with a small HA tag) (Karp and Greenwald 2003), indicating that the GFP moiety does not affect HLH-2 stability.
These results establish that the regulation of HLH-2 stability is a post-translational process, since hlh-2prox::GFP-HLH-2 lacks other potential hlh-2 regulatory sequences (UTRs and introns). Furthermore, GFP-HLH-2 is stabilized in VUs when proteasome activity is reduced by RNAi, consistent with degradation of HLH-2 in VUs (Fig. 1E).
HLH-2 functions as a homodimer for all aspects of early ventral uterine development
Heterodimerization partners are involved in all developmental events requiring hlh-2 in C. elegans described to date, and heterodimerization is the basis for regulating E-protein function in many cell contexts in C. elegans, Drosophila, and mammals. Thus, a simple hypothesis is that differential expression of a heterodimerization partner in the AC or the VUs is responsible for differential stability of HLH-2. However, our expression analysis and optimized functional assays indicated that no other bHLH gene is involved in any aspect of early ventral uterine development (Supplemental Figs. S2–S5), suggesting that HLH-2 functions as a homodimer in ventral uterine development and that differential stability of HLH-2 is not regulated by a bHLH heterodimer partner.
Two additional tests support the inference that HLH-2 functions as a homodimer. First, we analyzed the sequence of E boxes present in known transcriptional targets of HLH-2 in the AC. In vitro studies have shown that HLH-2 homodimers bind to CAGGTG-type E boxes but not CATATG E boxes, which bind heterodimers in vitro and mediate heterodimer function in vivo (Krause et al. 1997; Harfe et al. 1998; Zhang et al. 1999; Hwang and Sternberg 2004; Hwang et al. 2007; Zhao et al. 2007; Grove et al. 2009). Thus, if HLH-2 functions as a homodimer in the AC, its known direct targets should have evolutionarily conserved CAGGTG E boxes. Indeed, we found that all four validated targets of HLH-2 in the AC have highly conserved CAGGTG E boxes (Supplemental Fig. S6). When we altered the two CAGGTG E boxes of the AC-specific Enhancer of lin-3 (ACEL), the best characterized direct target of HLH-2 in the AC (Hwang and Sternberg 2004), to heterodimer-type CATATG E boxes, expression in the AC was lost (Fig. 2B), indicating that the homodimer-type binding sequence is important for function in the AC.
Figure 2.
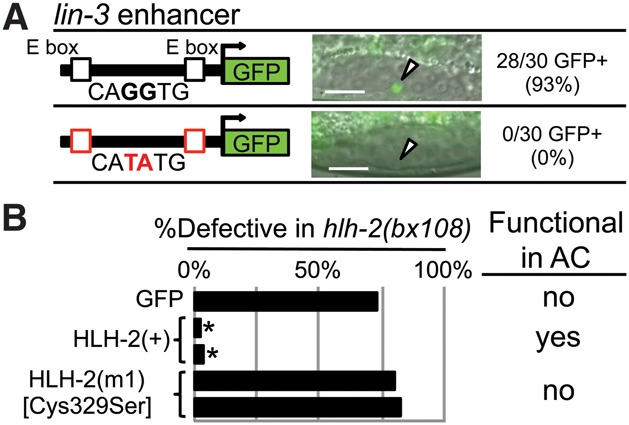
Assessing HLH-2 function as a homodimer in the AC. As shown in Supplemental Figures S2–S5, we analyzed the expression of 17 class II bHLH genes, potential heterodimerization partners of HLH-2; none were expressed in the α or β cells, the AC, or the VUs. In addition, we analyzed the function of all 42 C. elegans bHLH genes (the class II genes plus additional classes) by performing RNAi in a background sensitized for RNAi and for detection of a potential HLH-2 heterodimerization partner and saw no effect on AC number or function. These analyses suggested that HLH-2 functions as a homodimer in the AC, further tested here. (A) In ACEL, two homodimer-binding E boxes, CAGGTG, promote expression in the AC; mutation of these E boxes to the functional heterodimer-binding type CATATG abolishes expression in the AC. n = 30 for each of three transgenic lines; see Supplemental Figure S7 for details. An arrowhead marks the AC. Bar, 10 μm. (B) Function of HLH-2 mutants was assessed by rescue of abnormal vulval eversion (Evl), reflecting abnormal AC function, caused by hlh-2(bx108); details are in Supplemental Figure S8. GFP-HLH-2 has rescuing activity, whereas GFP-HLH-2(C329S) [HLH(m1)], predicted to have disrupted homodimerization, does not. (*) P < 0.0001 by two-tailed Fisher's exact test compared with GFP alone.
Second, we tested the ability of HLH-2(C329S) [HLH(m1)] to rescue abnormalities associated with ventral uterine defects of the hypomorphic allele hlh-2(bx108). In human E2A, the corresponding cysteine, C573, stabilizes the E homodimer through a disulfide bond between the two monomers but is not required to stabilize heterodimers in vitro (Benezra 1994; Markus and Benezra 1999). Although HLH-2(+) efficiently rescues hlh-2(bx108), HLH-2(C329S) does not (Fig. 2C), consistent with HLH-2 function as a homodimer in this cell context.
Dimerization of HLH-2 leads to its degradation in VUs
To identify features that mediate differential stability of HLH-2 in the AC and VUs, we used hlh-2prox to express GFP-tagged mutant forms in the α and β cells and assayed the effect of the mutations on the pattern of GFP-HLH-2 accumulation (Fig. 3A,B) and function (Supplemental Fig. S8).
Figure 3.
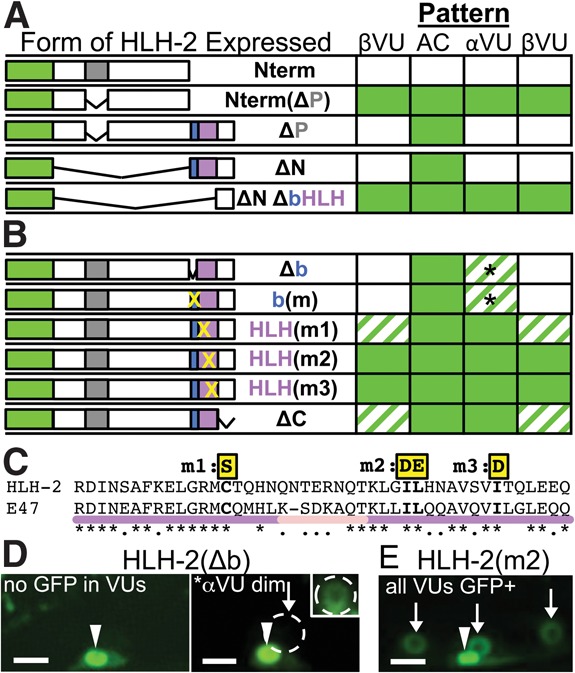
Dimerization directs HLH-2 degradation. (A) Deletion analysis implicates the bHLH domain in patterned regulation of HLH-2. (Top) The PEST region, when separated from the bHLH region, promotes general instability in the AC and VUs (N-terminal region [Nterm] compared with NtermΔP). However, when the PEST region is deleted from an otherwise intact HLH-2, the resulting protein (ΔP) is properly patterned. (Bottom) The bHLH domain is sufficient to confer patterned regulation. Domains are GFP (green), PEST region (gray), basic (blue), and HLH (purple). (B) Mutations in the DNA-binding or dimerization domain (X) correspond to changes shown in C, detailed in Supplemental Figure S10. Penetrance is indicated as in Figure 1 (full data sets are in Supplemental Fig. S9). The basic domain mutants are well regulated in the βVUs and better regulated in the αVUs than are the dimerization mutations: The basic domain mutants show lower penetrance of visible GFP and lower expressivity (i.e., they are dimmer, represented by an asterisk; see also D). (C) Alignment of the HLH regions of HLH-2 and its human ortholog, E47. Black asterisks mark identity, and dots mark similarity; Supplemental Figure S9 shows further sequence analysis. Yellow boxes highlight mutated residues, helices are purple, and the loop is pink. (D,E). Representative photomicrographs show the difference between a basic region mutant (incompletely penetrant weak stabilization) and a dimerization domain region mutant (highly penetrant strong stabilization). (D) Approximately 50% of animals show GFP-HLH-2(Δb) expression only in the AC (left); others (right; asterisk) show weak expression in the αVU, which is more readily apparent in the inset, where brightness and contrast were increased to show αVU expression (white circle). (E) GFP-HLH-2(I345D,L346E) [HLH(m2)], with arrows indicating visible GFP fluorescence evident using the same parameters as in D without adjustment. Bar, 5 μm.
Our initial analysis indicated that the bHLH domain is necessary and sufficient for differential stability (Fig. 3A). The N-terminal region (Nterm), which contains two adjacent predicted protein degradation (PEST) sequences (Fig. 1C; Supplemental Material; Rogers et al. 1986), destabilized GFP, as it resulted in GFP-HLH-2(Nterm) degradation in the AC as well as the VUs, whereas deletion of the PEST sequences from GFP-Nterm [GFP-Nterm(ΔP)] led to stability in all four cells. These results indicate that the PEST sequences promote general turnover when taken out of the HLH-2 structural context but do not regulate the differential stability of HLH-2. When the PEST sequence was deleted from otherwise intact HLH-2 [GFP-HLH-2(ΔP)], or the entire Nterm region was deleted [HLH-2(ΔN)], the wild-type pattern was observed, suggesting that the bHLH domain contains the information needed for proper ventral uterine patterning; i.e., stability in the AC and instability in the VUs. In Supplemental Figure S9, we further support this inference by showing that GFP-HLH-2(ΔP) is properly regulated and retains function in the background of hlh-2(tm1768), an in-frame deletion of the PEST sequences in the endogenous gene.
We then independently mutated either the basic region, which mediates DNA binding, or the HLH region, which mediates dimerization. Residues that mediate DNA binding of the E47 homodimer and residues that mediate dimerization are well characterized both in vitro and by crystal structure (Voronova and Baltimore 1990; Ellenberger et al. 1994); these residues are conserved in HLH-2 (Supplemental Fig. S10). As expected for mutations that abrogate such functions, these mutant forms did not rescue hlh-2(bx108) uterine defects (Supplemental Fig. S8). Mutation or deletion of residues required for E47 DNA binding had a relatively weak effect on HLH-2 accumulation in VUs, with low expressivity and variable penetrance; in contrast, mutations in conserved residues required for E47 dimerization led to strong HLH-2 accumulation in VUs, with high penetrance and high expressivity (Fig. 3B,D; Supplemental Fig. S9). Mutations that prevent DNA binding do not prevent dimerization, but dimerization is required for DNA binding (Voronova and Baltimore 1990). Therefore, the strong stabilization observed for the dimerization mutants but not for the basic region mutants suggests that the ability to dimerize per se, rather than a secondary effect on DNA binding, is the important determinant of stability.
The inference that dimerization is important to direct down-regulation of HLH-2 in VUs was further supported by analysis of two additional mutant forms. First, in addition to the HLH region, human E-protein dimerization also requires “domain C” (Goldfarb et al. 1998), which is conserved in HLH-2 (Supplemental Fig. S10). Deleting the region encompassing this conserved domain also stabilized HLH-2 in VUs and eliminated its rescuing activity (Fig. 3B; Supplemental Fig. S8). Second, GFP-HLH-2(C329S) [HLH(m1)], which should result in unstable E-protein homodimers without preventing dimer formation per se (Benezra 1994; Markus and Benezra 1999), also is stabilized in VUs (Fig. 3B).
In sum, the analysis of mutant forms indicates that the HLH-2 homodimer, rather than a specific sequence in the bHLH domain, is the key determinant of differential stability in the AC and VU. In addition, since dimerization domain mutant forms of HLH-2 are stable in the AC, we infer that (1) HLH-2 is not preferentially stabilized by dimerization in the AC, and (2) the dimerization-defective forms are not grossly unstructured such that the PEST region function “takes over” and leads to degradation. Thus, our results indicate that HLH-2 dimers are preferentially degraded in the VUs as they are being specified. The rapid and complete degradation of HLH-2 homodimers in the three presumptive VUs suggests that persistent HLH-2 activity would interfere with their fate specification. However, we cannot test this prediction by stabilizing HLH-2 by cis mutation and assaying the developmental consequences, as all tested mutations that block down-regulation also extinguish HLH-2 function.
Ectopic expression of other bHLH proteins and dimerization-dependent degradation in VUs
To ask whether down-regulation is a special property of HLH-2 homodimers, we expressed other bHLH proteins not normally found in α and β cells and examined their stability (Fig. 4A; Supplemental Fig. S11). All ectopically expressed bHLH proteins were stable in the AC and displayed different degrees of susceptibility to down-regulation. HLH-1/MyoD, HLH-6/ASCL4, and HLH-8/Twist were mostly stable in VUs. Strikingly, LIN-32/ATOH1 and NGN-1/Ngn3 were efficiently down-regulated in all three VUs, consistent with susceptibility to down-regulation comparable with that of HLH-2.
Figure 4.
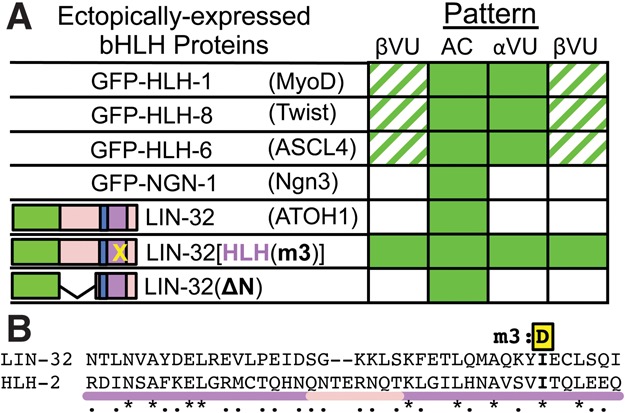
LIN-32/ATOH1 displays dimerization-dependent down-regulation in VUs. (A) Accumulation of ectopically expressed GFP-tagged bHLH proteins (full data sets are in Supplemental Fig. S11); parentheses show the nearest mammalian ortholog. Stability of a dimerization mutant LIN-32(I121D) [HLH(m3)] in VUs shows that down-regulation of LIN-32/ATOH1 is via a dimerization-dependent mechanism. Penetrance is indicated as in Figure 1. (B) Alignment of the HLH regions of LIN-32 and HLH-2. I121 of LIN-32, analogous to HLH-2 I353, was mutated to make LIN-32[HLH(m3)].
We further investigated LIN-32, the best-characterized dimerization partner of HLH-2 in C. elegans (Portman and Emmons 2000). We found that the bHLH region was sufficient and dimerization was necessary for down-regulation in VUs (Fig. 4A), indicating that the mechanism that degrades HLH-2 homodimers can regulate at least some bHLH heterodimers. We could not identify a specific shared sequence or structural feature in the monomers of the proteins that were more efficiently down-regulated or in the proteins that were not (Supplemental Material), suggesting that a general structural feature of bHLH dimers may be recognized by the dimerization-dependent mechanism, consistent with a dimerization-driven regulatory process.
Human E proteins can functionally substitute for hlh-2 and display dimerization-dependent degradation in VUs
The human E2A locus (also called TCF3) encodes two different isoforms: E47 and E12 (Massari and Murre 2000). Both can form homodimers and can form heterodimers with one another or with class II bHLH proteins (Murre et al. 1994). E47 readily forms homodimers; E12 does so less well due to an inhibitory domain in E12 that is not present in E47 or HLH-2 (Sun and Baltimore 1991; Benezra 1994).
We expressed GFP-tagged forms of E47 and E12 in the four α and β cells and observed that, like HLH-2, both E47 and E12 stably accumulate in the AC but not in the VUs (Fig. 5A,C; Supplemental Fig. S12). In addition, E47 and E12 each efficiently rescues defects associated with hlh-2(bx108) and hlh-2(RNAi) (Fig. 5B; Supplemental Figs. S8, S13). Finally, we tested the dimerization dependence of E47 stability and function by making two different mutations that disrupt dimerization in vitro (Voronova and Baltimore 1990) and found that the mutant proteins are stabilized and lose rescuing ability (Fig. 5A–C). Together, these results indicate that human E proteins and HLH-2 have retained both function and regulatory determinants through a billion years of evolution.
Figure 5.
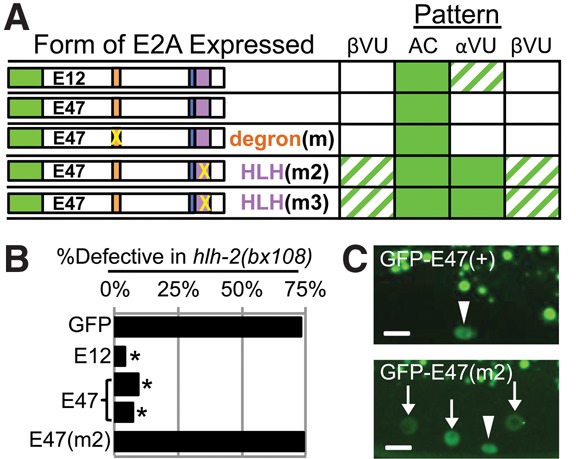
Human E2A can substitute for HLH-2 and displays dimerization-dependent down-regulation in VUs. (A) Summary of expression patterns (full data sets are in Supplemental Fig. S12). GFP-E12 and GFP-E47 are the two isoforms of E2A. Degron(m) is a triple point mutation known to abolish the function of a phosphodegron in E47 that promotes instability in human lymphocytes (see the text); HLH(m2) (I588D,L589E) and HLH(m3) (I596D) are analogous to HLH(m2) and HLH(m3) in HLH-2 (see Fig. 3). Penetrance is indicated as in Figure 1. (B) Rescue assay (full data are in Supplemental Fig. S8). Two transgenic lines expressing GFP-E2A proteins were tested for function in the AC by rescue of a hypomorphic mutant hlh-2(bx108ts). (*) P < 0.0001 by two-tailed Fisher's exact test compared with GFP alone. (C) Representative photomicrographs of GFP-E47 (top) and GFP-E47[HLH(m2)] (bottom). Bar, 5 μm.
In human lymphocytes, E47 protein is degraded in response to activation of Notch (Nie et al. 2003). Since LIN-12/Notch is also active in the VUs when HLH-2 is down-regulated, a simple hypothesis would be that down-regulation of HLH-2 and E47 in the VUs uses the same regulatory mechanism. Down-regulation of E47 in lymphocytes is mediated by phosphorylation of a degron (Nie et al. 2003) that is not present in HLH-2, so the equivalent phosphorylation is unlikely to be the mechanism by which HLH-2 is targeted. However, to test whether E47 instability in VUs may be mediated by phosphorylation of this degron rather than by dimerization, we expressed GFP-E47 with degron mutations [degron(m)] that stabilize E47 in lymphocytes. The degron mutant was down-regulated normally in the VU (Fig. 5A), indicating that these phosphorylation sites are not the major determinant of E47 instability in VUs. Since human E2A and C. elegans HLH-2 are targeted by a mechanism that is distinct from the mechanism targeting E47 in lymphocytes, we infer that there is a novel dimerization-dependent mechanism for regulating E-dimer stability.
Concluding remarks
Our analysis indicates that HLH-2 acts as a homodimer to mediate its known roles in early ventral uterine development—competence, the LIN-12/Notch-mediated AC/VU decision, and AC differentiation and function—even though these discrete events occur within the same cells in a short temporal window. There have been few roles identified for E homodimers, most notably the “classic” role in early B-lymphocyte development (Murre 2005). Indeed, study of E dimerization partners and HLH-only regulatory proteins (e.g., Id in mammals) has driven much research on the roles of post-translational regulation of bHLH activity in development. Our finding of E homodimers as the regulator of a critical signaling nexus in C. elegans organogenesis suggests that additional roles for E homodimers in mammalian development beyond the role defined in B lymphocytes are awaiting discovery.
The remarkable conservation of function and regulation of human E2A and HLH-2 revealed in our C. elegans assays suggests that the down-regulation mechanism that operates in C. elegans VUs may be conserved in humans as well. Furthermore, given our finding that the mechanism can operate on heterodimers as well as homodimers, there are a large number of possible cell contexts in which it may operate. We therefore suggest that dimerization-driven degradation of bHLH proteins will be a more common regulatory mechanism than is currently recognized.
Materials and methods
C. elegans mutants and transgenes
Strain names and full genotypes for all figures are listed in Supplemental Tables S1 and S2. Conditions used for C. elegans genetic analysis, RNAi, and generation of transgenes are described in the Supplemental Material and Supplemental Table S3.
Plasmid constructions and sequence analysis
Construction of hlh-2prox::cDNA constructs and methods used to analyze E-box and bHLH sequences are described in the Supplemental Material and Supplemental Figures S6 and S10.
Scoring GFP-bHLH protein stability and function
GFP-bHLH protein stability was assessed in multiple independent transgenic lines based on fluorescence visible at a 400-msec exposure time. GFP-HLH-2 and GFP-E2A protein function was assessed in rescue assays of the hypomorphic hlh-2 allele bx108 and of hlh-2(RNAi). Details of scoring and assays are described in the Supplemental Material and Supplemental Figures S8, S9, and S11–S13.
Supplementary Material
Acknowledgments
We gratefully acknowledge much expert advice from Xantha Karp. We also thank Oliver Hobert, Michelle Attner, Claire de la Cova, and Daniel Shaye for discussion and critical reading of this manuscript; Oliver Hobert, A. Marian Walhout, Cornelis Murre, David Dominguez-Sola, Douglas Portman, and TransgeneOme for reagents; Robert Townley and Holly Wolcott for specialized analysis of protein structures; Xinlan Zhou for microinjections; and Taner Aydin for valuable additional technical assistance. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). This study was supported by a grant from the National Institutes of Health (R01CA095389) and an Ellison Medical Foundation Senior Scholar Award (AG-SS-2951-12) to I.G., and M.D.S. was funded in part by training grants 5T32 GM007088 and 5T32 HD055165 from the National Institutes of Health.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.261917.115.
References
- Belle I, Zhuang Y. 2014. E proteins in lymphocyte development and lymphoid diseases. Curr Top Dev Biol 110: 153–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benezra R. 1994. An intermolecular disulfide bond stabilizes E2A homodimers and is required for DNA binding at physiological temperatures. Cell 79: 1057–1067. [DOI] [PubMed] [Google Scholar]
- Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. 1990. The protein Id: a negative regulator of helix–loop–helix DNA binding proteins. Cell 61: 49–59. [DOI] [PubMed] [Google Scholar]
- Chien CT, Hsiao CD, Jan LY, Jan YN. 1996. Neuronal type information encoded in the basic-helix–loop–helix domain of proneural genes. Proc Natl Acad Sci 93: 13239–13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberger T, Fass D, Arnaud M, Harrison SC. 1994. Crystal structure of transcription factor E47: E-box recognition by a basic region helix-loop-helix dimer. Genes Dev 8: 970–980. [DOI] [PubMed] [Google Scholar]
- Ellis HM, Spann DR, Posakony JW. 1990. extramacrochaetae, a negative regulator of sensory organ development in Drosophila, defines a new class of helix-loop-helix proteins. Cell 61: 27–38. [DOI] [PubMed] [Google Scholar]
- Goldfarb AN, Lewandowska K, Pennell CA. 1998. Identification of a highly conserved module in E proteins required for in vivo helix–loop–helix dimerization. J Biol Chem 273: 2866–2873. [DOI] [PubMed] [Google Scholar]
- Greenwald I. 2012. Notch and the awesome power of genetics. Genetics 191: 655–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove CA, De Masi F, Barrasa MI, Newburger DE, Alkema MJ, Bulyk ML, Walhout AJM. 2009. A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell 138: 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Gomes AV, Kenyon C, Liu J, Krause M, Fire A. 1998. Analysis of a Caenorhabditis elegans Twist homolog identifies conserved and divergent aspects of mesodermal patterning. Genes Dev 12: 2623–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang BJ, Sternberg PW. 2004. A cell-specific enhancer that specifies lin-3 expression in the C. elegans anchor cell for vulval development. Development 131: 143–151. [DOI] [PubMed] [Google Scholar]
- Hwang BJ, Meruelo AD, Sternberg PW. 2007. C. elegans EVI1 proto-oncogene, EGL-43, is necessary for Notch-mediated cell fate specification and regulates cell invasion. Development 134: 669–679. [DOI] [PubMed] [Google Scholar]
- Karp X, Greenwald I. 2003. Post-transcriptional regulation of the E/Daughterless ortholog HLH-2, negative feedback, and birth order bias during the AC/VU decision in C. elegans. Genes Dev 17: 3100–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp X, Greenwald I. 2004. Multiple roles for the E/Daughterless ortholog HLH-2 during C. elegans gonadogenesis. Dev Biol 272: 460–469. [DOI] [PubMed] [Google Scholar]
- Kimble J. 1981. Alterations in cell lineage following laser ablation of cells in the somatic gonad of Caenorhabditis elegans. Dev Biol 87: 286–300. [DOI] [PubMed] [Google Scholar]
- Kimble J, Hirsh D. 1979. The postembryonic cell lineages of the hermaphrodite and male gonads in Caenorhabditis elegans. Dev Biol 70: 396–417. [DOI] [PubMed] [Google Scholar]
- Krause M, Park M, Zhang JM, Yuan J, Harfe B, Xu SQ, Greenwald I, Cole M, Paterson B, Fire A. 1997. A C. elegans E/Daughterless bHLH protein marks neuronal but not striated muscle development. Development 124: 2179–2189. [DOI] [PubMed] [Google Scholar]
- Ling F, Kang B, Sun X-H. 2014. Id proteins: small molecules, mighty regulators. Curr Top Dev Biol 110: 189–216. [DOI] [PubMed] [Google Scholar]
- Markus M, Benezra R. 1999. Two isoforms of protein disulfide isomerase alter the dimerization status of E2A proteins by a redox mechanism. J Biol Chem 274: 1040–1049. [DOI] [PubMed] [Google Scholar]
- Massari ME, Murre C. 2000. Helix–loop–helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol 20: 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Miyazaki K, Chen S, Chandra V, Wagatsuma K, Agata Y, Rodewald H-R, Saito R, Chang AN, Varki N, et al. 2015. The E-Id protein axis modulates the activities of the PI3K–AKT–mTORC1–Hif1a and c-myc/p19Arf pathways to suppress innate variant TFH cell development, thymocyte expansion, and lymphomagenesis. Genes Dev 29: 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murre C. 2005. Helix–loop–helix proteins and lymphocyte development. Nat Immunol 6: 1079–1086. [DOI] [PubMed] [Google Scholar]
- Murre C, Bain G, van Dijk MA, Engel I, Furnari BA, Massari ME, Matthews JR, Quong MW, Rivera RR, Stuiver MH. 1994. Structure and function of helix-loop-helix proteins. Biochim Biophys Acta 1218: 129–135. [DOI] [PubMed] [Google Scholar]
- Nie L, Xu M, Vladimirova A, Sun X-H. 2003. Notch-induced E2A ubiquitination and degradation are controlled by MAP kinase activities. EMBO J 22: 5780–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portman DS, Emmons SW. 2000. The basic helix–loop–helix transcription factors LIN-32 and HLH-2 function together in multiple steps of a C. elegans neuronal sublineage. Development 127: 5415–5426. [DOI] [PubMed] [Google Scholar]
- Roark R, Itzhaki L, Philpott A. 2012. Complex regulation controls Neurogenin3 proteolysis. Biol Open 1: 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers S, Wells R, Rechsteiner M. 1986. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 234: 364–368. [DOI] [PubMed] [Google Scholar]
- Schindler AJ, Sherwood DR. 2011. The transcription factor HLH-2/E/Daughterless regulates anchor cell invasion across basement membrane in C. elegans. Dev Biol 357: 380–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seydoux G, Greenwald I. 1989. Cell autonomy of lin-12 function in a cell fate decision in C. elegans. Cell 57: 1237–1245. [DOI] [PubMed] [Google Scholar]
- Seydoux G, Schedl T, Greenwald I. 1990. Cell–cell interactions prevent a potential inductive interaction between soma and germline in C. elegans. Cell 61: 939–951. [DOI] [PubMed] [Google Scholar]
- Sun XH, Baltimore D. 1991. An inhibitory domain of E12 transcription factor prevents DNA binding in E12 homodimers but not in E12 heterodimers. Cell 64: 459–470. [DOI] [PubMed] [Google Scholar]
- Viñals F, Reiriz J, Ambrosio S, Bartrons R, Rosa JL, Ventura F. 2004. BMP-2 decreases Mash1 stability by increasing Id1 expression. EMBO J 23: 3527–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronova A, Baltimore D. 1990. Mutations that disrupt DNA binding and dimer formation in the E47 helix-loop-helix protein map to distinct domains. Proc Natl Acad Sci 87: 4722–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Chen L, Krause M, Fire A, Paterson BM. 1999. Evolutionary conservation of MyoD function and differential utilization of E proteins. Dev Biol 208: 465–472. [DOI] [PubMed] [Google Scholar]
- Zhao J, Wang P, Corsi AK. 2007. The C. elegans Twist target gene, arg-1, is regulated by distinct E box promoter elements. Mech Dev 124: 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.