

Abstract
Segmentation is a periodic and sequential morphogenetic process in vertebrates. This rhythmic formation of blocks of tissue called somites along the body axis is evidence of a genetic oscillator patterning the developing embryo. In zebrafish, the intracellular clock driving segmentation is comprised of members of the Her/Hes transcription factor family organized into negative feedback loops. We have recently generated transgenic fluorescent reporter lines for the cyclic gene her1 that recapitulate the spatio-temporal pattern of oscillations in the presomitic mesoderm (PSM). Using these lines, we developed an in vitro culture system that allows real-time analysis of segmentation clock oscillations within single, isolated PSM cells. By removing PSM tissue from transgenic embryos and then dispersing cells from oscillating regions onto glass-bottom dishes, we generated cultures suitable for time-lapse imaging of fluorescence signal from individual clock cells. This approach provides an experimental and conceptual framework for direct manipulation of the segmentation clock with unprecedented single-cell resolution, allowing its cell-autonomous and tissue-level properties to be distinguished and dissected.
Keywords: Developmental Biology, Issue 89, Zebrafish, Primary Cell Culture, Biological Clocks, Somitogenesis, Oscillator, In Vitro, Time-lapse Imaging, Primary Culture, Fluorescence
Introduction
The periodic formation of segments along the vertebrate body axis, or somitogenesis, is evidence of a spatial and temporal oscillator in the developing embryo. The favored mechanism controlling somitogenesis is conceptually described by a "clock and wavefront" model 1, wherein the "clock" consisting of cellular oscillators, now thought to be intracellularly driven by the rhythmic expression of a set of cyclic genes2, ticks off the formation of somites from the presomitic mesoderm (PSM). As the embryo develops, a maturation "wavefront" in the PSM moves in concert with the regressing tissue towards the posterior, slowing and arresting cellular oscillators as it passes 3. Together, this spatiotemporally dynamic system is termed the segmentation clock. Current approaches to study the segmentation clock span three increasing levels of organization from the genetic oscillator in single cells to local coupling that occurs between cells and finally to global regulation of positional information in the collective PSM tissue 4.
Previous studies suggest the cell-autonomous segmentation oscillator in zebrafish consists of genes and protein products from the her/hes transcription factorfamily, which are thought to form a negative feedback loop through transcriptional repression 5-7. The Delta/Notch signaling pathway synchronizes oscillations between neighboring cells and regulates the collective period of the population 8-10. Fgf signaling molecules produced in the tailbud appear to build a gradient across the zebrafish PSM, and are thereby hypothesized to contribute to slowing and arresting oscillating cells in the anterior 11. Until now, the functional roles of each of these molecules in somitogenesis have been investigated by genetic mutation, morpholino injection, heat-shock over-expression, and antagonist drug treatment of clock components and signaling between cells5,7,10,12 . Using these perturbations, segmentation clock function has been inferred from tissue-level descriptions of somite defects and the loss of uniform oscillations in expression of cyclic genes like her1, her7, and deltaC. However, nearly all of these data are from fixed embryos and fail to accurately capture changes that are inherent to the dynamic function of the segmentation clock. More recently, multiple-embryo time-lapse imaging has revealed the first mutants with altered oscillator period, but these observations were also made at the tissue level 7,13 . Thus, behavior of the hypothesized cell-autonomous oscillator during somitogenesis has not been observed.
Static snapshots of somitogenesis give an incomplete picture because, inherently, the process is driven by an oscillatory system. Earlier work in mouse and chick cells showed that levels of transcript and protein rise and fall, but sampling an approximately 2 hr oscillation every 30 or 45 min necessarily restricts the data collected and therefore the conclusions that can be drawn14,15 . Study of other biological oscillators, most notably, circadian clocks, has moved away from staged measurements of gene and protein expression to real-time monitoring using fluorescent and bioluminescent reporters 16,17. These tools are essential for demonstrating clock properties of single cells 18. A bioluminescent reporter of the Hes1 cyclic gene has been developed and briefly characterized in single mouse PSM cells 19. The average period and variance were calculated for a small number of cells, showing that oscillations persist for several cycles in vitro. However, these studies did not quantitatively address the stability and robustness of oscillator frequency and amplitude, whether cells can spontaneously enter or exit oscillations, and how the cells maintain their phase relations. Additionally, the effects of signaling molecules found in the embryo on the cell-autonomous clock have not been directly tested. Consequently, these fundamental properties of the single cell oscillator remain completely unknown.
We have recently developed transgenic fish lines using BAC recombineering 20 to drive Venus (YFP) fluorescence reporters of her1 expression30. Such lines exploit the regulatory cues of the intact chromosomal locus in which they are embedded and recapitulate the temporal dynamics and spatial pattern of her1. This breakthrough allows for real-time monitoring of gene expression in the developing zebrafish embryo in vivo. To study the fundamental properties of cell-autonomous oscillations and how such expression is regulated over time, we recently developed a reliable method to isolate and record from PSM cells in vitro. This protocol describes how we have utilized our transgenic reporter lines to generate dispersed cell cultures, from which we can characterize the oscillations of the zebrafish segmentation clock in single cells. We can thereby tackle outstanding questions in the field that were not accessible with static or tissue-level analysis, as well as directly manipulate the segmentation clock at the single cell level with signaling molecules and inhibitors.
Protocol
1. Before Dissection
- On the day before dissection, obtain embryos from an incross of zebrafish pairs heterozygous for the transgenic allele.
- Raise embryos in E3 medium without methylene blue at 28 oC until shield stage (6 hr post-fertilization).
- Transfer embryos in E3 medium without methylene blue to 20 oC O/N. At 20 oC, embryos will form 1 somite per hr once they reach tailbud stage. After 17-20 hr O/N at 20 oC, embryos should be at 5 to 8 somite stage the following morning at the beginning of the dissection protocol.
- Assemble tools and reagents needed for dissection.
- Fire-polished glass pipette for transfer of embryos and tissue
- Pair of fine forceps for removal of chorions from embryos
- Sylgard-coated 35-mm dish for dissection - Pour Sylgard polymer into a 35 mm dish and cure O/N in a 37 °C. Make the dissection well using a needle tip to remove a small volume of cured polymer. The dish can be cleaned and subsequently reused.
- Sharpened, flattened tungsten wire tools for PSM tissue manipulation
- Micro-scalpel for cutting desired pieces of PSM to culture
- 35-mm plastic dish for trypsin incubation
- L15 medium containing 10% fetal bovine serum
- 0.25% Trypsin/EDTA solution
- Sigmacoted gel-loading tips for dispersal
- Plastic petri dishes containing E3 medium without methylene blue.
Cover glass-bottom imaging dish with Fibronectin1 substrate (10 μg/ml in PBS). Leave dish on bench to coat during dissection.
- Use stereoscope with appropriate fluorescence filters to identify and sort positive transgenic embryos (5 to 8 somite stage).
- Identify transgenic embryos by examining the presomitic mesoderm (PSM) for YFP expression under fluorescence channel (Figure 1A). Fluorescence should be visible in the region from the last formed somite to the tailbud. Select embryos with brightest signal; 25% of offspring should be homozygous embryos carrying 2 copies of the transgene. The number of identified embryos needed varies based on the experiment. One dissected tailbud piece will yield 1,000 cells, on average. Typically, a few additional positive embryos are useful in case errors are made during the dissection.
- Use transmitted light as a positional reference to distinguish any autofluorescence, particularly in the yolk cell, from signal.
- Transfer positive embryos to a separate plastic petri dish containing E3 medium without methylene blue.
2. PSM Dissection and Dispersal
- Prepare embryos for dissection.
- Under a dissection microscope, using fine forceps, carefully remove the chorion from each embryo in the E3 medium. Be sure not to damage the embryo or disrupt the yolk cell.
- Fill Sylgard-coated dissecting dish with L15 medium with serum. Using forceps or another flat tool, remove any air bubbles from the surface of the Sylgard.
- Transfer dechorionated embryos using the fire-polished glass pipette to the dissection dish.
- Using the wire tools, move all embryos to one side of the dissecting dish.
- Dissect PSM from a single embryo.
- Orient a single embryo on its lateral side in the small well made in the Sylgard layer within the dissecting dish (Figure 1B).
- Using the micro-scalpel, slice through the embryo and yolk cell just anterior to the hindbrain and through the ventral pole of the embryo (Figure 1B, dotted red line).
- Remove the anterior piece of the embryo from the well, moving it away to the side of the dish, and then use the wire tools to scrape away remaining yolk cell granules from the posterior section including the PSM.
- Once the yolk has been scraped away, flatten and orient the PSM with the anterior end pointing away from the experimenter and the posterior pointing towards the experimenter (Figure 1C).
- If the thin layer of ectoderm has not pulled off by this point, use the wire tools to peel it away from the top of the PSM tissue.
- Using the micro-scalpel cut the tailbud, the most posterior tip of the PSM past the end of the notochord, away from the rest of the tissue (Figure 1C, dotted red line). NOTE: Other pieces of tissue from the PSM, for example anterior PSM closer to the last formed somite, can also be taken into culture, depending on the experimental question.
- Move the tailbud piece to one corner of the dish away from the dissecting area. Use the wire tools to clear any debris and unwanted embryo tissue from the dissecting field.
- Repeat with the next embryo.
Pool tailbud pieces from multiple embryos, depending on how many cells will be required for the experiment. On average, a single piece of tailbud tissue yields 1,000 cells.
Fill empty 35-mm plastic dish with a small volume of trypsin-EDTA.
Using the fire-polished glass pipette, transfer tailbud pieces from dissecting dish into dish containing trypsin/EDTA. Incubate tailbud pieces in trypsin/EDTA for 20 min at RT.
- While the tissue is being incubated in trypsin, remove the Fibronectin1 solution from the glass-bottomed imaging dish.
- Wash the solution from glass 3 times with MilliQ water. Use suction to remove each wash and ensure the dish is completely dry.
- Disperse tailbud pieces in medium for imaging.
- Pipette 100 μl of L15 medium with serum into imaging dish.
- Using a coated gel tip, remove the tailbud pieces from the trypsin/EDTA in as small a volume as possible.
- Pipette the tailbud pieces into the medium in the imaging dish. Pipette the pieces up and down multiple times to break them apart and suspend cells in the medium. Be careful not to introduce air bubbles. Check for cell clumps under the microscope and disperse more as needed.
- Allow dispersed cells in the suspension to settle onto the Fibronectin1-coated glass for 20 min at RT.
Add a small volume of additional L15 medium with serum to the cells before beginning imaging. Use care not to disrupt settled cells.
Given the experimental question, add any supplemental or drug treatments to the culture.
3. Imaging of Dispersed PSM cells
Set up imaging dish in temperature chamber on the time-lapse imaging microscope. Set Warner chamber to desired temperature for the experiment. Allow dish to equilibrate for at least 30 min before beginning image acquisition. Crosscheck temperature with external temperature probe placed into dish, if required. NOTE: Due to the relationship between temperature and somitogenesis rate 21 stable temperature control is essential for accurate measurements of period in single PSM cells.
Acquire test images in the fluorescence channel to check that the exposure time and gain provide a large dynamic range of intensities without saturation to ensure good signal to noise levels. A typical fluorescence image acquired from dispersed cells generated from our lines using this protocol requires 400 msec and 40 msec for a transmitted light image with an EMCCD camera operating at an EM Gain of 85. In addition, pre-amp gain and readout speed from the camera are also essential to maximize signal over noise.
Using the transmitted light channel, choose fields of cells for the time-lapse acquisition.
- Run time-lapse acquisition protocol for desired length of time.
- Acquire one transmitted light and one fluorescence image per field. NOTE: Set an interval between acquisition rounds that will capture temporal dynamics without photo-bleaching over extended imaging or inducing toxicity in the cells. This protocol uses a 2 min interval.
Check time-lapse set-up occasionally during recording to ensure that cells remain in focus, no software or hardware errors, etc.
4. Image Processing of Acquired Time-lapse Movies
- Open movie file for an acquired field in an image processing software. NOTE: Fiji was used for all processing in this protocol.
- Split transmitted light and fluorescence frames from one field to create 2 stacks of images.
- Track a single cell in the transmitted light channel.
- Place a circular ROI (region of interest) around the selected cell in the first frame in the transmitted light channel. NOTE: Only cells that 1. are healthy at the end of the recording, 2. do not move outside of the field, and 3. do not come into contact with other cells are tracked.
- Save an ROI every few frames to the ROI manager. Track cell until the last frame.
- Measure intensity using the saved ROIs on the fluorescence channel.
- Select the fluorescence stack and with the saved ROIs, use the custom circle interpolator plug-in and macro to measure the intensity of the tracked cell over time.
- Check the output trace from the macro. If the trace qualitatively captures features of the fluorescence time-lapse, export the values to an Excel worksheet.
- Save the ROI list for the cell.
Repeat tracking in transmitted channel and measuring in fluorescence channel for other cells in the field.
Repeat all steps for additional fields from the experiment.
Representative Results
This protocol produces cultures of viable, dispersed, single PSM cells for time-lapse imaging of fluorescence signal (Figure 2). Our transgene generates a reporter whose cycle of production and degradation occurs with similar dynamics to the endogenous gene and protein in the embryo, on the order of half an hour. Due to its fast turnover, the YFP signal in single cells should be detected quickly to minimize bleaching and with a high temporal resolution to capture the features of each oscillatory cycle. Also, given the relative dimness of the signal, culture and acquisition conditions have been carefully tuned to ensure sensitive and robust results. We have found the following factors to be important in generating optimal PSM cell cultures for imaging: 1. An ECM substrate used for coating the glass-bottom culture dishes. 2. Addition of serum to the dissection and imaging medium. 3. Dissection of PSM from identified embryos taken after mating heterozygous pairs, whose offspring potentially carry 2 copies of the transgene. 4. Image acquisition within an optimal magnification range, using a higher NA objective to ensure efficient capture of the fluorescence signal. 5. Illumination with a solid-state light source to minimize intensity fluctuations that could contribute to background noise. 6. Signal detection with a highly sensitive EM-CCD camera to maximize signal readout.
Sub-optimal cultures will contain cells that do not remain rounded and healthy throughout the recording. We hypothesized that our ECM substrate, a fragment of zebrafish fibronectin1 keeps cells in an undifferentiated PSM-like state, in comparison to other commonly used substrates like poly-lysine or laminin. Other substrates we tested caused cells to flatten on the glass and the loss of oscillating fluorescent signal over the course of the recording. We also found that the addition of serum to the medium during dissection, dispersal, and recording was not only important to quench the trypsin used for dissociation, but also increased fluorescence intensity over background, likely due to improved cell viability. To ensure optimal signal capture from single cells we used a 40x objective specifically designed for fluorescence imaging (Zeiss Plan NeoFluor series) with a high NA. We also found that a solid-state light source provided more stable illumination than traditional mercury lamps, which is critical to minimize background fluctuations that contribute to noisy images. These modifications are important to ensure robust results.
Using this protocol we expect cultures in which the majority of cells in a given field are fluorescent at some point during the recording. We find that fluorescent cells typically remain rounded and are sometimes quite motile during recordings. A few cells, including fluorescent cells, may become apoptotic during the recording. These cells are excluded from any analysis. We also exclude cells that come into contact with other cells or move outside of the field of view. On average, we see a 12% reduction in cell number per field by the end of the 10 hr recording as measured by counting the number of healthy cells in the transmitted light channel. This loss includes both cell death and cells that have moved out of the field. Given these caveats we can, on average, track 5 fluorescent cells per field that remain viable and visible, and typically record 6 fields per condition. For example, an experiment with 4 conditions will have 24 total fields acquired and typically about 30 tracked cells per condition. Under standard imaging conditions with L15 medium containing 10% fetal bovine serum, we find that PSM cells (n=101 cells from 4 experimental replicates) can produce between 2 and 7 peaks, with the mean and standard deviation of 3±1 peaks (2-3 cycles). The median peak number, as well as the 25% percentile, is 2 peaks and the 75% percentile is 3 peaks. Using our semi-automated tracking and analysis, we can quickly generate fluorescence intensity over time traces for individual PSM cells, with a representative cell trace shown in Figure 2C. These raw traces can then be used to make quantitative measurements of properties of the oscillating PSM cells, such as frequency, amplitude, number of cycles, and timing of peaks.
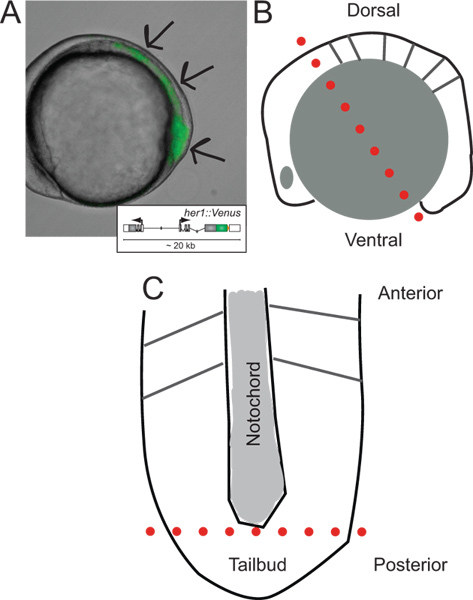 Figure 1. Identification of transgenic embryos and schematics of their dissection.(A) Lateral view of a transgenic zebrafish embryo expressing YFP fluorescence at ~ 5-somite stage. Image is an overlay of transmitted light and fluorescence channel. Arrows indicate areas of signal starting at the tip of the tailbud, throughout the PSM, towards the last formed somite. Inset shows a schematic of the reporter transgene (for more information on its development and in vivo behavior refer to Soroldoni et al.30. (B) Schematic of lateral view of embryo prior to first cut during dissection. Dotted red line indicates the first cut made across the hindbrain, though the yolk cell just behind the tailbud. (C) Schematic view of flattened PSM following removal of yolk granules and epidermal layer, oriented along its anterior-posterior axis. Dotted red line indicates the second cut to remove the tip of the tailbud for dispersal and culture. Please click here to view a larger version of this figure.
Figure 1. Identification of transgenic embryos and schematics of their dissection.(A) Lateral view of a transgenic zebrafish embryo expressing YFP fluorescence at ~ 5-somite stage. Image is an overlay of transmitted light and fluorescence channel. Arrows indicate areas of signal starting at the tip of the tailbud, throughout the PSM, towards the last formed somite. Inset shows a schematic of the reporter transgene (for more information on its development and in vivo behavior refer to Soroldoni et al.30. (B) Schematic of lateral view of embryo prior to first cut during dissection. Dotted red line indicates the first cut made across the hindbrain, though the yolk cell just behind the tailbud. (C) Schematic view of flattened PSM following removal of yolk granules and epidermal layer, oriented along its anterior-posterior axis. Dotted red line indicates the second cut to remove the tip of the tailbud for dispersal and culture. Please click here to view a larger version of this figure.
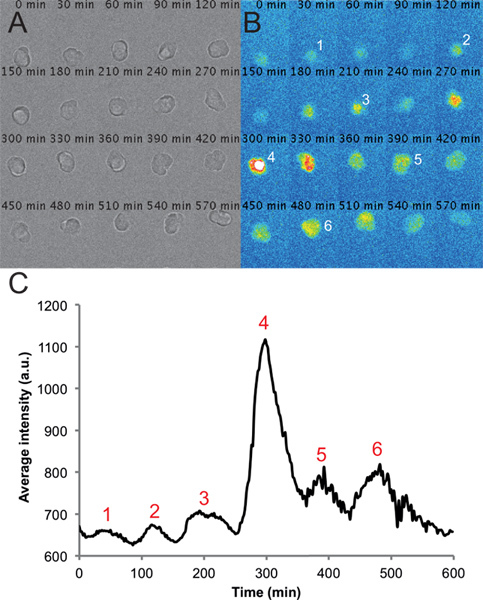 Figure 2. Representative images and traces from a single cell in a dispersed PSM culture.(A) Montage of transmitted light images of a single PSM cell in a 6-h time-lapse recording. Note that the cell remains rounded and healthy throughout the course of the recording. (B) Corresponding montage of fluorescence images from a single PSM cell. Peaks in intensity of the cell are numbered over the course of the recording. (C) Average intensity over time measured from an ROI placed on this cell. Values are taken from every frame from a movie with a frame rate of one per 2 min using the circle interpolator plug-in and custom macro written in Fiji. Peaks in intensity are again numbered throughout the trace. Please click here to view a larger version of this figure.
Figure 2. Representative images and traces from a single cell in a dispersed PSM culture.(A) Montage of transmitted light images of a single PSM cell in a 6-h time-lapse recording. Note that the cell remains rounded and healthy throughout the course of the recording. (B) Corresponding montage of fluorescence images from a single PSM cell. Peaks in intensity of the cell are numbered over the course of the recording. (C) Average intensity over time measured from an ROI placed on this cell. Values are taken from every frame from a movie with a frame rate of one per 2 min using the circle interpolator plug-in and custom macro written in Fiji. Peaks in intensity are again numbered throughout the trace. Please click here to view a larger version of this figure.
Discussion
To study a cellular process that takes place over the course of embryonic development, biologists typically utilize an approach in the context of the entire embryo. However, to fully understand how a single cell behaves within a developmental time frame, a method to examine and perturb individual cells in isolation is also highly beneficial. By generating dispersed PSM cell cultures using transgenic zebrafish embryos we now have a tool to directly study the cell autonomous nature of genetic oscillations in segmentation clock in a quantitative way. It is possible to measure dynamics of the oscillations in our fluorescent reporter in hundreds of cells under a variety of conditions.
The observed period of single cells in culture is longer than the somitogenesis period in the intact embryo. We observe that an intact PSM explant in culture also exhibits slower oscillations than the intact embryo (data not shown), suggesting that the longer period observed in isolated cells is not simply due to damage from dispersal. A variable period and amplitude is observed in most of our single-cell time series. The source of this variability is unknown, but should reveal important details about the segmentation clock's pace-making circuits.
With this method we can study the genetic components of segmentation at the cellular level, and address questions that are challenging to examine in the whole embryo. For example, by controlled addition of known signaling molecules found in the developing embryo to our cultures, we can examine their effects on the single PSM cellular oscillator in a robust and reproducible assay. Our PSM culture system opens the door to a rigorous evaluation of what factors, alone, or in combination promote oscillations in these cells, what factors inhibit these oscillations, and to test the interplay between such molecules. With these tools in hand, we aim to evaluate existing models of somitogenesis that are based on published tissue-level data, as well as use results in single cells to generate predictions that can be tested in the whole embryo.
To our knowledge, this is the first zebrafish primary cell culture protocol for acute time-lapse recording of fluorescence in single cells; further development and refinement of this protocol is no doubt possible. Other protocols often use zebrafish embryos to generate stable cell lines that can be transfected with reporters and used for long-term imaging 27-29. While stable lines are useful for imaging a process that is not tied to development timing, such as the circadian clock, the question of embryonic segmentation necessitates immediate imaging while cells are still in their oscillatory, progenitor state. Once cells stop oscillating, they assume a differentiated cell fate, and when in the tissue, would be incorporated into a somite. It is possible that with the right factor or factors present in vitro we could generate PSM-like zebrafish cell culture lines that would remain oscillatory, enabling significantly longer-term observations, multiple sequential perturbations, or high-throughput screening.
We expect that this method of preparing primary cultures of dispersed cells for time-lapse imaging is well-suited for the study of any cellular process that occurs within a developmental time frame that is not approachable using stable zebrafish cell lines. The isolation of distinct cell-types at distinct developmental stages from the appropriate transgenic reporter lines, either via dissection or via FACS after embryonic dissociation, could provide the starting cells. Some optimization of culture conditions, guided by the embryonic origin of the cells, may be required. By combining this flexible and sensitive protocol with the rapidly growing collection of transgenic zebrafish lines, we hope to facilitate an in vitro developmental biology approach that is complementary to classical genetic and embryological methods.
Disclosures
Author Contributions:
ABW developed and refined the dispersal, culture, imaging, and cell tracking protocols. DS generated the transgenic lines and oversaw the design of the time-lapse fluorescence microscope used in this protocol. AO pioneered initial proof-of-principle experiments to dissociate and image PSM cells in culture. JS wrote the circle interpolator plugin tool in Fiji used to measure fluorescence intensity from time-lapse movies. ABW and ACO wrote the manuscript.
Acknowledgments
This work was supported by an EMBO Long-term fellowship (ABW), a National Science Foundation International Postdoctoral Research Fellowship (ABW), the Max Planck Gesellschaft (ABW, DS, JS, ACO), a DIGS-BB fellowship (AO), and the European Research Council under the European Communities Seventh Framework Programme STG-207634 (DS, ACO). We thank Ravi Desai for helpful comments on the manuscript. We would also like to thank the MPI-CBG protein expression facility for production of the zebrafish Fibronectin1 fragment, the MPI-CBG fish facility staff for care and maintenance of our fish lines and the MPI-CBG light microscopy facility for imaging support.
References
- Cooke J, Zeeman EC. A clock and wavefront model for control of the number of repeated structures during animal morphogenesis. J Theor Biol. 1976;58:455–476. doi: 10.1016/s0022-5193(76)80131-2. [DOI] [PubMed] [Google Scholar]
- Oates AC, Morelli LG, Ares S. Patterning embryos with oscillations: structure, function and dynamics of the vertebrate segmentation clock. Development. 2012;139:625–639. doi: 10.1242/dev.063735. [DOI] [PubMed] [Google Scholar]
- Dequeant ML, Pourquie O. Segmental patterning of the vertebrate embryonic axis. Nat Rev Genet. 2008;9:370–382. doi: 10.1038/nrg2320. [DOI] [PubMed] [Google Scholar]
- Oates AC, Gorfinkiel N, Gonzalez-Gaitan M, Heisenberg CP. Quantitative approaches in developmental biology. Nat Rev Genet. 2009;10:517–530. doi: 10.1038/nrg2548. [DOI] [PubMed] [Google Scholar]
- Oates AC, Ho RK. Hairy/E(spl)-related (Her) genes are central components of the segmentation oscillator and display redundancy with the Delta/Notch signaling pathway in the formation of anterior segmental boundaries in the zebrafish. Development. 2002;129:2929–2946. doi: 10.1242/dev.129.12.2929. [DOI] [PubMed] [Google Scholar]
- Lewis J. Autoinhibition with transcriptional delay: a simple mechanism for the zebrafish somitogenesis oscillator. Curr Biol. 2003;13:1398–1408. doi: 10.1016/s0960-9822(03)00534-7. [DOI] [PubMed] [Google Scholar]
- Schroter C, Oates AC. Segment Number and Axial Identity in a Segmentation Clock Period Mutant. Curr Biol. 2010;20:1254–1258. doi: 10.1016/j.cub.2010.05.071. [DOI] [PubMed] [Google Scholar]
- Riedel-Kruse IH, Muller C, Oates AC. Synchrony dynamics during initiation, failure, and rescue of the segmentation clock. Science. 2007;317:1911–1915. doi: 10.1126/science.1142538. [DOI] [PubMed] [Google Scholar]
- Ozbudak EM, Lewis J. Notch signalling synchronizes the zebrafish segmentation clock but is not needed to create somite boundaries. PLoS Genet. 2008;4:e15. doi: 10.1371/journal.pgen.0040015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrgen L, et al. Intercellular Coupling Regulates the Period of the Segmentation Clock. Curr Biol. 2010;20:1244–1253. doi: 10.1016/j.cub.2010.06.034. [DOI] [PubMed] [Google Scholar]
- Sawada A, et al. Fgf/MAPK signalling is a crucial positional cue in somite boundary formation. Development. 2001;128:4873–4880. doi: 10.1242/dev.128.23.4873. [DOI] [PubMed] [Google Scholar]
- Giudicelli F, Ozbudak EM, Wright GJ, Lewis J. Setting the tempo in development: an investigation of the zebrafish somite clock mechanism. PLoS Biol. 2007;5:e150. doi: 10.1371/journal.pbio.0050150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrgen L, Schroter C, Bajard L, Oates AC. Multiple embryo time-lapse imaging of zebrafish development. Methods Mol Biol. 2009;546:243–254. doi: 10.1007/978-1-60327-977-2_15. [DOI] [PubMed] [Google Scholar]
- Hirata H, et al. Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science. 2002;298:840–843. doi: 10.1126/science.1074560. [DOI] [PubMed] [Google Scholar]
- Maroto M, Dale JK, Dequeant ML, Petit AC, Pourquie O. Synchronised cycling gene oscillations in presomitic mesoderm cells require cell-cell contact. Int J Dev Biol. 2005;49:309–315. doi: 10.1387/ijdb.041958mm. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Welsh DK, Imaizumi T, Kay SA. Real-time reporting of circadian-regulated gene expression by luciferase imaging in plants and mammalian cells. Methods Enzymol. 2005;393:269–288. doi: 10.1016/S0076-6879(05)93011-5. [DOI] [PubMed] [Google Scholar]
- Soroldoni D, Oates AC. Live transgenic reporters of the vertebrate embryo's Segmentation Clock. Curr Opin Genet Dev. 2011;21:600–605. doi: 10.1016/j.gde.2011.09.006. [DOI] [PubMed] [Google Scholar]
- Masamizu Y, et al. Real-time imaging of the somite segmentation clock: revelation of unstable oscillators in the individual presomitic mesoderm cells. Proc Natl Acad Sci USA. 2006;103:1313–1318. doi: 10.1073/pnas.0508658103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroldoni D, Hogan BM, Oates AC. Simple and efficient transgenesis with meganuclease constructs in zebrafish. Methods in molecular biology. 2009;546:117–130. doi: 10.1007/978-1-60327-977-2_8. [DOI] [PubMed] [Google Scholar]
- Schroter C, et al. Dynamics of zebrafish somitogenesis. Dev Dyn. 2008;237:545–553. doi: 10.1002/dvdy.21458. [DOI] [PubMed] [Google Scholar]
- Picker A, Roellig D, Pourquie O, Oates AC, Brand M. Tissue micromanipulation in zebrafish embryos. Methods in molecular biology. 2009;546:153–172. doi: 10.1007/978-1-60327-977-2_11. [DOI] [PubMed] [Google Scholar]
- Mould AP, et al. Identification of multiple integrin beta1 homologs in zebrafish (Danio rerio) BMC Cell Biol. 2006;7:24. doi: 10.1186/1471-2121-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould AP, Koper EJ, Byron A, Zahn G, Humphries MJ. Mapping the ligand-binding pocket of integrin alpha5beta1 using a gain-of-function approach. Biochem J. 2009;424:179–189. doi: 10.1042/BJ20090992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Liu X, Collodi P. Identification and characterization of a novel fibronectin in zebrafish. Exp Cell Res. 2001;268:211–219. doi: 10.1006/excr.2001.5291. [DOI] [PubMed] [Google Scholar]
- Westerfield M. In The zebrafish book : a guide for the laboratory use of zebrafish (Brachydanio rerio) 1993.
- Vallone D, Santoriello C, Gondi SB, Foulkes NS. Basic protocols for zebrafish cell lines: maintenance and transfection. Methods Mol Biol. 2007;362:429–441. doi: 10.1007/978-1-59745-257-1_35. [DOI] [PubMed] [Google Scholar]
- Carr AJ, Whitmore D. Imaging of single light-responsive clock cells reveals fluctuating free-running periods. Nat Cell Biol. 2005;7:319–321. doi: 10.1038/ncb1232. [DOI] [PubMed] [Google Scholar]
- Whitmore D, Foulkes NS, Sassone-Corsi P. Light acts directly on organs and cells in culture to set the vertebrate circadian clock. Nature. 2000;404:87–91. doi: 10.1038/35003589. [DOI] [PubMed] [Google Scholar]
- Soroldoni D, et al. Science. Vol. 345. DOI: 1126. A Doppler effect in embryonic pattern formation; pp. 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]