

Abstract
CDKN1B encodes the cyclin-dependent kinase inhibitor p27/Kip1. CDKN1B mutations and polymorphisms are involved in tumorigenesis; specifically, the V109G single nucleotide polymorphism has been linked to different tumours with controversial results. Multiple endocrine neoplasia type 1 (MEN1) is a rare autosomal dominant syndrome, characterized by the development of different types of neuroendocrine tumours and increased incidence of other malignancies. A clear genotype–phenotype correlation in MEN1 has not been established yet. In this study, we assessed whether the CDKN1B V109G polymorphism was associated with the development of aggressive tumours in 55 consecutive patients affected by MEN1. The polymorphism was investigated by PCR amplification of germline DNA followed by direct sequencing. Baseline and follow-up data of tumour types and their severity were collected and associated with the genetic data. MEN1-related aggressive and other malignant tumours of any origin were detected in 16.1% of wild-type and 33.3% of polymorphism allele-bearing patients (P = NS). The time interval between birth and the first aggressive tumour was significantly shorter in patients with the CDKN1B V109G polymorphism (median 46 years) than in those without (median not reached; P = 0.03). Similarly, shorter was the time interval between MEN1 diagnosis and age of the first aggressive tumour (P = 0.02). Overall survival could not be estimated as 96% patients were still alive at the time of the study. In conclusion, CDKN1B V109G polymorphism seems to play a role in the development of aggressive tumours in MEN1.
Keywords: CDKN1B, polymorphisms, MEN1, neuroendocrine tumours, prognosis
Introduction
Multiple endocrine neoplasia type 1 (MEN1) is an inherited tumour syndrome characterized by the occurrence of tumours of the parathyroid glands, anterior pituitary, pancreatic islets and the adrenal glands, as well neuroendocrine tumours (NETs) of the foregut (thymic, bronchial and gastric carcinoids). Other non-endocrine tumours of the skin (angiofibroma, lipoma, collagenoma) and central nervous system (ependimoma, meningioma) may be associated 1. According to the MEN consensus panel, the clinical diagnosis of this syndrome is based on the concomitant occurrence of at least two of the three MEN1-related endocrine tumours (parathyroid adenoma, pituitary adenoma, pancreatic NET). Familial MEN1 is defined by the presence of at least one MEN1-related NET plus at least one-first-degree relative with just one of the three classical tumours or a known MEN1 germline mutation 2. MEN1 is, in fact, caused by mutations of the MEN1 gene mapped on chromosome 11q13 3. More than 1336 different mutations (1133 germline and 203 somatic) have been reported 4, the majority of which are inactivating according to the notion that MEN1 is a tumour suppressor gene. No MEN1 mutational hot spots have been detected and, up to date, a clear correlation between genetic events and the variable clinical expression of the disease has not been established 4,5. The gene product, menin, is an adaptor protein that interacts with multiple partners involved in several cellular processes, including transcription regulation, DNA replication and repair and signal transduction. It is also essential for viability, as reported in Men1 null mutant mice 6. Menin targets several genes including homeobox domain (HOX), human telomerase (hTERT) and nuclear receptor genes and CDKN2C and CDKN1B 7–10. Specifically, CDKN1B encodes a regulatory protein that controls the progression from the G1 to the S phase of the cell cycle. Loss-of-function mutations of the gene have been described and contribute to tumorigenesis, in agreement with the notion that aberrant cell cycle control is one of the hallmarks of cancer 11,12. Cdkn1b mutations in rat are linked to the development of the MENX syndrome characterized by a clinical picture that overlaps MEN1 and MEN2 syndromes. Consistently, CDKN1B mutations are responsible for the MEN4 syndrome in humans that displays the same features as MENX 13. In addition to gene mutations, at least 21 gene polymorphisms have been reported and variably associated with tumour progression. Studies have mainly been focused on a single nucleotide polymorphism (SNP; database: rs2066827) that replaces a valine (V) with a glycine (G) residue at codon 109 and on its relationship with cancer risk. Controversial results have been reported: two studies showed that the G polymorphic allele is associated with an increased risk of oral squamous cell carcinoma and prostate cancer 14,15 whereas others suggested an association with a decreased risk of breast cancer 16 or a more favourable disease progression in sporadic medullary thyroid carcinoma 17. Other reports, finally, showed no association of the CDKN1B V109G polymorphism with breast 18,19 pancreatic 20,21 and prostate cancer risk 22.
The aim of this study was to verify whether the CDKN1B V109G polymorphism is linked to aggressive tumours in MEN1 patients.
Materials and methods
Patients
Fifty-five consecutive MEN1 patients (22 males and 33 females, age range 5–82 years, belonging to 20 families), followed at the Neuroendocrine Tumours Unit, Department of Clinical Medicine and Surgery of the ‘Federico II’ University of Naples, were prospectively enrolled in this study starting from 2005. Patients’ characteristics are reported in Table1. All patients had a diagnosis of MEN1 according to guidelines 2,23: 52 of them were positive and three negative at the genetic screening for MEN1 mutations 2. Twenty of 55 patients had clinically evident MEN1 manifestations, whereas 35 were their first-degree relatives, identified as gene mutations carriers through the genetic screening. Patients were screened for MEN1-related aggressive and other malignant tumours of any origin. MEN1-related aggressive tumours were defined as follows: (i) a pancreatic NET >2 cm in size 23, and (ii) a thoracic (bronchial or thymic) NET of any stage 23. Ninety cases, age- and gender-matched with the enrolled patients, served as control. Thirty-six male and fifty-four female individuals with no history of tumours were recruited among the medical and paramedical personnel of the institutions participating in this study. These criteria were designed to ensure that all of them had a minimal risk of having or ever developing MEN1. After informed consent was obtained, each participant was interviewed using a pretested questionnaire to obtain information on medical history, lifestyles and family history of cancer up to first-degree relatives. Based on this information, the control group does not include individuals with history of breast, hereditary prostate and pancreatic tumours, or any other cancer. All patients and controls were of European descent with a nationwide distribution. Tumour type, clinical manifestations and outcome data collected for all patients were related to the CDNK1B genetic profile. All MEN1 patients enrolled in this study received the same clinical, laboratory and imaging follow-up, according to the most recent MEN1 guidelines 23.
Table 1.
Patients’ characteristics
| Family | Patient no. | Sex | Age at CDKN1B analysis (years) | MEN1 mutation (NM.130803.2) | CDKN1B | MEN1-related manifestations | Age at diagnosis of the first MEN1 manifestation |
|---|---|---|---|---|---|---|---|
| 1 | 1 | F | 50 | c.303delC, exon 2 | POL | PAH, PA, pNET | 42 |
| 1 | 2 | F | 25 | c.303delC, exon 2 | POL | PAH, pNET | 18 |
| 1 | 3 | M | 52 | c.303delC, exon 2 | WT | PAH, PA, pNET | 44 |
| 1 | 4 | M | 26 | c.303delC, exon 2 | WT | PAH, PA | 20 |
| 1 | 5 | F | 23 | c.303delC, exon 2 | WT | PA | 16 |
| 2 | 6 | M | 41 | c.1046delC, exon 7 | POL | PAH, pNET | 32 |
| 2 | 7 | F | 56 | c.1046delC, exon 7 | POL | PAH, PA, pNET, AT | 47 |
| 2 | 8 | M | 35 | c.1046delC, exon 7 | POL | PAH, pNET | 29 |
| 3 | 9 | F | 49 | c.673T>A, p.W225R, exon 4 | POL | PAH | 32 |
| 3 | 10 | F | 26 | c.673T>A, p.W225R, exon 4 | POL | PAH, PA, pNET | 18 |
| 4 | 11 | M | 34 | c.451delAAG, exon 2 | POL | pNET, tNET | 32 |
| 4 | 12 | M | 62 | c.451delAAG, exon 2 | WT | PAH, PA, pNET, AT | 51 |
| 4 | 13 | F | 38 | c.451delAAG, exon 2 | WT | PAH, pNET | 36 |
| 4 | 14 | F | 46 | c.451delAAG, exon 2 | WT | PAH, pNET, AT | 42 |
| 4 | 15 | M | 12 | c.451delAAG, exon 2 | WT | – | – |
| 4 | 16 | F | 15 | c.451delAAG, exon 2 | WT | – | – |
| 4 | 17 | M | 38 | c.451delAAG, exon 2 | WT | PAH | 36 |
| 4 | 18 | F | 28 | c.451delAAG, exon 2 | WT | PAH, PA | 26 |
| 4 | 19 | F | 59 | c.451delAAG, exon 2 | WT | PAH, pNET | 57 |
| 4 | 20 | M | 58 | c.451delAAG, exon 2 | WT | PAH, pNET, AT | 57 |
| 5 | 21 | M | 49 | c.335delA, exon 2 | WT | PAH, PA, pNET, AT | 39 |
| 6 | 22 | M | 53 | c.502G>A,p.G168R exon 3 | POL | PAH, PA, pNET, AT | 49 |
| 7 | 23 | F | 36 | c.557C>A, p.H186R exon3 | POL | PAH, PA, pNET | 33 |
| 7 | 24 | M | 16 | c.557C>A, p.H186R exon3 | POL | – | – |
| 7 | 25 | M | 6 | c.557C>A, p.H186R exon3 | POL | – | – |
| 7 | 26 | F | 53 | c.557C>A, p.H186R exon3 | POL | pNET | 53 |
| 7 | 27 | M | 62 | c.557C>A, p.H186R exon3 | POL | PAH | 61 |
| 7 | 28 | M | 60 | c.557C>A, p.H186R exon3 | POL | PAH, PA | 59 |
| 7 | 29 | M | 66 | c.557C>A, p.H186R exon3 | WT | PAH | 45 |
| 8 | 30 | M | 51 | c.825+1G>A intr5 | POL | PAH, PA, pNET, AT | 49 |
| 8 | 31 | M | 20 | c.825+1G>A intr5 | POL | PAH, PA, pNET | 19 |
| 9 | 32 | F | 43 | c.95C>G, p.P32R exon2 | POL | PAH, PA, tNET | 29 |
| 9 | 33 | F | 82 | c.95C>G, p..P32R exon2 | WT | – | 81 |
| 10 | 34 | F | 60 | c.1576del11(delACTGTCGCTGG), exon 10 | WT | PAH, PA, pNET | 52 |
| 10 | 35 | F | 30 | c.1576del11(delACTGTCGCTGG), exon 10 | WT | PAH, PA, pNET | 17 |
| 11 | 36 | M | 35 | c.1576del11(delACTGTCGCTGG), exon 10 | WT | PAH, PA, pNET | 30 |
| 11 | 37 | F | 62 | c.1576del11(delACTGTCGCTGG), exon 10 | WT | PAH, PA, pNET | 57 |
| 12 | 38 | M | 42 | c.1065+1G>A, intron 7 | WT | PAH, PA, pNET | 37 |
| 13 | 39 | F | 32 | c.799-9G>A, intron 4 | WT | PAH, PA, pNET | 27 |
| 13 | 40 | M | 31 | c.799-9G>A, intron 4 | WT | pNET | 28 |
| 13 | 41 | F | 51 | c.799-9G>A, intron 4 | WT | PAH, pNET, AT | 48 |
| 13 | 42 | F | 8 | c.799-9G>A, intron 4 | WT | – | – |
| 14 | 43 | F | 53 | c.518T>C, p.L173P, exon 3 | WT | PAH, pNET | 36 |
| 14 | 44 | F | 30 | c.518T>C, p.L173P, exon 3 | WT | PAH, pNET | 28 |
| 14 | 45 | F | 49 | c.518T>C, p.L173P, exon 3 | WT | PAH, pNET | 47 |
| 14 | 46 | F | 24 | c.518T>C, p.L173P, exon 3 | WT | PAH | 24 |
| 15 | 47 | F | 50 | c.1061del C, exon 7 | WT | PAH, pNET | 40 |
| 16 | 48 | M | 42 | c.1339C>T, p.Q447X, exon 9 | WT | PAH, PA, pNET, AT | 41 |
| 17 | 49 | F | 39 | c.1258C>T, p.R420X, exon 9 | WT | PAH, PA, pNET | 38 |
| 17 | 50 | F | 5 | c.1258C>T, p.R420X, exon 9 | POL | – | – |
| 17 | 51 | F | 19 | c.1258C>T, p.R420X, exon 9 | POL | – | – |
| 17 | 52 | F | 12 | c.1258C>T, p.R420X, exon 9 | POL | – | – |
| 18 | 53 | F | 58 | Negative | POL | PAH, PA, pNET, renal carcinoma | 45 |
| 19 | 54 | F | 34 | Negative | POL | PAH, pNET | 25 |
| 20 | 55 | F | 44 | Negative | POL | PAH, PA | 43 |
PAH: parathyroid adenoma/hyperplasia; PA: pituitary adenoma; pNET: pancreatic neuroendocrine tumour; tNET: thoracic neuroendocrine tumour; AT: adrenal tumour.
DNA sequencing
Patients were tested for the presence of MEN1 point mutations on the germline DNA extracted from peripheral blood. MEN1 exons 2–10 were PCR amplified and automatically sequenced as previously described 24. Patients tested negative were subjected to the multiplex ligation-dependent probe amplification (MLPA) technique using the MEN1 MLPA kit (MRC Holland, Amsterdam, The Netherlands) to identify and evaluate possible MEN1 large deletions. Patients and controls were also tested for the CDKN1B V109G polymorphism on germline DNA by PCR amplification of exon 1 followed by direct sequencing using the primers and conditions previously described 17. Indeed, both CDKN1B exons 1 and 2 were screened for the detection of any other genetic variations in this gene, as reported 17. Written informed consent for all genetic screenings, blood samples handling and processing and other clinical procedures was provided by all investigated participants in accordance with the guidelines approved by the local ethical committee and with the Helsinki declaration.
Statistical analysis
The statistical analysis was performed by SPSS for Windows version 20.0 (SPSS, Inc., Chicago, IL, USA). Data are reported as mean ± SEM. The significance was set at 5% (P < 0.05). Comparison between the numerical data was performed by the anova test and Student’s t-test was used to assess differences between two groups. The comparison between the categorical data was performed by the chi-squared test with the Yates correction, Fisher exact test or McNemar test as appropriate. The time interval between date of birth and appearance of the first aggressive tumour as well as that between MEN1 diagnosis and first aggressive tumour was calculated according to the Kaplan–Meier method and log rank test 25.
Results
Clinical and genetic findings
Taking into account the main MEN1-related manifestations, 42 patients (76%) had primary hyperparathyroidism, 23 (42%) pituitary adenoma, 33 (60%) pancreatic NETs, 2 (4%) thoracic NET and 7 (13%) adrenocortical tumour (Table1). The entire CDKN1B gene was investigated testing 55 patients and 90 matched controls. No mutations or other polymorphisms were detected along the entire gene (exons 1 and 2) with the exception of the T/G transversion at nucleotide 326 of exon 1 (rs2066827) that causes a valine (GTC) for glycine (GGC) substitution at position 109 of the mature protein. A significant difference in the frequency of the wild-type (T/T, 56.4%) and polymorphic alleles (T/G 38.2%, G/G 5.4%) was found between patients and controls (P = 0.001, 95% CI = 0.287–0.076; Table2). The frequency of the wild-type (T/T 56%) versus the polymorphic alleles (T/G and G/G 44%) among our MEN1 patients was not different from that reported for other types of tumours 15,16. The CDKN1B V109G polymorphism was detected in 24 of 55 MEN1 patients (44%). On this basis, patients were divided into two groups: group A (24 patients, 10M and 14F, bearing the polymorphism) and group B (31 patients, 12M and 19F, bearing the wild-type allele).
Table 2.
Relative frequency of the T/G (V109G) polymorphism in MEN1 patients and control individuals
| CDKN1B Genotype | |||
|---|---|---|---|
| DNA | T/T | T/G | G/G |
| Protein | V109V | V109G | G109G |
| Cases (n = 55) | 31 (56.4%) | 21 (38.2%) | 3 (5.4%) |
| Controls (n = 90) | 56 (62.2%) | 28 (31.1%) | 6 (6.7%) |
No differences between group A and B were detected taking into account either gender or diagnostic circumstances (clinical diagnosis versus genetic screening; P = NS). A strong association was, instead, found with the MEN1 genotype (P = 0.0001); in particular, three types of MEN1 mutations [c.502G>A, p.G168R in exon 3; c.673T>A, p.W225R in exon 4; c.825+1G>A in intron 5] were only found in group A, that included also the patients tested negative for MEN1 mutations. In contrast, six MEN1 mutations [c.335delA in exon 2; c.518T>C, p.L173P in exon 3; c.1339C>T, p.Q447X in exon 9; c.1576del11 (delACTGTCGCTGG) in exon 10; c.799-9G>A in intron 4; c.1065+1G>A in intron 7] were only found in group B.
Genotype/phenotype correlation
No significant differences in the frequency of the typical MEN1-related manifestations (primary hyperparathyroidism, pituitary adenoma, pancreatic NET) were detected between the two groups. At the time of the study, 6/24 (25%) patients of group A and 4/31 (13%) of group B had not yet clinical or radiological evidence of MEN1-related manifestations (P = NS). The age at diagnosis of the first MEN1-related manifestation was similar in group A (37.6 ± 3.1 years, range 18–61) and in group B (39.3 ± 2.7 years, range 16–81; P = NS). In contrast, the age at diagnosis of the first aggressive tumour was lower in group A (35.2 ± 3.9 years, range 19–50) than in group B (44.0 ± 1.6 years, 41–48), although there was no statistical difference (P = NS). MEN1-related aggressive tumours or other malignancies were more frequent in participants with the CDKN1B genetic variant (8/24 patients in group A versus 5/31 in group B; 33.3 versus 16.1%), although this test did not result in statistical significance (P = NS) (Table3). The time interval between the date of birth and age of the first aggressive tumour was significantly shorter in group A (median 46 years) than in group B (median not reached; P = 0.03). Similarly, the time interval between MEN1 diagnosis and age of the first aggressive tumour was significantly shorter in group A (median 36 months) than in group B (median not reached; P = 0.02). Overall survival could not be evaluated as 53/55 patients (96%) were still alive at the time of the study.
Table 3.
Patients with MEN1-related aggressive tumours and other malignant tumours
| Family | Patient no. | CDKN1B | Aggressive NET | Other malignancies |
|---|---|---|---|---|
| 1 | 1 | POL | Pancreas >2 cm + LN, L metastases | – |
| 1 | 2 | POL | Pancreas | – |
| 1 | 3 | WT | Pancreas >2 cm + LN metastases | – |
| 2 | 8 | POL | Pancreas >2 cm | – |
| 4 | 11 | POL | Thymus | – |
| 4 | 14 | WT | Pancreas >2 cm | – |
| 7 | 26 | POL | Pancreas >2 cm | – |
| 9 | 32 | POL | Bronchi | – |
| 13 | 41 | WT | Pancreas >2 cm | – |
| 14 | 43 | WT | Pancreas >2 cm | – |
| 16 | 48 | WT | Pancreas >2 cm | – |
| 18 | 53 | POL | Pancreas >2 cm | Renal carcinoma |
| 19 | 54 | POL | Pancreas >2 cm | – |
LN: lymph node; L: liver.
Discussion
The preferred approach to search for low-penetrance cancer susceptibility genes, especially ‘driver’ genes, involves genetic association studies that compare the frequencies of common variants between cases and unaffected controls. The identification of such genes that can be used as prognostic markers is particularly compelling for tumours that do not have a clear genotype/phenotype correlation. MEN1 is a rare syndrome, with inter- and intra-familial variability, without a known genotype–phenotype correlation. Novel genetic prognostic markers that could help in a better management and in predicting the outcome of this syndrome are thus largely needed.
Figure 1.
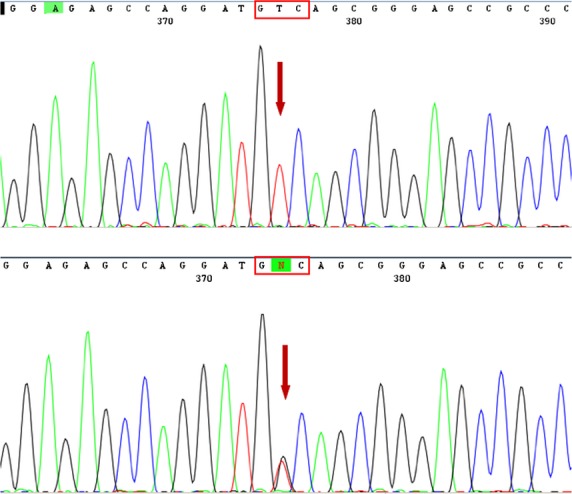
Identification of the CDKN1B V109G single nucleotide polymorphism. Direct sequencing of CDKN1B-amplified exon 1 from two patients’ DNA shows either the wild-type sequence or the T/G transversion at nucleotide 326. The arrow in the lower electropherogram denotes two overlapping peaks, indicative of the presence of two different nucleotides at that position of the DNA sequence. The change causes a valine (GTC) for glycine (GGC) substitution at position 109 of the mature protein.
In this study, we investigated the possible association of MEN1 malignant tumours with CDKN1B V109G polymorphism. Indeed, functional polymorphisms in this gene contribute to the susceptibility and severity of different types of cancers 26,27; in particular, the V109G SNP has extensively been associated with the risk of cancer although inconsistent results have been reported.
Patients selected for this study had all a clinical diagnosis of MEN1, according to the published guidelines 2. The polymorphism in MEN1 patients was as frequent as other types of tumours, whereas it was significantly different from healthy controls. No significant differences were found between the polymorphism and wild-type allele carrying groups according to frequency of MEN1 manifestations as well as diagnostic circumstances.
As previously reported, it should be noted that the genetic screening is helpful to anticipate the identification of MEN1 participants so detecting MEN1-related tumours at an early stage 28,29. This aspect impacts on patients’ prognosis and quality of life but it does not influence the proportion of aggressive tumours that are equally distributed between participants with initial clinical diagnosis and those identified at the genetic screening. The aggressiveness of the tumour seems to be rather an intrinsic characteristic of the tumour. The CDKN1B V109G polymorphism could in part explain this characteristic, as it positively correlates with the onset of aggressive tumours. In particular, polymorphism-positive patients displayed a shorter time interval before the occurrence of an aggressive tumour than those negative.
Finally, the polymorphism was associated with specific MEN1 mutations [c.502G>A, p.G168R in exon 3; c.673T>A, p.W225R in exon 4; c.825+1G>A in intron 5] and patients carrying both genetic variants had a worse prognosis, providing additional support to the association with a more aggressive disease course. Based on these data, it is tempting to speculate that a mutated menin could have an impaired transactivation potential on the target gene CDKN1B, as reported 7–10, reducing the amount of the p27/Kip1 produced. Moreover, the V109G polymorphism has been correlated with low levels of p27/Kip1 as it is located in a protein region that physically interacts with Jun activation domain-binding protein 1, an event that could enhance nuclear export and p27/Kip1 degradation 30,31. Disrupting the binding between these two proteins would affect the p27/Kip1 fate and hence cell growth. Thus, the simultaneous presence of specific MEN1 mutations and the CDKN1B polymorphism could potentiate the negative effects of a mutated menin, further reducing p27/Kip1 and its regulatory function. This reasoning could explain at least in part the association found in this tumour syndrome.
In conclusion, we provide the first evidence that among patients who develop MEN1-related tumours, those bearing the CDKN1B V109G polymorphism have a higher frequency to develop aggressive tumours and hence a more aggressive behaviour, emphasizing the added value of this variant. Thus, the polymorphism may play a role in dictating the evolution of the malignancy whereby MEN1 acts as a ‘driver’ gene and the CDKN1B polymorphism as a ‘modifier’ of tumour progression. While further studies are needed to better elucidate the mechanisms underlying the interplay of this polymorphism with the MEN1-gene pathways, we suggest that the association of the polymorphism with a severe prognosis might be used as a novel and early prognostic marker to direct more selective and targeted therapies.
Acknowledgments
This study was in part supported by Multidisciplinary Group for NeuroEndocrine Tumours of Naples.
Conflicts of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Marx SJ, Agarwal SK, Kester MB, et al. Multiple endocrine neoplasia type 1: clinical and genetic features of the hereditary endocrine neoplasias. Recent Prog Horm Res. 1999;54:397–438. [PubMed] [Google Scholar]
- Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86:5658–71. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru S, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia type 1. Science. 1997;76:404–7. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29:22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- Wautot V, Vercherat C, Lespinasse J, et al. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat. 2002;20:35–47. doi: 10.1002/humu.10092. [DOI] [PubMed] [Google Scholar]
- Zhang HL, Li WY, Zhang CP, et al. Differentially expressed genes in Men1 knockout and wild type embryoid bodies for pancreatic islet development. Mol Med Report. 2011;4:301–5. doi: 10.3892/mmr.2011.409. [DOI] [PubMed] [Google Scholar]
- Dreijerink KM, Hoppener JW, Timmers HM, et al. Mechanism of disease: multiple endocrine neoplasia type 1-relation to chromatin modifications and transcription regulation. Nat Clin Pract Endocr Metab. 2006;2:562–70. doi: 10.1038/ncpendmet0292. [DOI] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–97. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Lin SY, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. 2003;113:881–9. doi: 10.1016/s0092-8674(03)00430-6. [DOI] [PubMed] [Google Scholar]
- Milne TA, Hughes CM, Lloyd R, et al. Menin and MLL cooperatively regulate expression of cyclindependent kinase inhibitors. Proc Natl Acad Sci USA. 2005;102:749–54. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon E. The sweet secrets of p27 regulation and function in cell migration. Cell Cycle. 2011;10:3429. doi: 10.4161/cc.10.20.17529. [DOI] [PubMed] [Google Scholar]
- Itamochi H, Yoshida T, Walker CL, et al. Novel mechanism of reduced proliferation in ovarian clear cell carcinoma cells Cytoplasmic sequestration of CDK2 by p27. Gynecol Oncol. 2011;122:641–7. doi: 10.1016/j.ygyno.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Pellegata NS, Quintanilla-Martinez L, Siggelkow H, et al. Germline mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA. 2006;103:15558–63. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibel AS, Suarez BK, Belani J, et al. CDKN1A and CDKN1B polymorphisms and risk of advanced prostate carcinoma. Cancer Res. 2003;63:2033–6. [PubMed] [Google Scholar]
- Li G, Sturgis EM, Wang LE, et al. Association between the V109G polymorphism of the p27 gene and the risk and progression of oral squamous cell carcinoma. Clin Cancer Res. 2004;10:3996. doi: 10.1158/1078-0432.CCR-04-0089. [DOI] [PubMed] [Google Scholar]
- Figueiredo JC, Knight JA, Cho S, et al. Polymorphisms cMyc-N11S and p27-V109G and breast cancer risk and prognosis. BMC Cancer. 2007;7:99. doi: 10.1186/1471-2407-7-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquali D, Circelli L, Faggiano A, et al. CDKN1B V109G polymorphism a new prognostic factor in sporadic medullary thyroid carcinoma. Eur J Endocrinol. 2011;164:397–404. doi: 10.1530/EJE-10-0929. [DOI] [PubMed] [Google Scholar]
- Naidu R, Har YC, Taib NA. p27 V109G Polymorphism is associated with lymph node metastases but not with increased risk of breast cancer. J Exp Clin Cancer Res. 2007;26:133–40. [PubMed] [Google Scholar]
- Spurdle AB, Deans AJ, Duffy D, et al. No evidence that CDKN1B (p27) polymorphisms modify breast cancer risk in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat. 2009;115:307–13. doi: 10.1007/s10549-008-0083-5. [DOI] [PubMed] [Google Scholar]
- Chen J, Killary AM, Sen S, et al. Polymorphisms of p21 and p27 jointly contribute to an earlier age at diagnosis of pancreatic cancer. Cancer Lett. 2008;272:32–9. doi: 10.1016/j.canlet.2008.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Amos CI, Merriman KW, et al. Genetic variants of p21 and p27 and pancreatic cancer risk in non-Hispanic Whites: a case-control study. Pancreas. 2010;39:1–4. doi: 10.1097/MPA.0b013e3181bd51c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SP, Yu CC, Liu CC, et al. CDKN1B V109G polymorphism frequency and prostate cancer risk in Taiwan. Urol Int. 2008;81:36–40. doi: 10.1159/000137638. [DOI] [PubMed] [Google Scholar]
- Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) J Clin Endocrinol Metab. 2012;97:2990–3011. doi: 10.1210/jc.2012-1230. [DOI] [PubMed] [Google Scholar]
- Ramundo V, Del Prete M, Marotta V, et al. Impact of long-acting octreotide in patients with early-stage MEN1-related duodeno-pancreatic neuroendocrine tumors. Clin Endocrinol. 2014;80:850–5. doi: 10.1111/cen.12411. [DOI] [PubMed] [Google Scholar]
- Kaplan EL, Maier P. Non-parametric estimation for uncompleted observations. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- Hershko DD. Cyclin-dependent kinase inhibitor p27 as a prognostic biomarker and potential cancer therapeutic target. Future Oncol. 2010;6:1837–47. doi: 10.2217/fon.10.144. [DOI] [PubMed] [Google Scholar]
- Wander SA, Zhao D, Slinqerland JM. p27: a Barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res. 2011;17:12–8. doi: 10.1158/1078-0432.CCR-10-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieterman CR, Schreinemakers JM, Koppeschaar HP, et al. Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. Clin Endocrinol. 2009;70:575–81. doi: 10.1111/j.1365-2265.2008.03324.x. [DOI] [PubMed] [Google Scholar]
- Ramundo V, Milone F, Severino R, et al. Clinical and prognostic implications of the genetic diagnosis of hereditary NET syndromes in asymptomatic patients. Horm Metab Res. 2011;43:794–800. doi: 10.1055/s-0031-1286324. [DOI] [PubMed] [Google Scholar]
- Tomoda K, Kubota Y, Arata Y, et al. The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. Biol Chem. 2002;277:2302–10. doi: 10.1074/jbc.M104431200. [DOI] [PubMed] [Google Scholar]
- Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398:160–5. doi: 10.1038/18230. [DOI] [PubMed] [Google Scholar]