

Abstract
Usher syndrome type II (USH2) is an autosomal recessive disorder characterized by moderate to severe hearing impairment and progressive visual loss due to retinitis pigmentosa (RP). To identify novel mutations and determine the frequency of USH2A mutations as a cause of USH2, we have carried out mutation screening of all 72 coding exons and exon–intron splice sites of the USH2A gene. A total of 20 USH2 American probands of European descent were analyzed using single strand conformational polymorphism (SSCP) and direct sequencing methods. Ten different USH2A mutations were identified in 55% of the probands, five of which were novel mutations. The detected mutations include three missense, three frameshifts and four nonsense mutations, with c.2299delG/p.E767fs mutation, accounting for 38.9% of the pathological alleles. Two cases were homozygotes, two cases were compound heterozygotes and one case had complex allele with three variants. In seven probands, only one USH2A mutation was detected and no pathological mutation was found in the remaining eight individuals. Altogether, our data support the fact that c.2299delG/p.E767fs is indeed the most common USH2A mutation found in USH2 patients of European Caucasian background. Thus, if screening for mutations in USH2A is considered, it is reasonable to screen for the c.2299delG mutation first.
Keywords: mutations, usherin, Usher syndrome, USH2, USH2A
INTRODUCTION
Combined deafness and blindness is a feature of at least 40 recognized human syndromes. Usher syndrome (USH) accounts for over 50% of these and has an estimated incidence of 4.4:100 000.1,2 USH is the most frequent cause of recessive retinitis pigmentosa (RP).3 The disorder is both clinically and genetically heterogeneous. Three clinical types have been defined, USH1, USH2 and USH3 on the basis of the severity and age during the onset of hearing impairment, vestibular dysfunction and retinal phenotypes. USH1 is the most severe with congenital profound hearing loss and vestibular dysfunction. USH2 is defined as moderate to severe, early onset, bilateral and symmetric sensorineural hearing loss. The symptomatic onset of RP is often in late adolescence and the vestibular system is normal. USH3 is distinguished from USH2 by the progressive nature of its hearing loss and variable impairment of vestibular function. Although USH3A is relatively rare in most populations, it may account for approximately 40% of USH cases in Finland4–6 and in Ashkenazi Jews.7–9 Progressive pigmentary retinopathy is usually typical for RP in all clinical types, but has been reported as occuring earlier in USH1; however, because of the considerable overlap between the ages of onset of visual disturbance in the USH subtypes, the onset of night blindness cannot be regarded as a reliable diagnostic discriminator.10–15 Further subdivision of USH has been possible based on the genomic loci and causative genes. To date, 11 loci have been mapped for USH and genes for 10 of them have been cloned.16 USH1 is genetically heterogeneous. Five corresponding genes of the seven USH1 loci (USH1B–USH1H) have been identified so far. Known USH1 molecules are the molecular motor myosin VIIa (USH1B), the two cell–cell adhesion proteins, cadherin 23 (USH1D) and protocadherin 15 (USH1F) and the scaffold proteins, harmonin (USH1C), which contains post-synaptic density, disc-large, Zo-1 protein domains (PDZ) domains and SANS (scaffold protein containing ankyrin repeats and SAM domain, USH1G). There are three genetic loci now reported for USH2 (USH2A, USH2C and USH2D). The affected genes at these loci have been determined. The USH2 protein whirlin (USH2D) is also a PDZ domain-containing scaffold protein, whereas the remaining two USH2 proteins, usherin (USH2A) and GPR98 (G protein-coupled receptor 98 also known as VLGR1b or MASS1) (USH2C), are large transmembrane proteins and have similar pentraxin (PTX) homology domains,17 suggesting that they may share an affinity for a common binding partner. Only one locus for USH3 (USH3A) has been reported so far. USH3A transcripts encode clarin-1, a membrane glycoprotein with four transmembrane domains from the clarin family.
Molecular analysis of USH protein function revealed that all the identified USH proteins are integrated into a protein network.18–24 PDZ domains are protein modules that mediate protein–protein interactions.25 In the inner ear, the PDZ containing proteins harmonin and whirlin are thought to facilitate such protein interactions within the supramolecular USH-protein network.21 Previous molecular analyses and localization studies in mouse models have suggested roles for the USH1 transmembrane proteins, cadherin 23 and protocadherin 15, in the extracellular interstereocilia links in the inner ear sensory cells, specifically in formation of the tip links,26–29 whereas the three USH2 proteins usherin, GPR98 and whirlin were proposed as molecular components of ankle-links between adjacent stereocilia of mechanosensitive hair cells.30–32
Usher syndrome type II is the most frequent type of USH, accounting for over half of the reported cases.12,33 Mutations in the other two known USH2 genes, whirlin (USH2D) and GPR98 (USH2C), are rare.34,35 Mutations in the USH2A gene on chromosome 1q41, encoding usherin, appear to be responsible for up to 85% of the USH2 cases.36,37 Usherin transcript was originally reported to be 5 kb, encoding a putative protein of 170 kDa38 (Figure 1a). A much larger USH2A transcript, expanding the length of coding sequence to 15 kb, encoding a putative 580-kDa protein was subsequently reported.39 The long isoform contains, in addition to the previously known extracellular domains, 2 laminin G and 31 fibronectin type III repeats, as well as a transmembrane region followed by an intracellular domain with a PDZ-binding motif at the C-terminal end, that interacts with the PDZ domain of harmonin and whirlin22,30 (Figure 1b). Although the function of these gene products remains to be fully elucidated, the short variant is predicted to be a secreted, extracellular protein that is found expressed in both the retina and the inner ear.40 The long variant is anchored on the cell membrane with a large extracellular domain that may serve to connect with the USH1 protein network.20 Evidence suggests that this network links the actin core of the hair cell, stereocilia, within the inner ear.18,41
Figure 1.
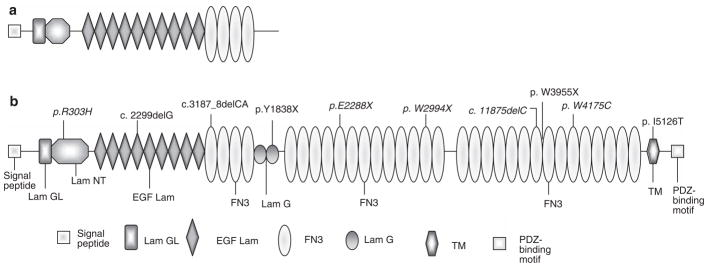
Schematic representation of the short (a) and long (b) isoforms of the Usher syndrome type II (USH2)A protein. The mutations identified in the present study are depicted along the USH2A protein domains (b). Novel mutations are in bold, italic letters. EGF, epidermal growth factor; FN, fibronectin; GL, laminin G like; NT, N-terminal; TM, transmembrane.
Mutations in USH2A have been associated with a wide spectrum of phenotypes including typical USH2, atypical USH and can also be responsible for non-syndromic autosomal recessive RP.42,43 We and others have shown that mutation analysis of the short isoform of usherin encoded by exons 1–21 revealed only 45–61% of the expected USH2A mutations.44–46 The identification of additional 51 exons at the 3′ end of USH2A39 has provided obvious candidates for further mutation screening for molecular diagnosis of USH2A. At present, approximately 110 pathogenic mutations have been reported in the short isoform encoded by exons 1–21 and nearly 80 putative pathogenic mutations were reported in exons 22–72 of the long isoform.47,48 It is interesting to note that with the exception of the c.2299delG/ p.E767fs, many mutations are private and there are few common mutations among probands of different ancestry. In this study, we report the results of USH2A exons 1–72 mutation screening in a cohort of American probands of European ancestry with USH2 syndrome.
MATERIALS AND METHODS
Subjects
A total of 20 American probands of European ancestry were investigated. The clinical and family history was obtained on each proband. Patients were diagnosed with USH2 based on audiological, ophthalmological and vestibular evaluations. A Caucasian diversity panel of 150 unrelated individuals (Coriell cell repositories, Camden, NJ, USA) comprised the control genomic DNA samples. The study was conducted according to the tenets of the Declaration of Helsinki and written informed consent was obtained from all of the participants before donation of blood sample.
Clinical evaluation
Records of audiological examinations were available on many of the patients and were reviewed. Clinical ocular examinations were carried out on all patients. Visual function studies included kinetic perimetry (V-4e target) and full-field electroretinography (ERG). Techniques and details of the data analysis methods have previously been described.49–52
DNA isolation and mutation screening
Blood samples were collected and genomic DNA was extracted using a standard extraction method. The single-strand conformation polymorphism (SSCP) and direct sequencing techniques were used to screen genomic DNA for mutation in USH2A exons 1–72. A total of 86 primer sets were designed to screen the entire 790 kb of genomic DNA using SSCP analysis. The sequences were derived from the cDNA (GenBank accession number AY481573). For exons 2, 6, 13, 17, 41, 42, 61 and 65, two to five overlapping sets of primers, spanning the complete exon, were used to generate suitable polymerase chain reaction (PCR) products for SSCP analysis (primer sequences are available on request). PCR was carried out in a 12.5μl reaction with 40 ng of genomic DNA, 10 pmol of each primer, 200 μM dNTPs, 1.5 mM MgCl2 and 1 U of TaqDNA polymerase. The amplification conditions were as follows: 95 °C for 5 min, then 30 cycles of 95 °C for 1 min, 60 °C for 1 min and 72 °C for 1 min, with final extension for 5 min at 72 °C. For mutation analysis, the PCR products were initially run on a 1 mm thick 8% non-denaturing polyacrylamide gel (PAGE) (acrylamide–N,N′-methylene bisacrylamide 49:1) at 4 °C. The gels were developed using a previously described silver staining procedure.53 Samples with altered migration in the SSCP analysis were sequenced using an automated sequencer (ABI 3100).
RESULTS
In total, 10 different USH2A mutations were identified in 60% (12 out of 20) of the probands, 5 of which have not been previously reported. Two novel missense mutations were identified. Three frameshifts and four nonsense mutations were detected in our cohort. One of the frameshift mutations and two of the nonsense mutations are novel. In 5 out of 20 probands (25%), biallelic USH2A mutations were identified that were likely pathological. Two cases were homozygotes for the c.2299delG/p.E767fs mutation,44,54 two cases were compound heterozygotes (one, p.R303H/p.W3955X;39 and one, p.W2994X/ p.W4175C), and one case had complex allele with three variants (p.E2288X/p.E2288X/c.3187-3188delCA43). In 7 out of 20 (35%), only one USH2A mutation was detected. No pathological mutations were found in the remaining eight probands. The USH2A genotypes of those patients are summarized in Table 1. A schematic representation of the distribution of the mutations identified in the present study, along the USH2A protein domains, is shown in Figure 1b. Three mutations causing frameshifts with premature termination were detected in this study. A previously identified single-base deletion of a guanosine residue in exon 13, at nucleotide position 2299 (c.2299delG/p.E767fs),44,54 caused a frameshift at amino acid position 767, followed by a stop codon 20 amino-acid residues downstream. This mutation was predicted to give rise to a truncated protein of approximately 85% of the size of normal transcript. We identified five individuals with the c.2299delG; two of them were homozygous, and three were heterozygous with only one identifiable mutation. Thus, the c.2299delG mutation was the most prevalent mutation, accounting for 38.9% (7 out of 18) of the pathological alleles. One previously reported USH2A deletion c.3187_3188delCA43 was identified in our cohort. The deletion is a two-base deletion of CA at position 3187–3188 (c.3187_3188delCA) in exon 16. This mutation caused a frameshift at codon 1063 resulting in a premature stop at codon 1077 in the first fibronectin type III (FN3) repeat of USH2A. One homozygous proband for p.E2288X was found to carry the c.3187_3188delCA mutation in a complex allele state (p.E2288X/p.E2288X/c.3187_3188delCA), suggesting that p.E2288X and c.3187_3188delCA can occur in cis. Family members were not available for us to carry out segregation analyses to determine on which parental chromosome the two mutations are in cis. One new USH2A deletion was identified in our cohort. The new mutation consists of a deletion of a C at nucleotide position 11875 in exon 61, that results in a frameshift, starting at codon 3959, with a subsequent premature stop codon of 25 amino acids residues downstream. This mutation was identified in a heterozygous state in one proband. Furthermore, we have identified four different nonsense mutations. Of these, two are new. One of the four nonsense mutations occurs in the second laminin G (LamG) domain and the remaining three are located in the FN3 repeats. The previously described c.11864G>A/p.W3955X mutation in exon 61,39 was detected in a compound heterozygous state with the c.908G>A/p.R303H mutation. The identified nonsense mutation c.5514T>A/p.Y1838X48 was found in a heterozygous state with a second unidentified allele. The first novel nonsense mutation c.6862G>T/p.E2288X was detected in a complex allele state with three variants with the c.3187_3188delCA mutation (p.E2288X/p.E2288X/c.3187-8delCA). The second novel mutation c.8981G> A/p.W2994X was found in a compound heterozygous state with a missense c.12525G>C/p.W4175C mutation. In addition, three novel missense mutations were identified in the present study, with one of them found in the laminin N-terminal (Lam NT) domain, one in the FN3 and one in the transmembrane region. The c.908G>A/p.R303H and c.12525G>C/p.W4175C mutations were detected in compound heterozygous states with the c.11864G>A/p.W3955X and c.8981G>A/p.W2994X mutations, respectively. The missense (c.15377T>C/p.I5126T) mutation was observed in a heterozygous state with the mutation on the second allele that could not be detected. This change occurs within the transmembrane domain, suggesting that it may interfere with the function of Usherin in cell adhesion and maintenance of the cellular cytoskeleton. The p.I5126T caused by the T–C transition, affecting nucleotide 15377 in exon 71, results in the replacement of isoleucine, a non-polar/large/nonaromatic amino acid, with an uncharged polar amino acid threonine. These substitutions thus could have consequences that are unpredictable unless the structure of each of the mutant proteins is determined. The three missense changes might be pathogenic, on the basis of the following criteria: (1) these substitutions were absent in 150 unrelated control DNA samples of Caucasian background; (2) the three newly identified changes are located within functional domains of the USH2A protein (Figure 1b); (3) interspecies comparison of USH2A shows that three of the identified missense substitutions (p.R303, p.W4175 and p.I5126) are evolutionary conserved between human, chimpanzee (P. troglodytes), domestic dog (C. familiaris) and mouse (Figure 2). Finally, we identified a total of 21 variants or polymorphisms in the USH2A gene that we categorized as non-pathogenic. Seven of these were novel. With the exception of the c.373G>A/p.A125T (20 out of 40 USH2A alleles) and the c.4457G>A/p.R1486K (24 out of 40 USH2A alleles), the polymorphisms detected in the present investigation were found to be rare among 20 patients and 50 normal controls tested for their presence (Table 2).
Table 1.
Likely pathologic mutations in USH2A detected in 20 Usher type II patients
| Location in exon | Base change | Predicted codon change | Number of alleles | Individual’s genotype(s) | References |
|---|---|---|---|---|---|
| Exon 6 | c.908G>Aa | p.R303H | 1 | Compound heterozygote with p.W3955X (1) | This study |
| Exon13 | c.2299delG | p.E767fs | 7 | Homozygous (2)/Heterozygotes (3) | 44,54 |
| Exon16 | c.3187_3188delCA | p.Q1063fs | 1 | Complex allele with p.E2288X homozygous (1) | 43 |
| Exon 27 | c.5514T>A | p.Y1838X | 1 | Heterozygote (1) | 48 |
| Exon 36 | c.6862G>Ta | p.E2288X | 2 | Complex allele with c.3187delCA (1) | This study |
| Exon 45 | c.8981G>Aa | p.W2994X | 2 | Compound heterozygote with p.W4175C (1) Heterozygote (1) | This study |
| Exon 61 | c.11864G>A | p.W3955X | 1 | Compound heterozygote with p.R303H (1) | 39 |
| Exon 61 | c.11875delCa | p.Q3959fs | 1 | Heterozygote (1) | This study |
| Exon 63 | c.12525G>Ca | p.W4175C | 1 | Compound heterozygote with p.W2994X (1) | This study |
| Exon 71 | c.15377T>C | p.I5126T | 1 | Heterozygote (1) | 48 |
Numbers in the parentheses are the number of individuals with the mutant allele identified.
Novel changes identified in the present study.
Figure 2.

Multiple sequence alignment showing conservation of residues R303, W4175 and I5126.
Table 2.
Polymorphisms detected in the USH2A gene in Usher type II patients and in normal controls
| Polymorphisms | Location in exon | Predicted codon change | Alleles frequency in USH2A patients | Alleles in control chromosomesa |
|---|---|---|---|---|
| c.373G>A, | 2 | p.A125T | 20/40 | |
| c.b IVS19-44delG | 19 | 2/40 | 0/100 | |
| c.4457G>A | 21 | p.R1486K | 24/40 | |
| c.4714C>T | 22 | p.L1572F | 4/40 | |
| c.b IVS23+5G>T | 23 | 3/40 | 0/100 | |
| c.4994T>G | 25 | p.I1665T | 2/40 | |
| c.5013C>A | 25 | p.G1671G | 2/40 | |
| c.b IVS28-34delC | 28 | 4/40 | 8/100 | |
| c.6506T>C | 34 | p.I2169T | 2/40 | |
| c.b IVS36+8T>G | 36 | 1/40 | 11/100 | |
| c.8624G>A | 43 | p.R2875Q | 1/40 | |
| c.8656C>T | 43 | p.I2886E | 1/40 | |
| c.b IVS47-79G>A | 47 | 1/40 | 4/100 | |
| c.b IVS49+51-3delA | 49 | 1/40 | 3/100 | |
| c.9595A>Gb | 49 | p.N3199D | 1/40 | |
| c.10232A>C | 52 | p.E3411A | 12/40 | |
| c.b12093C>T | 62 | p.Y4031Y | 1/40 | 2/100 |
| c.b12612A>G | 63 | p.T4204T | 7/40 | 0/100 |
| c.13191G>A | 63 | p.E4397E | 2/40 | |
| c. 13297G>Tb | 63 | p.V4433L | 1/40 | 4/100 |
| c.b IVS71-24T>C | 71 | 1/40 | 0/100 |
Only newly detected polymorphisms are tested in normal controls for their presence.
New changes in this study.
Clinical characteristics of 12 patients with USH2A gene mutations are provided in Table 3. In these patients the sensorineural hearing loss was bilateral, ranged from mild in the lower frequencies and moderate to severe in the higher frequencies and, in two cases, was progressive. There was a range of visual acuities and refractive errors in the patients. Visual field extent with a large bright target (V-4e) could be normal in early disease stages and reduced to a small central island in more advanced stages. There was a spectrum of ERG abnormalities but most patients showed rod more than cone dysfunction, in keeping with our earlier reports using these and other methods in this USH genotype.51,52 ERGs were not detectable in five patients, all of whom were in the fourth decade of life. In younger patients, ERGs could show a non-detectable rod b-wave and reduced cone responses. One patient (P11), at 17 years of age had normal ERG amplitudes, whereas her fraternal twin (P12) had more severe disease expression. Details of retinal phenotypes of the dizygotic twin sisters have previously been described.51 Major discordances in retinal dysfunction between the twins add further support to the contention that there may be involvement of modifier genes and environmental factors in USH2A.
Table 3.
Clinical characteristics of the USH2A patients
| Patient | Mutation | Age at First Visit (y)/Gender | Hearing impairment | Eye | Visual acuity | Refraction | Kinetic visual field extent (V-4e)a | Electroretinograms
|
|
|---|---|---|---|---|---|---|---|---|---|
| Rod b-Wave Amplitude | Cone Flicker Amplitude | ||||||||
| P1 | R303H/W3955X | 44/F | NA | RE | 20/20 | −3.75+0.50×041 | 64 | Not detectable | Not detectable |
| LE | 20/40 | −2.00 | 39 | ||||||
| P2 | W4175C/W2994X | 46/F | Moderate/Severe | RE | 20/60 | Plano | <1 | NP | NP |
| LE | 20/100 | Pseudophakia | <1 | ||||||
| P3 | 3187-8delCA/E2288X/E2288X | 42/M | NA | RE | 20/60 | −2.50+3.50×090 | 18 | Not detectable | Not detectable |
| LE | 20/50 | −2.25+3.50×090 | 20 | ||||||
| P4 | I5126T/+ | 41/M | NA | RE | 20/60 | −6.00+0.50×090 | 3 | Not detectable | Not detectable |
| LE | 20/50 | −6.25+0.50×090 | 6 | ||||||
| P5b | E767fs/+ | 56/F | NA | RE | 20/30 | −0.75+0.50×85 | 1 | NP | NP |
| LE | 20/40 | −0.50 | 1 | ||||||
| P6b | E767fs/+ | 44/M | Moderate/Severe | RE | 20/40 | −2.50+1.00×105 | <1 | NP | NP |
| LE | 20/50 | −4.00+2.50×105 | <1 | ||||||
| P7 | Y1838X/+ | 31/M | Moderate/Severe | RE | 20/30 | −2.00+1.00×090 | 10 | NP | NP |
| LE | 20/200 | −9.25+4.25×010 | 9 | ||||||
| P8 | Q3959fs/+ | 15/M | Moderate/Severe | RE | 20/20 | −1.75+1.75×097 | 100 | Not detectable | Reduced |
| LE | 20/20 | −1.50+1.25×086 | 88 | ||||||
| P9b | E767fs/+ | 38/F | Moderate/Severe | RE | 20/25 | −0.50 | 62 | Not detectable | Not detectable |
| LE | 20/25 | −0.50 | 61 | ||||||
| P10 | W2994X/+ | 38/F | NA | RE | 20/30 | −1.00 | 7 | Not detectable | Not detectable |
| LE | 20/25 | −0.75+0.75×093 | 6 | ||||||
| P11b | E767fs/E767fs | 17/F | Moderate/Severe | RE | 20/20 | −0.50 | 93 | Normal | Normal |
| LE | 20/20 | Plano | 93 | ||||||
| P12b | E767fs/E767fs | 17/F | Moderate/Severe | RE | 20/20 | −0.25+0.75×105 | 96 | Not detectable | Reduced |
| LE | 20/20 | −0.25+0.75×090 | 100 | ||||||
Abbreviations: LE, left eye; NP, not performed; RE, right eye.
Expressed as a percentage of the normal mean for the V-4e target; 2 s.d. below the mean is 90% (Jacobson et al.50).
Patients with some data previously reported in Schwartz et al.
DISCUSSION
To date, approximately 190 mutations in the USH2A gene have been reported in patients with USH2.47,48 In this study, altogether a total of 18 mutated alleles were identified in a screening of 20 American USH2 patients of European ancestry. A high diversity of mutations was found in our cohort of patients: three missense, three frameshift, and four nonsense mutations. Of the 40 independent USH2A alleles, 18 were shown to carry a mutation (45%). Mutations in USH2A have been identified in 12 of 20 (60%) probands with USH2 phenotype. In seven individuals, only one mutation was detected (7/20=35%), two patients carried two pathogenic mutations (E767fs homozygotes) and two carried two possibly pathogenic mutations (compound heterozygotes with a nonsense and missense) and one case had three potentially pathogenic changes (E2288X/E2288X/3187-3188delCA) in the USH2A gene. In 7 of the 20 patients studied, we were unable to identify the second mutation. No pathological mutations were found in eight probands. It is very likely that we could have missed the disease-causing mutations in the USH2A gene in those cases because of the amplicons analysis, as neither SSCP nor direct sequencing, the two methods used for mutation screening, even combined, detects all mutations present. It is also known that certain types of heterozygous mutations such as exon-spanning deletions or inversions cannot be detected if single-exon amplification is carried out. It is also possible that the mutations are located in the promotor region, the intronic regions and the 3′ and 5′ untranslated regions, which we did not screen. Misdiagnosis could be an alternative reason for failure of mutation detection. Finally, the USH2A locus may be in digenic interactions. In this case, the second mutation might involve a gene whose product interacts with usherin.
The six new mutations described in this study were found to be distributed along the USH2A gene (Figure 1b): the c.908G>A/ p.R303H mutation occurs in the laminin N-terminal domain; Four of the mutations (c.6862G>T/p.E2288X, c.8981G>A/p.W2994X, c.11875delC/p.Q3959fs, c.12525G>C/p.W4175C) are detected in the FN3 repeats tandem of fibronectin type III domains. Finally, the c.15377T>C/p.I5126T was identified within the transmembrane domain of Usherin. The c.2299delG/p.E767fs mutation, located in the EGF Lam domain of the USH2A protein seems to be extremely common in many populations. Prevalence ranging from 0.16–0.44 among patients with USH2A have been reported, depending on the geographic origin of the patients. The c.2299delG has been detected in 25% of families with USH2 from a Spanish population55 and in upto 50% of USH2A families from Europe and the United States of America.37,54,56 Our previous mutation analysis of the USH2A gene have shown that the 2299delG mutation can also lead to atypical USH.54 In this study, the c.2299delG mutation occurred in 5 probands in a group of 20, thereby accounting for 25% (5/20) of USH2A mutant alleles. Our present data further support the finding that c.2299delG is indeed the most common mutation found in the USH2A gene in USH2 patients of European descent. Our study confirms the absolute necessity to screen the entire USH2A gene consisting of 72 exons, as 10 out of 18 mutated alleles identified in this study are located in exons 22–72 of the gene. It also indicates that, if screening for mutations in USH2A is considered, it is reasonable to screen for the c.2299delG mutation first.
Mutations in the USH2A gene display a wide phenotypic spectrum. This latter underlines how the relationship between pathogenetic mutations and disease phenotype is becoming increasingly complex. The most common mutation in USH2A, 2299delG, causes typical USH and we have shown that it can also lead to atypical USH syndrome.54 Another frequent variant in the USH2A gene, the C759F mutation, has been described in 4.5% of patients with non-syndromic recessive RP.57 Compound heterozygosity for C759F and nonsense, splicing, or missense mutations in the USH2A gene have been reported in cases with recessive RP without hearing loss57,58 and in atypical USH,59 as well as in asymptomatic cases.58 It is also known that there is both interfamilial and intrafamilial phenotypic variation among subjects with the same USH2A mutated genotype.58,60 In accordance with previous reports, we were not able to assign any phenotypic variation to the particular mutations detected in this study. This observation implies that the clinical manifestations of USH2A may be modulated by environmental or genetic modifiers or stochastic factors. This hypothesis is corroborated by our previous data which have shown that phenotypic differences in USH2A are also detected in monozygotic as well as in dizygotic twins.51,54 There are a number of instances whereby identical mutations in a given gene can give rise to diverse phenotypes and, in many diseases, only a subset of all mutations reliably predicts the phenotype.61 The USH2A gene represents another example of such phenotypic variation of a Mendelian disease. Mutation analysis and careful clinical investigation of additional patients with USH2, combined with the study of mutated proteins, should allow a more accurate prediction of the phenotype based on the genotype. Perhaps more importantly, these studies should also provide the basis for a genome wide-scale segregation analyses for mapping and identification of ‘gene modulators’. From this knowledge, we should gain insight into the pathogenic mechanisms underlying deafness and/or blindness in USH. In summary, lack of strong phenotype–genotype correlations and clinical differences between the twin cases indicate that there may be important roles for modifier genes and environmental factors, other than mutations in USH2A gene, and they may have contributed to the development of phenotype in USH2.
Acknowledgments
We thank all the patients for their participation and Ms Sharon B Schwartz for clinical coordination of the study. This work is supported by NIH RO1 DC005575 to Liu XZ.
References
- 1.Vernon M. Usher’s syndrome—deafness and progressive blindness. Clinical cases, prevention, theory and literature survey. J Chronic Dis. 1969;22:133–153. doi: 10.1016/0021-9681(69)90055-1. [DOI] [PubMed] [Google Scholar]
- 2.Boughman JA, Vernon M, Shaver KA. Usher syndrome: definition and estimate of prevalence from two high-risk populations. J Chronic Dis. 1983;36:595–603. doi: 10.1016/0021-9681(83)90147-9. [DOI] [PubMed] [Google Scholar]
- 3.Keats BJ, Corey DP. The Usher syndromes. Am J Med Genet. 1999;89:158–166. [PubMed] [Google Scholar]
- 4.Karjalainen S, Pakarinen L, Terasvirta M, Kaariainen H, Vartiainen E. Progressive hearing loss in Usher’s syndrome. Ann Otol Rhinol Laryngol. 1989;98:863–866. doi: 10.1177/000348948909801106. [DOI] [PubMed] [Google Scholar]
- 5.Pakarinen L, Karjalainen S, Simola KO, Laippala P, Kaitalo H. Usher’s syndrome type 3 in Finland. Laryngoscope. 1995;105:613–617. doi: 10.1288/00005537-199506000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Joensuu T, Hamalainen R, Lehesjoki AE, de la Chapelle A, Sankila EM. A sequence-ready map of the Usher syndrome type III critical region on chromosome 3q. Genomics. 2000;63:409–416. doi: 10.1006/geno.1999.6096. [DOI] [PubMed] [Google Scholar]
- 7.Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, et al. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur J Hum Genet. 2002;10:339–350. doi: 10.1038/sj.ejhg.5200831. [DOI] [PubMed] [Google Scholar]
- 8.Fields RR, Zhou G, Huang D, Davis J, Möller C, Jacobson S, et al. Usher syndrome type III: revised genomic structure of the USH3 gene and identification of novel mutations. Am J Hum Genet. 2002;71:607–617. doi: 10.1086/342098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ness SL, Ben-Yosef T, Bar-Lev A, Madeo A, Brewer C, Avraham K, et al. Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J Med Genet. 2003;40:767–772. doi: 10.1136/jmg.40.10.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar A, Fishman G, Torok N. Vestibular and auditory function in Usher’s syndrome. Ann Otol Rhinol Laryngol. 1984;93:600–608. doi: 10.1177/000348948409300613. [DOI] [PubMed] [Google Scholar]
- 11.Moller CG, Kimberling WJ, Davenport SL, Priluck I, White V, Biscone-Halterman K, et al. Usher syndrome: an otoneurologic study. Laryngoscope. 1989;99:73–79. doi: 10.1288/00005537-198901000-00014. [DOI] [PubMed] [Google Scholar]
- 12.Hope CI, Bundey S, Proops D, Fielder AR. Usher syndrome in the city of Birmingham–prevalence and clinical classification. Br J Ophthalmol. 1997;81:46–53. doi: 10.1136/bjo.81.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsilou ET, Rubin BI, Caruso RC, Reed GF, Pikus A, Hejtmancik JF, et al. Usher syndrome clinical types I and II: could ocular symptoms and signs differentiate between the two types? Acta Ophthalmol Scand. 2002;80:196–201. doi: 10.1034/j.1600-0420.2002.800215.x. [DOI] [PubMed] [Google Scholar]
- 14.Iannaccone A, Kritchevsky SB, Ciccarelli ML, Tedesco SA, Macaluso C, Kimberling WJ, et al. Kinetics of visual field loss in Usher syndrome type II. Invest Ophthalmol Vis Sci. 2004;45:784–792. doi: 10.1167/iovs.03-0906. [DOI] [PubMed] [Google Scholar]
- 15.Cohen M, Bitner-Glindzicz M, Luxon L. The changing face of Usher syndrome: clinical implications. Int J Audiol. 2007;46:82–93. doi: 10.1080/14992020600975279. [DOI] [PubMed] [Google Scholar]
- 16.Reiners J, Nagel-Wolfrum K, Jurgens K, Marker T, Volfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res. 2006;83:97–119. doi: 10.1016/j.exer.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Foord SM, Jupe S, Holbrook J. Bioinformatics and type II G-protein-coupled receptors. Biochem Soc Trans. 2002;30:473–479. doi: 10.1042/bst0300473. [DOI] [PubMed] [Google Scholar]
- 18.Boëda B, El Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, et al. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21:6689–6699. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weil D, El Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet. 2003;12:463–471. doi: 10.1093/hmg/ddg051. [DOI] [PubMed] [Google Scholar]
- 20.Reiners J, van Wijk E, Marker T, Zimmermann U, Jurgens K, te Brinke H, et al. Scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type 2. Hum Mol Genet. 2005;14:3933–3943. doi: 10.1093/hmg/ddi417. [DOI] [PubMed] [Google Scholar]
- 21.Adato A, Michel V, Kikkawa Y, Reiners J, Alagramam KN, Weil D, et al. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14:347–356. doi: 10.1093/hmg/ddi031. [DOI] [PubMed] [Google Scholar]
- 22.van Wijk E, van der Zwaag B, Peters T, Zimmermann U, te Brinke H, Kersten FFJ, et al. The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum Mol Genet. 2006;15:751–765. doi: 10.1093/hmg/ddi490. [DOI] [PubMed] [Google Scholar]
- 23.Senften M, Schwander M, Kazmierczak P, Lillo C, Shin JB, Hasson T, et al. Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J Neurosci. 2006;26:2060–2071. doi: 10.1523/JNEUROSCI.4251-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maerker T, van Wijk E, Overlack N, Kersten FF, McGee J, Goldmann T, et al. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum Mol Genet. 2008;17:71–86. doi: 10.1093/hmg/ddm285. [DOI] [PubMed] [Google Scholar]
- 25.Fanning AS, Anderson JM. PDZ domains: fundamental building blocks in the organization of protein complexes at the plasma membrane. J Clin Invest. 1999;103:767–772. doi: 10.1172/JCI6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lagziel A, Ahmed ZM, Schultz JM, Morell RJ, Belyantseva IA, Friedman TB. Spatiotemporal pattern and isoforms of cadherin 23 in wild type and waltzer mice during inner ear hair cell development. Dev Biol. 2005;280:295–306. doi: 10.1016/j.ydbio.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 27.Michel V, Goodyear RJ, Weil D, Marcotti W, Perfettini I, Wolfrum U, et al. Cadherin 23 is a component of the transient lateral links in the developing hair bundles of cochlear sensory cells. Dev Biol. 2005;280:281–294. doi: 10.1016/j.ydbio.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 28.Siemens J, Lillo C, Dumont RA, Reynolds A, Williams DS, Gillespie PG, et al. Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature. 2004;428:950–955. doi: 10.1038/nature02483. [DOI] [PubMed] [Google Scholar]
- 29.Muller U. Cadherins and mechanotransduction by hair cells. Curr Opin Cell Biol. 2008;20:557–566. doi: 10.1016/j.ceb.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adato A, Lefevre G, Delprat B, Michel V, Michalski N, Chardenoux S, et al. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum Mol Genet. 2005;14:3921–3932. doi: 10.1093/hmg/ddi416. [DOI] [PubMed] [Google Scholar]
- 31.McGee J, Goodyear RJ, McMillan DR, Stauffer EA, Holt JR, Locke KG, et al. The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J Neurosci. 2006;26:6543–6553. doi: 10.1523/JNEUROSCI.0693-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michalski N, Michel V, Bahloul A, Lefèvre G, Barral J, Yagi H, et al. Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J Neurosci. 2007;27:6478–6488. doi: 10.1523/JNEUROSCI.0342-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg T, Haim M, Hauch AM, Parving A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin Genet. 1997;51:314–321. doi: 10.1111/j.1399-0004.1997.tb02480.x. [DOI] [PubMed] [Google Scholar]
- 34.Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, et al. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet. 2007;121:203–211. doi: 10.1007/s00439-006-0304-0. [DOI] [PubMed] [Google Scholar]
- 35.Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet. 2004;74:357–366. doi: 10.1086/381685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pieke-Dahl SA, Weston MD, Kimberling WJ. Genetics heterogeneity of the Usher syndromes. Assoc Res Otolaryngol. 1997;20:A870. [Google Scholar]
- 37.Weston MD, Eudy JD, Fujita S, Yoo SF, Usami S, Cremers C, et al. Genomic structure and identification of novel mutations in Usherin, the gene responsible for Usher syndrome Type IIa. Am J Hum Genet. 2000;66:1199–1210. doi: 10.1086/302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998;280:1753–1757. doi: 10.1126/science.280.5370.1753. [DOI] [PubMed] [Google Scholar]
- 39.van Wijk E, Pennings RJ, te Brinke H, Claassen A, Yntema HG, Hoefsloot LH, et al. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet. 2004;74:738–744. doi: 10.1086/383096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhattacharya G, Miller C, Kimberling WJ, Jablonski MM, Cosgrove D. Localization and expression of usherin: a novel basement membrane protein defective in people with Usher’s syndrome type IIa. Hear Res. 2002;163:1–11. doi: 10.1016/s0378-5955(01)00344-6. [DOI] [PubMed] [Google Scholar]
- 41.El-Amraoui A, Petit C. Usher I syndrome: unraveling the mechanisms that underlie the cohesion of the growing hair bundle in inner ear sensory cells. J Cell Sci. 2005;118:4593–4606. doi: 10.1242/jcs.02636. [DOI] [PubMed] [Google Scholar]
- 42.Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene. Am Hum Genet. 2000;66:1975–1978. doi: 10.1086/302926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seyedahmadi BJ, Rivolta C, Keene JA, Berson EL, Dryja TP. Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp Eye Res. 2004;79:167–173. doi: 10.1016/j.exer.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 44.Dreyer B, Tranebjaerg L, Rosenberg T, Weston MD, Kimberling WJ, Nilssen O. Identification of novel USH2A mutations. Eur J Hum Genet. 2000;8:500–506. doi: 10.1038/sj.ejhg.5200491. [DOI] [PubMed] [Google Scholar]
- 45.Ouyang XM, Yan D, Hejtmancik JF, Jacobson SG, Li AR, Du LL, et al. Mutational spectrum in Usher syndrome type II. Clin Genet. 2004;65:288–293. doi: 10.1046/j.1399-0004.2004.00216.x. [DOI] [PubMed] [Google Scholar]
- 46.Pennings RJ, Te Brinke H, Weston MD, Claassen A, Orten DJ, Weekamp H, et al. USH2A mutation analysis in 70 Dutch families with Usher syndrome type II. Hum Mutat. 2004;24:185. doi: 10.1002/humu.9259. [DOI] [PubMed] [Google Scholar]
- 47.Baux D, Faugère V, Larrieu L, Le Guédard-Méreuze S, Hamroun D, Béroud C, et al. UMD-USHbases: a comprehensive set of databases to record and analyse pathogenic mutations and unclassified variants in seven Usher syndrome causing genes. Hum Mutat. 2008;29:E76–E87. doi: 10.1002/humu.20780. [DOI] [PubMed] [Google Scholar]
- 48.Genediagnostic Homepage. http://www.gendia.net.
- 49.Jacobson SG, Voigt WJ, Parel JM, Apáthy PP, Nghiem-Phu L, Myers SW, et al. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology. 1986;93:1604–1611. doi: 10.1016/s0161-6420(86)33522-x. [DOI] [PubMed] [Google Scholar]
- 50.Jacobson SG, Yagasaki K, Feue WJ, Roman AJ. Interocular asymmetry of visual function in heterozygotes of X-linked retinitis pigmentosa. Exp Eye Res. 1989;48:679–691. doi: 10.1016/0014-4835(89)90009-2. [DOI] [PubMed] [Google Scholar]
- 51.Schwartz SB, Aleman TS, Cideciyan AV, Windsor EA, Sumaroka A, Roman AJ, et al. Disease expression in Usher syndrome caused by VLGR1 gene mutation (USH2C) and comparison with USH2A phenotype. Invest Ophthalmol Vis Sci. 2005;46:734–743. doi: 10.1167/iovs.04-1136. [DOI] [PubMed] [Google Scholar]
- 52.Herrera W, Aleman TS, Cideciyan AV, Roman AJ, Banin E, Ben-Yosef T, et al. Retinal disease in Usher syndrome III caused by mutations in the Clarin-1 gene. Invest Ophthalmol Vi Sci. 2008;49:2651–2660. doi: 10.1167/iovs.07-1505. [DOI] [PubMed] [Google Scholar]
- 53.Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJTV, Steel KP, et al. Mutations in the myosin VII A gene cause nonsyndromic recessive deafness. Nat Genet. 1997;16:188–190. doi: 10.1038/ng0697-188. [DOI] [PubMed] [Google Scholar]
- 54.Liu XZ, Hope C, Liang CY, Zou JM, Xu LR, Cole T, et al. A mutation (2314 delG) in the Usher syndrome type IIA gene: high prevalence and phenotypic variation. Am J Hum Genet. 1999;64:1221–1225. doi: 10.1086/302332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beneyto M, Cuevas JM, Millán JM, Espinós C, Mateu E, González-Cabo P, et al. Prevalence of 2314delG mutation in Spanish patients with Usher syndrome type II (USH2) Ophthalmic Genet. 2000;21:123–128. [PubMed] [Google Scholar]
- 56.Leroy BP, Aragon-Martin JA, Weston MD, Bessant DA, Willis C, Webster AR, et al. Spectrum of mutations in USH2A in British patients with Usher syndrome type II. Exp Eye Res. 2001;72:503–509. doi: 10.1006/exer.2000.0978. [DOI] [PubMed] [Google Scholar]
- 57.Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000;66:1975–1978. doi: 10.1086/302926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bernal S, Ayuso C, Antiñolo G, Gimenez A, Borrego S, Trujillo MJ, et al. Mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa: high prevalence and phenotypic variation. J Med Genet. 2003;40:e8. doi: 10.1136/jmg.40.1.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aller E, Najera C, Millan JM, Oltra JS, Pérez-Garrigues H, Vilela C, et al. Genetic analysis of 2299delG and C759F mutations (USH2A) in patients with visual and/or auditory impairments. Eur J Hum Genet. 2004;12:407–410. doi: 10.1038/sj.ejhg.5201138. [DOI] [PubMed] [Google Scholar]
- 60.Bernal S, Medà C, Solans T, Ayuso C, Garcia-Sandoval B, Valverde D, et al. Clinical and genetic studies in Spanish patients with Usher syndrome type II: description of new mutations and evidence for a lack of genotype—phenotype correlation. Clin Genet. 2005;68:204–214. doi: 10.1111/j.1399-0004.2005.00481.x. [DOI] [PubMed] [Google Scholar]
- 61.Dipple KM, McCabe ERB. Phenotypes of patients with ‘simple’ Mendelian disorders are complex traits: Thresholds, modifiers and systems dynamics. Am J Hum Genet. 2000;66:1729–1735. doi: 10.1086/302938. [DOI] [PMC free article] [PubMed] [Google Scholar]