

Abstract
Background
Further advances of targeted cancer therapy require comprehensive in-depth profiling of somatic mutations that are present in subpopulations of tumor cells in a clinical tumor sample. However, it is unclear to what extent such intra-tumor heterogeneity is present and whether it may affect clinical decision making. To unravel this challenge, we established a deep targeted sequencing platform to identify potentially actionable DNA alterations in tumor samples.
Methods
We assayed 515 FFPE tumor samples and matched germline (475 patients) from 11 disease sites by capturing and sequencing all the exons in 201 cancer related genes. Mutations, indels and copy number data were reported.
Results
We obtained a 1000-fold average sequencing depth and identified 4794 non-synonymous mutations in the samples analyzed, which 15.2% were present at less than 10% allele frequency. Most of these low level mutations occurred at known oncogenic hotspots and are likely functional. Identifying low level mutations improved identification of mutations in actionable genes in 118 (24.84%) patients, among which 47 (9.8%) would otherwise be unactionable. In addition, acquiring ultra-high depth also ensured a low false discovery rate (less than 2.2%) from FFPE samples.
Conclusion
Our results were as accurate as a commercially available CLIA-compliant hotspot panel, but allowed the detection of a higher number of mutations in actionable genes. Our study revealed the critical importance of acquiring and utilizing high depth in profiling clinical tumor samples and presented a very useful platform for implementing routine sequencing in a cancer care institution.
Keywords: cancer, sequencing, high-depth, mutations, copy number
INTRODUCTION
Next generation sequencing (NGS) can significantly facilitate personalized cancer therapy approaches by identifying actionable somatic events in tumor samples (1). Furthermore, high-quality sequencing data can reveal associations with sensitivity or resistance that can inform the development and implementation of targeted therapeutics and, in particular, aid in the design of future trials to validate findings and actionability. Critical alterations include mutations (single nucleotide variations [SNVs] and insertions and deletions [indels]), copy number variations (CNVs) and rearrangements that can potentially predict response and resistance to targeted agents. Whole genome (WGS) and whole exome sequencing (WES) allow the detection of SNVs, indels, CNVs, and rearrangements. However, the relatively low coverage of WGS and WES, as currently implemented in most of the sequencing laboratories (100–250x), may have limited ability to cost-effectively detect aberrations that are present in a subpopulation of tumor cells while identifying a myriad of aberrations of unknown clinical significance (2). Somatic aberrations present at low allele frequencies across different types of tumors (3, 4) can potentially impact patient prognosis or response (5) and thus are important to be reliably detected. Targeted sequencing to depth that allows detection of relatively low mutant allele frequency (MAF) may represent an alternative or a complement to WGS and WES to detect clinically relevant alterations. Additionally, in most clinical and research settings, the amount of DNA that can be isolated from tumor samples is limited and the DNA is often damaged due to fixation and storage procedures, such as formalin fixed paraffin embedded (FFPE) samples. Therefore a multiplexed targeted platform that can generate reliable data with high sensitivity from limited amounts of DNA from FFPE is needed. Several targeted sequencing panels have been successfully implemented (6, 7). However, the details of a platform’s design and parameterization will influence the precision and reliability of the molecular profiling results, impacting both translational research and clinical decision-making. Thus, it is of great value to explore multiple potential solutions in a real patient care environment until a community wide solution is established, validated, and well accepted.
To identify such a solution, we implemented a deep targeted sequencing platform designed to identify actionable and clinically relevant DNA alterations in 201 cancer-relevant genes (about 5,000 exons and 1 Megabase [Mb] of sequence) in clinical samples. Our platform, called T200, was optimized for FFPE specimens and low-input DNA. We also optimized the mutation detection approach to reliably detect low-frequency mutations as well CNVs. The data presented here demonstrate the feasibility, challenges and advantages of a targeted high-depth platform over broader sequencing approaches using clinical specimens and its relevance for cancer research and care.
METHODS
1. Selection of the genes
We selected 201 genes (Supplemental Table 1) that are biologically relevant in cancer, based on mutational data in the Catalogue of Somatic Mutations in Cancer (COSMIC) (8) and The Cancer Genome Atlas (TCGA). Using those databases, we included genes found mutated in 5% or more of the samples tested across all cancer types (all diseases combined) and in 3% or more of the samples in one specific cancer type (e.g. breast cancer) when there was at least 50 samples analyzed. We gave priority to genes or pathways targeted by a drug that was commercially available, in clinical trials, or under late stage preclinical development. Prior to implementation of the T200 platform, input on cancer relevant genes was sought from faculty across The University of Texas MD Anderson Cancer Center. Because depth was important and we desired a panel no larger than 1Mb, a number of large genes previously shown mutated in cancer but with no direct clinical implications were not included, such as TTN (titin), NSD2 (Wolf-Hirschhorn syndrome candidate 1) and MACF1 (microtubule-actin crosslinking factor 1). Altogether, the T200 panel is comprised of 4,874 exons encoding 938,607 bases. Our sequencing pipeline consisted of DNA extraction from the sample, library preparation, target enrichment, sequencing and data analysis (Supp. Fig. 1). Data analysis was optimized for mutation calling from deep coverage (see section 8 of Methods).
Additional experimental details are described in Supplemental Information.
RESULTS
1. Relevance of high-depth and DNA input
We aimed to determine a minimum sequencing depth at which we were confident in detecting very low-frequency mutations (as low as 1%). Achieving such sensitivity with high confidence is challenging given the degraded nature of FFPE samples and the errors intrinsic to the next generation sequencing (9, 10). According to our power estimation (described in Methods section 6.2), about 1,000 unique reads are required to ensure detection of 1% frequency alleles with a false discovery rate (FDR) of no more than 1% (Figs. 1A, B). In contrast, at a sequence-coverage of 50 reads, the 1% alleles can merely be detected with a 10% chance and with limited confidence (Fig. 1A).
Figure 1. Technical characterization of T200.

(A) Chance of detecting (i.e., observing 2 or more reads from) a 1% frequency variant allele (Y axis) from a sequencing coverage (X axis) of 10 to 2000 folds. (B) The minimal allele frequency (Y axis) that can be confidently (FDR < 1%) detected at a given sequencing coverage (X axis). (C,D) DNA input and duplicate rates (C) and depth of coverage (D). Each dot represents one tumor sample. Areas circled in red: high duplicate rate and low coverage in samples with low DNA input; areas circled in blue and green: ideal range of DNA input to obtain low duplicate rate and desired coverage; areas circled in gray: high duplicate rate and low coverage despite the DNA input.
Since the amount of DNA available in clinical settings is limited, we assessed the impact of DNA input in the coverage. We established a minimum of 170 nanograms (ng) of DNA input for our pipeline, although we were able to generate data with as little as 50 ng. This cutoff was established based on our data from FFPE clinical specimens showing that less than 170 ng of DNA drastically increased duplicate rate, consequently decreasing sequencing depth (Fig. 1C and 2D, respectively). Furthermore, with less than 100 ng of input DNA, the duplicate rate was higher than 70% while the highest coverage achieved was 347x (red rectangles in Figs. 1C, D), which would make it impossible to call 1% frequency mutations from 15% of target sites (according to Fig. 1A). Interestingly, our data shows that DNA input higher than 500 ng did not necessarily improve the performance in terms of either duplicate rate or coverage. Therefore, the ideal range for DNA input on this platform was between 170 and 500 ng (blue and green rectangles in Figs. 1C and 1D). Samples highlighted in gray rectangles showed high duplicate rate (Fig. 1C) and low depth (Fig. 1D) despite the amount of DNA input, which was probably an effect of different levels of DNA damage in the FFPE sample pool.
Figure 2. Concordance between fresh-frozen and FFPE samples.
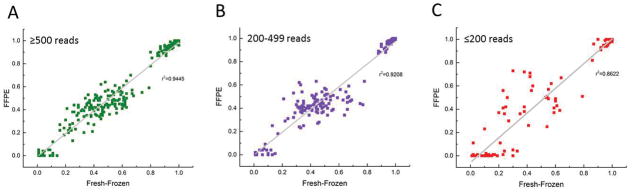
Each dot represents an SNV in a matched pair of fresh-frozen and FFPE tumor samples detected at above 500 reads (high correlation) (A) between 200 and 499 reads (medium to high correlation) (B) and below 200 reads (low correlation) (C).
2. Platform performance, sensitivity and specificity
As most of the tumor samples in our institution and many others around the world are FFPE derived, it was important to ensure that our platform was prepared to handle this type of material, despite the DNA damage that the fixation process introduces (10). To test the robustness of the T200 pipeline in FFPE samples, we sequenced a pair of matched fresh-frozen and FFPE tumor samples and compared the variant allele frequencies estimated for them. We found high concordances between the fresh-frozen and FFPE tissues in sites with more than 200 reads (r2 ≥ 0.92, Figs. 2A, B). For sites with less than 200 reads, the correlation between fresh-frozen and paraffin decreases markedly with r2=0.86 (Fig. 2C). Another pair of matched fresh-frozen-FFPE pair was independently processed, captured and sequenced showing similar results (Supp. Fig. 2), demonstrating the reproducibility of our platform.
We also examined the accuracy of copy number prediction using our platform. We compared the ERBB2 (Her2) copy number status measured in tumor samples by the T200 platform with those obtained from fluorescent in situ hybridization (FISH) or immunohistochemistry (IHC) for the same samples. We found an accurate (98.3%) classification rate for the high copy number (>=5) amplifications (Supp. Fig. 3). In addition, the high-depth allowed robust detection of focal alterations, as demonstrated by a sharp dip in read counts across exons removed by a homozygous EGFR vIII deletion in a brain and a breast cancer sample (Supp. Fig. 4). We also validated the copy number data for six breast cancer samples using the Oncoscan copy number platform from Affymetrix. Our level of concordance for H. DEL. was 100% for 5 of the samples analyzed. For H. AMP. we found 100% of concordance for 4 out of 6 samples (Supplemental Table 2).
3. Detection of Genomic Alterations in Clinically Actionable Genes in FFPE Tumor Samples
In this study we defined genes as potentially actionable if alterations in the gene may potentially direct treatment options due to: 1) the availability of approved drugs that directly or indirectly target the gene, 2) predicted resistance to existing treatment options, and/or 3) clinical trials selecting for genomic alterations in the gene of interest. Based on this rationale, there are 112 actionable genes in the T200 panel (Supplemental Table 3).
3.1. Depth of coverage in clinical samples
We analyzed 515 tumors (all FFPE) and matched normal (blood) samples of 475 patients in 11 disease sites: breast (22%), skin (20%), brain (16%), colon (15%), sarcoma (9%) as well as ovarian, stomach, kidney, head and neck, lung and prostate (3% or less; Fig. 3A). After mapping and removing duplicate reads, we obtained a median average haploid coverage of 906x on the targeted region of the tumors (Fig. 3B). The median coverage appeared to be consistent in the majority of targeted exons (≥ 200x and ≤ 1400x in ≥ 98.2% of the exons; Supp. Fig. 5A). Not surprisingly, the exons with extremely low coverage (<200x) reside in GC-rich (>70%) regions which are difficult to amplify and sequence (see Supplemental Fig. 5B, C). Most of the exons with extremely high coverage (>1400x) appeared to come a few genes (HYDIN, MLL3) and were likely caused by incompleteness (homologous regions not represented) in the hg19 reference genome (Supplemental Table 4); we therefore excluded mutations detected in those exons. Overall, 98.7% of target sites were covered by at least 200x (Fig. 3C) and 87.6% were covered by at least 500x (not shown).
Figure 3. Mutation profiling of 515 tumor samples using T200.

(A) Distribution of disease sites, (B) mean coverage (Y axis) in each sample (X axis, in no particular order), (C) percentage of target bases (Y axis) that are covered by at least 200x in each sample (X axis in no particular order), (D) median mutation rate per Mb in nine major (>10 samples) cancer types, (E) the mean variant allele frequency cutoff (Y axis) applied in variant calling as a function of mean coverage (X axis) in each sample, (F) number of mutations (Y axis) detected in various allele frequency bins (X axis) for four cancer types.
3.2. Detection of somatic mutations and copy number alterations
We found 4794 non-synonymous somatic mutations (including 4525 SNVs and 269 indels) from 515 tumor samples, with a median mutation rate of one per Mb in sarcoma, three in breast, ovarian and brain cancers, six in colorectal cancer, and seventeen in skin cancer (Fig. 3D). No non-synonymous somatic mutations in the 201 genes were found in 25 (5%) of the tumors pairs. The uniformly deep coverage across exons allowed sensitive and accurate detection of mutations with MAF as low as 1% at most of the target sites. To achieve a low false-discovery rate (around 1%), we applied stringent MAF cutoffs on our calls, which averaged from 0.4% to 11% with a median of 2.1% (Fig. 3E). We found 730 (15.23%) low-frequency (<10% MAF) mutations. Among those, 98 (13.4%) were found in the COSMIC, which is similar (p≥0.22, Fisher’s Exact Test (FET)) to the percentage 632/4064 (15.6%) of mutations found in COSMIC among high-frequency (≥10% MAF) mutations.
The low-frequency mutation landscape was similar to those of high-frequency mutations. For example, six of seven low-frequency BRAF mutations occurred at amino-acid position 600 while the seventh occurred at position 594 (not shown). Overall, similar total numbers of mutations were detected between 5–10%, 10–15%, 15–20% and 20–25% MAF (Fig. 3F). In comparison to skin tumors, more mutations were found between 5% and 10% MAF in breast (p<5.7e-9, FET), brain (p<2.8e-8, FET) and colorectal (p<2.5e-12, FET) tumors, indicating a potentially higher degree of intra-tumor heterogeneity or contaminating normal tissue cells in these tumor types.
We identified 35 significantly mutated genes (SMGs) that we defined as those that harbored significantly more non-synonymous mutations than expected by chance from their exon sizes (p<0.01, Poisson test, FDR corrected; Supplemental Table 5). At the top of the SMGs list were well-known driver genes TP53, KRAS, BRAF, NRAS, PTEN, IDH1, PIK3CA, KCNB2, CDKN2A, and EGFR, which were previously shown to be significantly mutated in cancer (11, 12). In our analysis, many genes were significantly mutated only in specific tumor types such as BRAF and NRAS in skin, IDH1 and EGFR in brain, and PIK3CA in breast cancer. These data well recapitulated previous cancer genomics studies (11–13) (Fig. 4).
Figure 4. Significantly mutated genes.
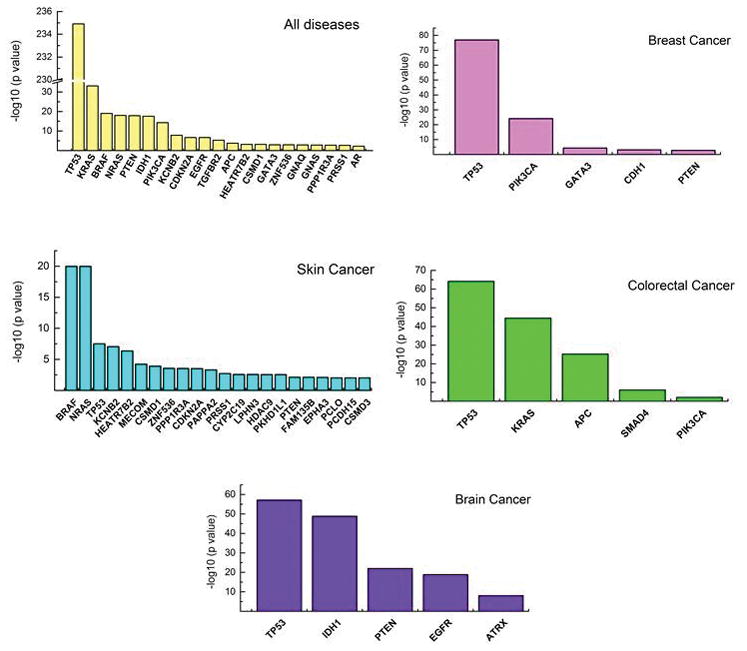
Negative log10 p values (Y axis) of significantly mutated genes (X axis) in five disease categories: all diseases and breast, skin, colorectal and brain cancers.
We found at least one highly amplified (H.AMP) or highly deleted (H.DEL) gene in 175 (34.0%) tumor samples. We define a gene as highly amplified (H.AMP) if its estimated copy number (ECN) is greater or equal to 5. We define a gene as highly deleted (H.DEL) if its ECN is less than or equal to 0.6. From these, we derived a list of 30 genes with significant copy-number alterations (p<0.01, Poisson test, FDR corrected; Supplemental Table 6). The majority of these genes (including NOTCH1, EGFR, RB1, PTEN, CDK4, ERBB2, FGFR1, TP53, NF1 and KIT) were known to have significantly altered copy numbers in cancer (14). Tumor suppressor genes such as RB1, PTEN, TP53, and NF1 were frequently deleted, and oncogenes including EGFR, CDK4, ERBB1, FGFR1, and KIT were frequently amplified. Like the genes with non-synonymous mutations, these genes were altered in a cancer-type specific fashion. For example, EGFR was only significantly amplified in brain while RB1 was only significantly deleted in sarcoma.
Three hundred nineteen of the 475 patients (67%) assessed had high-frequency aberrations in at least one potentially clinically actionable gene (PCAG) (Supplemental Table 3) and 118 (24.84%) patients had low-frequency mutations in PCAG. Forty-seven (9.8%) patients had only low-frequency mutations in PCAGs. Overall 336 (70.73%) patients had non-synonymous mutations in at least one potentially clinically actionable gene (PCAG). In addition to SNVs and indels, we also found that 155 (32.63%) patients had high copy number alterations (CNA) in at least one PCAG. Taken both mutations and CNVs into account, we found alterations in at least one PCAG in 387 (81.47%) of the 475 patients using T200.
The identified non-synonymous mutations appeared to affect the function of PCAGs in a variety of ways (Supplemental Table 7). Most are missense mutations that may not be functional. To obtain more precise characterization, we developed a cancer driver annotator (CanDrA) (15), which estimates the likelihood that a missense SNV in a given cancer type is a driver based on a large set of evidence summarizing aspects of sequence conservation, protein structure, sequence context, mutational spectrum, and mutation prevalence (96 features) in the COSMIC, TCGA and Cancer Cell Line Encyclopedia [CCLE] (16) databases. We annotated each missense SNV in our set as either functional or non-functional using CanDrA, in conjunction with Mutation Assessor, SIFT, PolyPhen, and ConDel among others (17). When we excluded SNVs that were annotated as non-functional by any of these annotators, 216 (45.47%) patients had potential functional mutations in at least one PCAG (Supplemental Table 8). Among these patients, 60 (27.8%) had low-frequency functional mutations; 77 (35.6%) had two or more potentially functional mutations, which may pose both opportunities and challenges for clinical decision-making.
Among the 475 patients, 25 had both primary and metastatic tumors sequenced. Although derived from the same patients, these tumor samples may differ considerably due to tumor evolution (3). Nonetheless, 548 (78.4%) of the 699 of mutations in the metastasis samples were present in the corresponding primary tumors. This result not only demonstrated the reproducibility of our assay but also suggested that clonal heterogeneity may be developed in only a subset of patients. Indeed, more than half (89/151) of non-recurring mutations were found in 15 tumor samples derived from eight patients with colorectal cancer. Among them, at least 4 primary-metastasis pairs from different patients demonstrated a microsatellite instability (MSI) phenotype with high mutation rates (7–48 mutations per Mb) and harbored functional somatic mutations in TGFBR2 and BRAF which are suggestive of MSI or germline deleterious mutations in mismatch repairing genes MSH2 and MSH6 (18, 19) (not shown). Mutations in these samples clustered at different allele frequencies (3 pairs shown in Supplemental Fig. 6). Unlike previous studies (3, 20), we could confidently identify mutations clustering, around 5% allele frequencies, even though we only sequenced the exons of 201 genes. Some of these low-frequency mutations were found in important cancer genes such as MAP2K4, DDR2 and MLL2 (see Supplemental Fig. 6A, B). A nonsense mutation APC E1309* was detected at a mere 2.33% allele frequency (see Supplemental Fig. 6C). All of the frame-shift mutations in TGFBR2 occurred in low-frequency, suggesting the presence of MSI sub-clones in these tumors. Overall, significantly more non-recurring mutations (61/151 vs. 29/548, p<2.2e-16, FET) were in lower frequency than recurring mutations, suggesting that a subgroup of the non-recurring mutations may be present in both the primary and the metastasis but was below the sensitivity of detection at that allele in either sample.
4. Concordance with other sequencing platforms
Validation of next-generation sequencing data is critical especially when this technology is used to screen. All the samples tested in this study were tested on the AmpliSeq46 gene panel in a CLIA accredited clinical laboratory at The University of Texas MD Anderson Cancer Center (21). The AmpliSeq46 was designed by Ion Torrent (Life Technologies) and it is an amplicon based panel of 46 cancer-related genes (Supplemental Table 9). All the genes in this panel except ERBB4 and SRC are also in the T200 gene panel. We found that 98.2% of mutations identified by CMS46 were also identified by T200 (Fig. 5A). It is important to mention that the DNA samples used in both platforms (T200 and AmpliSeq46) were extracted from different batches of slides from the same paraffin block for each patient. We also compared the MAFs for the most common PIK3CA mutations detected by both sequencing platforms and found similar frequencies for the sites compared (Fig. 5B). Mutations found at lower than 10% MAF and identified by both platforms are represented in Fig. 5C, showing the reliability of the T200 platform to detect the low-frequency mutations.
Figure 5. Concordance with other sequencing platforms.

All the samples tested in this study were also tested in the AmpliSeq46 gene panel in a CLIA accredited clinical laboratory. (A) 98.2% of mutations identified by AmpliSeq46 were also identified by T200. (B) Allele frequency of PIK3CA mutations identified in different patients by AmpliSeq46 (pink bars) and T200 (blue bars). (C) Mutations found at lower than 10% MAF by T200 in different cancer types validated by AmpliSeq46. (D) Peaks obtained from Sequenom validation of two new mutations identified by T200 (NF1_Q554* and SMARCA4_R1665*). A complete list of all sites tested on Sequenom can be found in the supplemental material.
To estimate the false discovery rate of T200, we randomly selected 98 novel T200 mutations that were not covered by the AmpliSeq46 platform, not found in the COSMIC v63, and having allele frequencies greater than 10%. We re-tested them using Sequenom MassArray™ (Supplemental Table 10). Out of the 98 new sites tested, 96 were validated, which indicated a 2.0% false discovery rate. The wild type and mutant peaks for two of the sites tested and validated are shown in Fig. 5D.
DISCUSSION
Our study describes a newly developed targeted DNA sequencing platform to screen 201 genes (all exons) in tumor DNA samples. The platform is suitable to be implemented in clinical settings and can greatly contribute to the tailoring of cancer treatment to specific patients.
We first evaluated the effects of sequencing depth and DNA input on the sensitivity of our platform. Our data clearly showed that although we could obtain data from as low as 50 ng of input genomic DNA, amounts less than 100 ng significantly increased the duplicate rate, consequently decreasing the depth – probably owing to the limited library diversity - and thus limiting our ability to call rare events. Importantly, although we had access to only two to four FFPE slides for most of the patients, only a small percentage of patients (<5%) did not have enough DNA for sequencing. Furthermore, despite the relatively low-input and the degraded nature of the FFPE DNA our failure rate, defined by less than 10 median non redundant reads, was lower than 2%.
Another goal of our study was to compare fresh-frozen and matched FFPE samples and assess how the depth of sequencing could impact the performance of more challenging samples such as FFPE. The correlation between MAF in fresh-frozen and matched paraffin samples was expressively high at medium and high-depth, but was significantly lower at sequencing depth lower than 200x. Thus, using approaches that do not routinely reach the required depth for FFPE samples can be challenging and may be risky regarding mutation identification. Importantly, the difference between assessment of paraffin and fresh-frozen samples, which by necessity are from different parts of the tumor, may represent intratumoral heterogeneity. However, at least in these cases, very little intratumoral heterogeneity in this set of cancer genes was present (maximum of 3%).
We then sequenced 515 tumor samples and their matched normal samples across several disease sites. The average coverage with our platform was higher 900x, with some regions with lower coverage, most of which were found to be CG enriched. The mutational landscape revealed across the disease sites was similar to those found by other large sequencing databases such as TCGA, demonstrating the robustness and reliability of the T200 platform. Our data also show that this platform can identify potential sub-clones and heterogeneity between primary and metastatic samples. Additionally, the fact that the low-frequency mutation landscape was similar to those of high-frequency mutations suggests that the low-frequency mutations are unlikely false positives, and reinforces the importance of a high-depth sequencing platform. In our study, the deep coverage enabled sensitive discovery of mutations in as low as 1% MAF, which would be important either when tumor content is low or when tumors are heterogeneous. Consequently, we were able to detect mutations in potentially clinically actionable genes in 81.5% of patients, which was substantially more than the percentage of patients with mutations in actionable genes based on a CLIA compliant hotspot assay (AmpliSeq46) (not shown). However it should be emphasized that many of the alterations identified with T200 were missense mutations with unknown biological and clinical implications; thus not all mutations in actionable genes may be clinically actionable.
Although our results were obtained in a preclinical research environment, through an IRB-approved protocol, they were used to guide clinical decision-making. All the T200 results were returned to treating physicians, who were able to order additional CLIA-compliant assays to confirm alterations that were relevant for decision-making. Notably the reported prospective laboratory protocol was focused on discovery of somatic alterations. However, our sequencing of matched germline DNA made it possible not only to reliably distinguish somatic and germline mutations but also, enabled us to uncover pathogenic germline mutations that may indicate the potential risks for and heritability of a wide spectrum of genetic diseases, so-called “incidental findings” (22). Therefore, a companion germline analysis protocol with prospective informed consent has been activated to facilitate both discovery of germline alterations, and to solicit patient preferences of return of incidental results. Germline variant annotation algorithms have just been established and identification and validation of deleterious findings is still ongoing.
The deep coverage ensured a very low false-discovery rate (<2%), as validated by our side-by-side comparison of our results with results obtained from multiple orthogonal assays. Therefore, these highly accurate results could be routinely obtained in important cancer genes from FFPE tumor samples at a reasonable cost. Although a smaller panel of known mutations based on specific cancer types could be an alternative to T200, our platform can potentially identify new unexpected genomic aberrations in specific disease sites which can potentially lead to the development of new clinical trials or increase the enrollment of patients in existing ones.
Our study also revealed a few limitations of T200. One of the main limitations is the turnaround time. The current turnaround time of T200 is 3–4 weeks in the best case scenario, which is not ideal in a clinical environment and demands further improvement. Another limitation is that, as with any other targeted panel, we could not identify mutations outside of our target region. This limitation can be improved by routinely updating the panel on the basis of new discoveries from inside and outside our group, such as those from recent large-scale pan-cancer studies (11, 12, 14) and studies of unusual responders and markers of sensitivity and resistance. We are now in the process of updating our T200 platform by adding new potentially actionable genes and a whole genome copy number scan to allow analysis of regional copy number changes while keeping the panel at no more than 2 Mb to achieve the coverage desired and without dramatically inflating costs.
Taken together, the advantages of targeted exome sequencing in sensitivity, clinical relevance, robustness, and cost, have outweigh its limitations and made it one of the most promising approaches for clinical cancer genomic profiling. The aspects emphasized here seem to be crucially important in clinical scenarios and should be taken in consideration when making treatment decisions based on sequencing data.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by the Sheikh Bin Zayed Al Nahyan Foundation, NCI R01 CA172652 to Ken Chen, 1U01 CA180964, NCATS grant UL1 TR000371 (Center for Clinical and Translational Sciences), the Bosarge Foundation, the MD Anderson Cancer Center Support grant (NIH/NCI P30 CA016672), and the MD Anderson Moon Shot Program. We also thank Sarah J Bronson from MD Anderson Scientific Publications Department for reviewing this manuscript.
Footnotes
DISCLOSURE (Conflict of interest)
GBM: SAB/Consultant: Illumina, AstraZeneca, Blend, Critical Outcome Technologies, HanAl Bio Korea, Nuevolution, Pfizer, Provista Diagnostics, Roche, Signalchem Lifesciences, Symphogen, Tau Therapeutics.
References
- 1.Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, et al. Personalized oncology through integrative high-throughput sequencing: A pilot study. Sci Transl Med. 2011;3:111ra21. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao BY, Pritchard JR, Lauffenburger DA, Hemann MT. Addressing genetic tumor heterogeneity through computationally predictive combination therapy. Cancer Discov. 2014;4:166–74. doi: 10.1158/2159-8290.CD-13-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature biotechnology. 2013;31:1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The cosmic (catalogue of somatic mutations in cancer) database and website. British journal of cancer. 2004;91:355–8. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mardis ER. A decade’s perspective on DNA sequencing technology. Nature. 2011;470:198–203. doi: 10.1038/nature09796. [DOI] [PubMed] [Google Scholar]
- 10.Kerick M, Isau M, Timmermann B, Sultmann H, Herwig R, Krobitsch S, et al. Targeted high throughput sequencing in clinical cancer settings: Formaldehyde fixed-paraffin embedded (ffpe) tumor tissues, input amount and tumor heterogeneity. BMC medical genomics. 2011;4:68. doi: 10.1186/1755-8794-4-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Futreal A, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nature reviews Cancer. 2004;4:177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nature genetics. 2013;45:1134–40. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao Y, Chen H, Liang H, Meric-Bernstam F, Mills GB, Chen K. Candra: Cancer-specific driver missense mutation annotation with optimized features. PloS one. 2013;8:e77945. doi: 10.1371/journal.pone.0077945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey AM, Mao Y, Zeng J, Holla V, Johnson A, Brusco L, et al. Implementation of biomarker-driven cancer therapy: Existing tools and remaining gaps. Discovery medicine. 2014;17:101–14. [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, et al. Germline mutations affecting the proofreading domains of pole and pold1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–44. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, et al. Clonal architecture of secondary acute myeloid leukemia. The New England journal of medicine. 2012 doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh RR, Patel KP, Routbort MJ, Reddy NG, Barkoh BA, Handal B, et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. The Journal of molecular diagnostics: JMD. 2013;15:607–22. doi: 10.1016/j.jmoldx.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Gutmann A. Ethics. The bioethics commission on incidental findings. Science. 2013;342:1321–3. doi: 10.1126/science.1248764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.