

Abstract
Epigenetic changes play a significant role in leukaemia pathogenesis, therefore histone deacetylases (HDACis) are widely accepted as an attractive strategy for acute promyelocytic leukaemia (APL) treatment. Belinostat (Bel, PXD101), a hydroxamate-type HDACi, has proved to be a promising cure in clinical trials for solid tumours and haematological malignancies. However, insight into molecular effects of Bel on APL, is still lacking. In this study, we investigated the effect of Bel alone and in combination with differentiation inducer retinoic acid (RA) on human promyelocytic leukaemia NB4 and HL-60 cells. We found that treatment with Bel, depending on the dosage used, inhibits cell proliferation, whereas in combination with RA enhances and accelerates granulocytic leukaemia cell differentiation. We also evaluated the effect of used treatments with Bel and RA on certain epigenetic modifiers (HDAC1, HDAC2, PCAF) as well as cell cycle regulators (p27) gene expression and protein level modulation. We showed that Bel in combination with RA up-regulates basal histone H4 hyperacetylation level more strongly compared to Bel or RA alone. Furthermore, chromatin immunoprecipitation assay indicated that Bel induces the accumulation of hyperacetylated histone H4 at the p27 promoter region. Mass spectrometry analysis revealed that in control NB4 cells, hyperacetylated histone H4 is mainly found in association with proteins involved in DNA replication and transcription, whereas after Bel treatment it is found with proteins implicated in pro-apoptotic processes, in defence against oxidative stress and tumour suppression. Summarizing, our study provides some novel insights into the molecular mechanisms of HDACi Bel action on APL cells.
Keywords: APL, HDACi, belinostat, epigenetics, granulocytic differentiation
Introduction
Acute promyelocytic leukaemia (APL) is an acute myeloid leukaemia (AML) subtype, characterized by block of granulocytic differentiation and accumulation of promyelocytes in the bone marrow and blood 1. Acute promyelocytic leukaemia patients possess specific reciprocal chromosomal translocations involving the retinoic acid receptor α (RARA) gene and one of its gene fusion partners. The pathogenesis of this disease in most cases is associated with the formation of chimeric PML-RARA protein (>98%) 2. It has been demonstrated that in APL, fusion proteins of the RARA recruit histone deacetylases (HDACs) containing co-repressor complexes 3, which in turn deacetylate and silence genes crucial for haematopoietic differentiation 4. Treatment with pharmacological doses of all-trans retinoic acid (RA) has been shown to force APL cells differentiation into mature granulocytes 5. However, resistance to the cytodifferentiating effects of RA is frequently acquired during drug therapy 6. Therefore, APL treatment requires other clinical approaches. Numerous investigations showed that there is a rationale to use RA in combination with epigenetic drugs such as HDAC inhibitors (HDACis) 7,8.
One of the most promising agents in this category—belinostat, which is a novel and potent hydroxamate-type HDACi, was shown to inhibit 1-st and 2-nd class HDACs enzymatic activity in vitro 9. Belinostat exerts its anti-deacetylase action via its hydroxamic acid moiety binding to zinc ion in enzymes’ catalytic domains and blocking substrate access 10. Previous studies have shown its activity resulting in cell cycle arrest, apoptosis and inhibition of cell proliferation 11,12. Belinostat has been already tested in phase I and II clinical trials against solid tumours, such as malignant pleural mesothelioma 13, thymic epithelial tumours 14, unresectable hepatocellular 15, ovarian, fallopian tube or primary peritoneal carcinoma 16,17. It should be emphasized that in solid tumours belinostat demonstrated more promising effects in combination with traditional chemotherapy, rather than applied as a single therapy 18. Belinostat also has been used in phase II trials as monotherapy in newly diagnosed AML 19. However, as a single agent it was shown to have minimal effect. In contrast, belinostat in combination with the proteasome inhibitor bortezomib elicited pro-apoptotic effect in AML and ALL cell lines and primary blasts, whereas analogous treatment was non-toxic to normal CD34(+) cells 20. In addition, belinostat in combination with decitabine, theophyline and RA has shown to exert anti-proliferative effect on AML blasts 21.
All this available data suggest that belinostat in combination with other drugs may be a valuable strategy for APL treatment. Therefore, a further more profound investigation is necessary to determine its applicability for APL differentiation therapy and to decipher belinostat’s molecular effects on APL cells.
In this study, we investigated the application of belinostat for leukaemia cell granulocytic differentiation using APL cell line NB4 (FAB-M3) and promyelocytes resembling HL-60 cells (FAB-M2), although not bearing typical APL translocation t(15;17). To unravel molecular mechanisms involved in belinostat’s action, we further examined its effect on APL cells gene and protein expression (HDAC1, HDAC2, PCAF, p27), as well as on histone H4 hyperacetylation level. Furthermore, we examined belinostat’s effect on composition modulation of protein complexes associated with hyperacetylated histone H4.
Materials and methods
Cell culture
The human APL cells NB4 and HL-60 (from DSMZ, GmbH, Braunschweig, Germany) were maintained in RPMI 1640 medium supplemented with 10% foetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin (Gibco, Grand Island, NY, USA) in a humidified incubator at 37°C with 5% CO2. For each experiment, logarithmically growing cells were seeded at a density of 0.5 × 106 cells/ml in 5 ml of medium. According to previous publication 22 cells were exposed to 0.2 and 2.0 μM Belinostat (Selleck Chemicals, Houston, TX, USA) alone or in combination with 1 μM RA (Sigma-Aldrich, St. Louis, MO, USA). The agents were left in the cell media for the duration of the experiment.
Assessment of granulocytic cell differentiation and cell cycle analysis
The degree of granulocytic differentiation was evaluated by cells ability to reduce soluble nitro blue tetrazolium (NBT) to insoluble blue-black formazan after stimulation with phorbolmyristate acetate. Nitro blue tetrazolium positive stained cells were counted in five consecutive non-overlapping microscopic fields at a magnification of 400. The average percent of NBT positive cells per high power field was calculated. Three independent experiments were performed and their results were averaged. Flow cytometric analysis of cell cycle distribution was performed as described earlier 22.
RNA extraction, cDNA synthesis and RT-qPCR assay
All procedures were performed as indicated earlier 22. The primer sets for the tested genes are listed in the Table1.
Table 1.
Primer sets of tested genes
| Analysis type | Phenotypic end-points | Gene | Sequence | Product size (bp) | Tm (°C) | Resource |
|---|---|---|---|---|---|---|
| RT-qPCR | Epigenetic modifiers | HDAC1 | F: CAAGCTCCACATCAGTCCTTCC | 102 | 60 | 26 |
| R: TGCGGCAGCATTCTAAGGTT | ||||||
| HDAC2 | F: AGTCAAGGAGGCGGCAAAA | 103 | 60 | 26 | ||
| R: TGCGGATTCTATGAGGCTTCA | ||||||
| PCAF | F: GGCCGAGGAGTCTTGTAAAT | 649 | 60 | Primer Bank | ||
| R: AGTGAAGACCGAGCGAAGCA | ||||||
| Cell cycle regulators | P27 | F: TAATTGGGGCTCCGGCTAACT | 116 | 60 | Primer Bank | |
| R:TGCAGGTCGCTTCCTTATTCC | ||||||
| Reference gene | GAPDH | F: TCCATGACAACTTTGGTATCG | 471 | 60 | 27 | |
| R: TGTAGCCAAATTCGTTGTCA | ||||||
| ChIP-qPCR | Cell cycle regulators | P27 | F: GGCCTCCCCCGCAGACCAC | 382 | 60 | Self-designed based on Ref. 28 |
| R: GTTCCGCCACCTCCCCTCGTTCC | ||||||
| Transcription factors | C/EBPα | F: GTGCAGCCTCGGGATACTC | 70 | 60 | Self-designed based on Ref. 29 | |
| R: CTCCTCCTGCCTGCCCTA | ||||||
| C/EBPε | F: GCTAACCGGAATATGCTAATCAG | 296 | 60 | Self designed based on Ref. 30 | ||
| R: CCTTTCAGAGACACCTGCTC |
Total protein isolation and Western blot analysis
For total protein extraction control and treated cells (2 × 106) were washed twice with PBS, incubated with Benzonase® Nuclease (Novagen, Merck KGaA, Darmstadt, Germany) 1 to 10 μl of pellet for 30 min. on ice, later resuspended in 10 volumes of 2× SDS protein loading buffer and heated for 5 min. in 95°C. Protein lysate then was centrifuged at 11,904 g. for 5 min. at 10°C and used for successive analysis. Five microlitre of protein specimens was run on gradient (7.5–15%) polyacrylamide gel. Electrophoresed proteins were transferred to ImmobilonTM PVDF transfer membrane (Millipore, Bedford, MA, USA). Immunoblotting was performed with antibodies against PCAF (Abcam, Cambridge, UK), HDAC1, HDAC2, GAPDH and p27 (Cell Signaling, Danvers, MA, USA). Immunoreactive bands were visualized with an enhanced chemiluminescence (WesternBrightTM ECL kit, Advansta Corporation, Menlo Park, CA, USA), according to the manufacturer’s instructions. Blots were scanned and optical density evaluated using ImageJ software (1.45s) (NIH, Bethesda, MD, USA).
Evaluation of global DNA methylation
For global DNA methylation analysis control and treated NB4 cells (1 × 106) were washed with PBS and genomic DNA extracted using ZR Genomic DNA™ – Tissue MiniPrep (Zymo Research, Irvine, CA, USA). Extracted genomic DNA was used for a subsequent 5-mC DNA quantity evaluation, using 5-mC DNA ELISA Kit (Zymo Research), according to the manufacturer’s instructions. Data were represented as a fold change compared with control.
Chromatin immunoprecipitation for qPCR analysis
Chromatin immunoprecipitation (ChIP) assay was performed with a previously described method with specific modifications 23. For ChIP assay 5–10 mg of antibody to hyperacetylated histone H4 (Upstate Biotechnology, Lake Placide, NY, USA) was used per 15–20 mg DNA. qPCR analysis of immunoprecipitated DNA was performed with Maxima® SYBR Green qPCR Master Mix on the Rotor-Gene 6000 system. The primer sets for the tested genes are listed in the Table1. For data evaluation, the percent input was calculated, according to the formula: 100 · 2(Adjusted input−Ct (IP).
Chromatin immunoprecipitation for mass spectrometry analysis
All ChIP procedures were carried out as discussed in the previous section. Protein A/G PLUS-Agarose –Antibody–Protein complexes were denatured in 7 M Urea, 2 M Thiourea, 40 mM DTT solution, with continuous rotation at 50 r.p.m. in the temperature controlled shaker for 0.5 hrs at 20°C. Complexes were centrifuged (180 g, 7 min., 20°C) and extraction repeated additional three times. All four extracted fractions were combined and subjected to further mass spectrometry (MS) analysis.
Mass spectrometry and data analysis
Extracted proteins were applied on Amicon Ultra-0.5 mL 30 kD centrifugal filter unit (Sigma-Aldrich). Trypsin digestion was carried out according to a modified Filter-aided sample preparation (FASP) protocol as described by Wisniewski et al. 24. Liquid chromatography separation of trypsin cleaved peptides and mass spectrometric analysis were performed as described earlier 25.
Raw data files were processed and searched using ProteinLynx Global SERVER (PLGS) version 2.5.2 (Waters Corporation, Manchester, UK). The following parameters were used to generate peak lists: (i) minimum intensity for precursors was set to 100 counts, (ii) minimum intensity for fragment ions was set to 30 counts, (iii) intensity was set to 500 counts. Processed data were analysed using trypsin as the cleavage protease, one missed cleavage was allowed and fixed modification was set to carbamidomethylation of cysteines, variable modification was set to oxidation of methionine. Minimum identification criteria included two fragment ions per peptide, five fragment ions per protein and minimum of two peptides per protein. The false discovery rate (FDR) for peptide and protein identification was determined based on the search of a reversed database, which was generated automatically using PLGS when global FDR was set to 4%. Functional protein association networks were constructed using STRING database (http://string-db.org/).
Statistical analysis
Unless otherwise specified, all experiments were repeated at least three times. Data were expressed as mean values with SDs. For statistical analysis two-sample Student’s t-test was used.
Results
Effects of belinostat alone and in combined treatment with RA on NB4 and HL-60 cells growth and cell cycle arrest
We determined the impact of treatment with Bel (0.2 and 2 μM) alone and its combined treatment together with RA (0.2 μM Bel + 1 μM RA) on NB4 and HL-60 cells growth. We demonstrated (Fig.1A), that 0.2 μM Bel alone has no observable inhibitory effect neither on NB4 nor on HL-60 cells proliferation. However, 2 μM Bel suppressed both cell lines growth and down-regulated their viability tremendously (Fig.1B; P < 0.01). Noticeably, upon combined treatment with 0.2 μM Bel + 1 μM RA no loss in cell viability was detected (Fig.1B). However, combined treatment inhibited NB4 and HL-60 cells proliferation more efficiently compared with treatments with 0.2 μM Bel and 1 μM RA alone. Statistically significant differences between treatment with RA alone and with combined treatment Bel + RA were observed in NB4 cells (upon 24 hrs treatment P < 0.05, upon 48 hrs treatment P < 0.01). Cell cycle analysis (Fig.1C) showed that treatments with 0.2 μM Bel + 1 μM RA, 1 μM RA and, to a lesser extent, with 0.2 μM Bel, arrest NB4 and HL-60 cells in G0/G1 cell cycle stage (P < 0.01), whereas treatment with 2 μM Bel blocks cell cycle in S phase (P < 0.01).
Figure 1.

Effects of Bel alone and in combined treatment with RA on NB4 and HL-60 cells growth, viability and cell cycle arrest. Control and NB4, HL-60 cells treated with 0.2 μM, 2 μM Bel, 1 μM RA and with 0.2 μM Bel + 1 μM RA, were subjected to evaluation of cell growth (A), cell viability (B) and cell cycle distribution (C). At each time-point indicated, aliquots of the cultures were subjected to 0.2% trypan blue staining for cell growth (A) and viability (B) determination. Results are given as mean (± SD, n = 3). Statistically significant differences between treatment with RA alone and with combined treatment Bel + RA were observed in NB4 cells (upon 24 hrs treatment P < 0.05, upon 48 hrs treatment P < 0.01; A). The cell cycle phase distribution (%; C) was assayed flow cytometrically from the DNA frequency distribution histograms of PI stained cells. Results are given as mean (±SD, n = 3). *P < 0.01 (difference between untreated and treated samples).
Belinostat enhances RA-induced NB4 and HL-60 cells granulocytic differentiation
For granulocytic differentiation evaluation cells were treated with 1 μM RA, 0.2 μM Bel alone and their combination (1 μM RA + 0.2 μM Bel). Nitro blue tetrazolium test revealed that Bel alone is not sufficient to induce NB4 and HL-60 cells differentiation. However, we showed that it enhances and accelerates RA-induced granulocytic differentiation (Fig.2), although not statistically significantly. The most pronounced effect was visible on NB4 cell line, therefore NB4 cells were chosen for subsequent gene and protein expression, as well as ChIP analysis.
Figure 2.
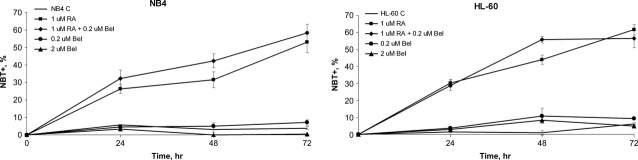
Effects of Bel, RA and their combined treatments on NB4 and HL-60 cells granulocytic differentiation. Control and NB4, HL-60 cells treated with 0.2 μM, 2 μM Bel, 1 μM RA and with 0.2 μM Bel + 1 μM RA at indicated time-points (24–72 hrs) were subjected to granulocytic differentiation analysis, using NBT+ test. Results are given as mean (± SD, n = 3).
Belinostats effect on NB4 cells’ gene expression
RT-qPCR analysis revealed the down-regulation of HDAC1 gene expression (Fig.3A) after treatment with 0.2 μM Bel, 1 μM RA and combined treatment 0.2 μM Bel + 1 μM RA. The strongest effect was induced by Bel alone, as Bel instantly decreased HDAC1 gene expression. Effect was still observable after 24 hrs incubation, whereas later the HDAC1 gene expression level has been restored. Consistently with this data, Bel alone was the most prominent in sudden HDAC2 gene expression reduction (Fig.3B). Although after longer incubation periods with Bel HDAC2 gene expression was restored, combined treatment Bel + RA restricted HDAC2 expression even after 72 hrs incubation.
Figure 3.
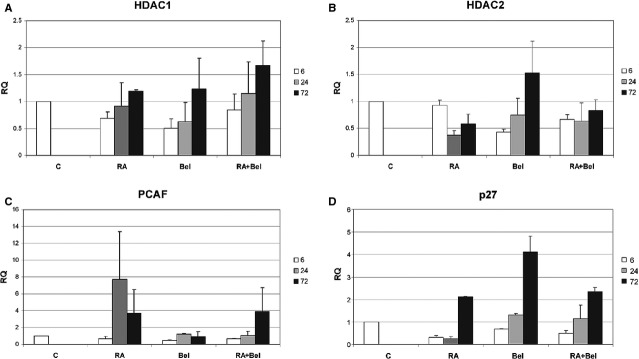
Effects of Bel, RA and their combined treatments on NB4 cells gene expression. (A–D) Control NB4 cells (C) and cells treated with 1 μM RA (RA), 0.2 μM Bel (Bel) and 1 μM RA + 0.2 μM Bel (RA + Bel) were subjected to RT-qPCR analysis. Target gene expression: HDAC1 (A), HDAC2 (B), PCAF (C) and p27 (D) was normalized to GAPDH reference gene. Fold change in gene expression (denoted as RQ – relative quantity) data is represented as mean (± SD, n = 3).
We were interested, if Bel also has an impact on PCAF gene expression. From data presented in Figure3C it is evident, that Bel alone had no effect on PCAF gene expression, whereas RA dramatically up-regulated PCAF mRNA level. Only Combined treatment Bel + RA had reached RA effect after 72 hrs incubation. In contrast, Bel alone, and to a lesser extent in combination with RA, compared with RA alone, was more efficient in p27 gene expression induction (Fig.3D).
Belinostat influence on NB4 cells’ protein level modulation
To evaluate molecular mechanisms that Bel modulates in greater detail, we investigated the effect of 0.2 μM Bel treatment (alone or in combination with 1 μM RA) on histone H4 hyperacetylation level, as well as HDAC1 and HDAC2 protein level regulation (Fig.4).
Figure 4.
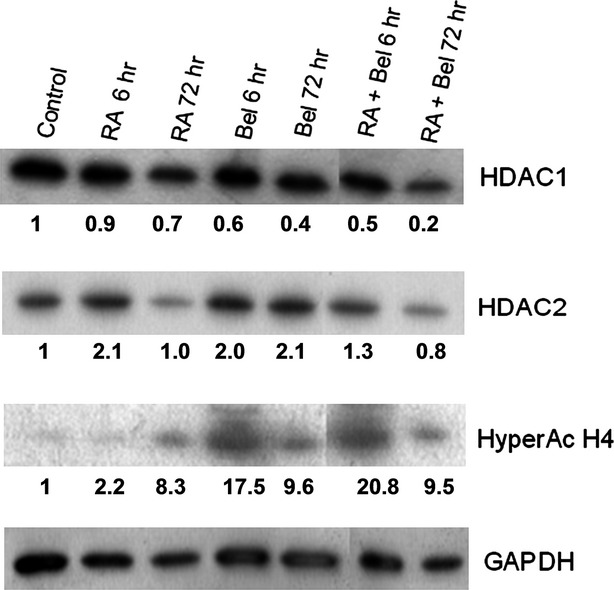
Effects of Bel, RA and their combined treatments on NB4 cells protein level regulation. NB4 cells were treated with 1 μM RA (RA), 0.2 μM Bel (Bel) and 1 μM RA + 0.2 μM Bel (RA + Bel) for 6–72 hrs. At indicated time-points total protein was isolated from control and treated cells. Identical amount of proteins were separated by SDS/PAGE electrophoresis in 7–15% acrylamide gradient gel, transferred onto PVDF membrane and subjected to western blot analysis, using antibodies against examined proteins (Hyper Ac H4, HDAC1, HDAC2) and GAPDH as a loading control. Blots were scanned and optical density evaluated using ImageJ software. The data are representative of three independent experiments showing similar results. The fold change values compared with control are indicated; SDs <15%.
The most efficient increase in NB4 cells’ histone H4 hyperacetylation level was observed after combined treatment 0.2 μM Bel + 1 μM RA. The effect was rapid (evident after 6 hrs incubation) and highly pronounced (histone H4 hyperacetylation level increased 21-fold compared with control cells). After a sudden initial increase, histone H4 hyperacetylation level later declined. However, after 72 hrs treatment with Bel + RA it was still approximately 10-fold greater compared with control cells. The similar effect was observable after 72 hrs treatment with RA or Bel alone.
Combined treatment of Bel + RA also had the most obvious effect on HDAC1 protein level down-regulation. After 6 hrs incubation with combination of these agents HDAC1 protein level has dropped more than twofold, after 72 hrs incubation HDAC1 protein level was reduced up to five times compared with untreated cells. From the presented data it is evident that RA alone is insufficient to reduce HDAC1 protein level, however, it is capable to enhance Bel effect on HDAC1 protein level down-regulation. In contrast to HDAC1, HDAC2 protein level was shown to be up-regulated immediately upon treatment with Bel, RA or their combination. Later HDAC2 was restored to its previous level (except after treatment with Bel).
Combined treatment with belinostat and RA effects NB4 cells global DNA methylation
We evaluated the effect of treatment with RA and Bel, as single agents, and their combined treatment Bel + RA on NB4 cells’ global DNA methylation patterns (data not shown). RA alone did not exert any significant activity towards global DNA methylation modulation, whereas the augmentation in global DNA methylation % was evident upon treatment with Bel (global DNA methylation increased by 15–38% after 6–24 hrs treatment with 0.2 μM Bel, whereas in later time-points the previous methylation level was restored). In contrast, the increase in DNA methylation level upon combined treatment Bel + RA was more sudden and more pronounced compared with treatment with Bel alone (DNA methylation increased by 40% after 6 hrs treatment). It should be highlighted that prolonged treatment with Bel + RA (72 hrs) down-regulated the global DNA methylation more than 14%.
Treatment with belinostat induces increased association of acetylated histone H4 with p27 promoter
To examine, if the increase in p27 gene expression after treatment with Bel can be attributed to or correlated with histone H4 hyperacetylation, ChIP analysis was performed. Indeed, upon NB4 cells treatment with 2 μM Bel for 6 hrs, histone H4 in p27 promoter region was almost twice more hyperacetylated compared with untreated cells (Fig.5). This suggests, that histone H4 hyperacetylation may be one of the factors leading to increased p27 mRNA level.
Figure 5.
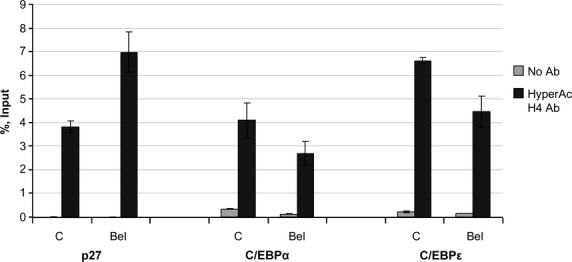
Bel effect on acetylated histone H4 association with p27, C/EBPα and C/EBPε promoter regions. ChIP with antibody against hyperacetylated histone H4 was performed with control (C) and NB4 cells treated with 2 μM Bel for 6 hrs (Bel). Specimens were further tested using qPCR analysis. Data are represented as percent input (± SD, n = 2).
It is widely admitted, that transcription factors C/enhancer binding protein alpha (EBPα) and C/EBPε play a crucial role in granulocytic differentiation 31,32. Our group previously demonstrated 22 that 24 hrs NB4 cells treatment with Bel induced C/EBPα mRNA expression twofold, whereas later C/EBPα mRNA level decreased. In addition, only low effect on C/EBPε mRNA expression was detected after treatment with Bel as a single agent. Combined treatment of 0.2 μM Bel + 1 μM RA has also demonstrated to be less effective in C/EBPε mRNA level up-regulation compared with RA alone, despite the evidence that Bel in combination with RA enhances and accelerates RA-induced granulocytic differentiation.
In this study we investigated, whether Bel treatment has an effect on histone H4 hyperacetylation level at C/EBPα and C/EBPε promoter regions. However, in accordance to gene expression data, no increase in H4 hyperacetylation at C/EBPα and C/EBPε promoter regions was detected, which also coincided with NBT test data, demonstrating that Bel alone is unable to induce granulocytic differentiation.
Belinostat modulates protein complexes associated with hyperacetylated histone H4
We were interested, if the increase in basal histone H4 hyperacetylation level after NB4 cells treatment with Bel is accompanied by compositional changes in protein complexes that are found in association with this epigenetic mark. Therefore, we used Co-IP and subsequent MS analysis that helped us to reveal proteins that are associated with hyperacetylated histone H4 in control and Bel-treated NB4 cells. Quantitative changes of identified proteins in control and Bel-treated cells are presented in Table2 as C/Bel ratio (the mark ‘C’ denotes that protein was detected only in control cells, whereas mark ‘Bel’ indicates that protein was seen only in treated cells).
Table 2.
Summary of identified NB4 cells proteins found in complexes with hyperacetylated histone H4 in control and Bel-treated cells
| No. | Accession | Gene name | Score | C/Bel ratio* | Function |
|---|---|---|---|---|---|
| 1 | Q5QNW6 | HIST1H2AH | 9505.53 | 0.77105 | Core component of nucleosome |
| 2 | Q99878 | HIST1H2AJ | 8305.59 | 0.59452 | Core component of nucleosome |
| 3 | P33778 | HIST1H2BB | 42,815.45 | 1 | Core component of nucleosome |
| 4 | P58876 | HIST1H2BD | 10,401.13 | 0.51171 | Core component of nucleosome |
| 5 | P57053 | HIST2H2BF | 45,639.36 | Bel | Core component of nucleosome |
| 6 | P84243 | H3F3A | 10,974.41 | 1.10517 | Core component of nucleosome |
| 7 | Q6NXT2 | H3F3C | 828.6 | 0.69768 | Core component of nucleosome |
| 8 | P62805 | HIST1H4A | 17,505.7 | 0.84366 | Core component of nucleosome |
| 9 | P16401 | HIST1H1B | 615.33 | 1.1853 | Nucleosomal condensation |
| 10 | P16403 | HIST1H1C | 2448.72 | 1.05127 | Nucleosomal condensation |
| 11 | Q71UI9 | H2AFV | 5543.68 | 1.23368 | Replaces conventional H2A in a subset of nucleosomes |
| 12 | P57053 | H2BFS | 10,401.13 | 0.5886 | Replaces conventional H2A in a subset of nucleosomes |
| 13 | P0C0S5 | H2AFZ | 7646.1 | Bel | Replaces conventional H2A in a subset of nucleosomes |
| 14 | Q14181 | POLA2 | 152.92 | C | DNA replication |
| 15 | Q9BZD3 | GCOM1 | 224.06 | C | Component of Pol II(G) complex |
| 16 | P0CAP2 | POLR2M | 284.39 | C | Component of Pol II(G) complex |
| 17 | P18615 | NELFE | 266.18 | C | Represses RNA polymerase II transcript elongation |
| 18 | P51504 | ZNF80 | 444.63 | Bel | Transcriptional regulation |
| 19 | P18124 | RPL7 | 235.63 | C | Translation apparatus regulation |
| 20 | P47914 | RPL29 | 622.4 | 0.92312 | Translation apparatus regulation |
| 21 | Q9BWG6 | SCNM1 | 620.47 | C | RNA splicing |
| 22 | Q8WXA9 | SREK1 | 148.46 | Bel | Regulation of alternative splicing |
| 23 | P19338 | NCL | 132.66 | C | Pre-rRNA transcription and ribosome assembly |
| 24 | P02788 | LTF | 275.73 | C | Antimicrobial and anti-inflammatory activity |
| 25 | P61626 | LYZ | 751.9 | 3.56085 | Bacteriolysis |
| 26 | P06702 | S100A9 | 2362.67 | Bel | Antimicrobial activity. Phagocyte migration promotion. Apoptosis |
| 27 | P05109 | S100A8 | 1869.7 | Bel | Antimicrobial activity. Phagocyte migration promotion. Apoptosis |
| 28 | P60709 | ACTB | 3806.68 | 1.82212 | Cell motility |
| 29 | P63261 | ACTG1 | 1713.96 | 0.34301 | Cell motility |
| 30 | Q562R1 | ACTBL2 | 664.65 | Bel | Cell motility |
| 31 | A6NHL2 | TUBAL3 | 110.36 | C | Microtubule element |
| 32 | Q71U36 | TUBA1A | 452.48 | C | Microtubule element |
| 33 | P07437 | TUBB | 620.75 | 2.2034 | Microtubule element |
| 34 | Q9BQS8 | FYCO1 | 61.54 | C | May mediate microtubule plus end-directed vesicle transport |
| 35 | Q13326 | SGCG | 315.83 | C | Component of sarcoglycan complex |
| 36 | Q9NY65 | TUBA8 | 123.57 | Bel | Microtubule element |
| 37 | Q9BQE3 | TUBA1C | 54.26 | Bel | Microtubule element |
| 38 | O15144 | ARPC2 | 666.79 | Bel | Regulation of actin polimerization |
| 39 | Q96A32 | MYLPF | 737.84 | Bel | Myosin light chain |
| 40 | Q6UY14 | ADAMTSL4 | 247.27 | C | Positive regulation of apoptosis |
| 41 | P47929 | LGALS7 | 392.7 | Bel | Apoptosis regulation. Pro-apoptotic |
| 42 | Q08378 | GOLGA3 | 7.44 | Bel | Golgi str. maintenance. Cleavage product necessary for apoptotic response |
| 43 | P50897 | PPT1 | 124.76 | Bel | Lysosomal degradation. DNA fragmentation during apoptosis |
| 44 | Q5M775 | SPECC1 | 380.22 | C | Proto-oncogene |
| 45 | P25054 | APC | 30.36 | Bel | Tumour suppressor |
| 46 | Q04760 | GLO1 | 253.31 | C | Involved in the regulation of TNF-induced transcriptional activity of NF-κB |
| 47 | Q5T200 | ZC3H13 | 22.03 | C | Down-regulation of NF-κB pathway |
| 48 | O95989 | NUDT3 | 215.22 | C | Signal transduction. Negatively regulates ERK1/2 pathway |
| 49 | Q99665 | IL12RB2 | 269.83 | C | Signalling component coupling to the JAK2/STAT4 pathway. Promotes the proliferation of T-cells as well as NK cells |
| 50 | Q8IV04 | TBC1D10C | 834.75 | 0.8781 | Ras signalling pathway inhibition |
| 51 | Q96NH3 | C6orf170 | 1777.55 | C | Controls ciliary morphology. Involved in Hedgehog signal transduction |
| 52 | P06748 | NPM1 | 612.64 | 1.85893 | Regulates tumour suppressors TP53/p53 and ARF. Chaperone |
| 53 | Q9NNW7 | TXNRD2 | 158.36 | Bel | Implication in the defences against oxidative stress |
| 54 | P29762 | CRABP1 | 133.35 | Bel | Regulates access of retinoic acid to the nuclear retinoic acid receptors |
| 55 | P17066 | HSPA6 | 121.58 | Bel | Chaperone |
| 56 | P48741 | HSPA7 | 78.01 | Bel | Chaperone |
| 57 | P11142 | HSPA8 | 144.69 | Bel | Chaperone. Repressor of transcriptional activation |
| 58 | P55735 | SEC13 | 122 | C | May be involved in protein transport |
| 59 | P62987 | UBA52 | 755.27 | 0.77105 | Proteosomal degradation, chromatin structure maintenance, gene expression regulation and stress response |
| 60 | P0CG47 | UBB | 241.72 | C | Proteosomal degradation, chromatin structure maintenance, gene expression regulation and stress response |
| 61 | Q6ZMR5 | TMPRSS11A | 860.74 | Bel | Probable serine protease |
| 62 | P00738 | HP | 1190.67 | Bel | Makes haemoglobin accessible to degradative enzymes |
| 63 | Q6S8J3 | POTEE | 346.61 | C | Protein and ATP binding |
| 64 | A5A3E0 | POTEF | 369.2 | 2.13828 | Protein and ATP binding |
| 65 | P0CG39 | POTEJ | 107.66 | 1.46228 | Protein and ATP binding |
| 66 | Q9BTF0 | THUMPD2 | 238.14 | C | RNA binding. Methyltransferase activity |
| 67 | Q68CQ7 | GLT8D1 | 206.26 | C | Glycosyltransferase |
| 68 | A6NIV6 | LRRIQ4 | 145.7 | Bel | Leucine-rich repeats and IQ motif containing |
‘C’ denotes that protein is seen in control only. ‘Bel’ – detected in treated cells only.
In untreated NB4 cells hyperacetylated histone H4 was found to associate with 45 different proteins (Table2). The network of proteins associated with hyperacetylated histone H4 in control NB4 cells is presented in Figure6. In control cells only, hyperacetylated histone H4 was found associated with proteins that are involved in DNA replication (POLA2), transcription (GCOM1, POLR2M, NELFE, NCL), translation (RPL7) and RNA splicing (SCNM1). Also hyperacetylated histone H4 was identified, associated with proto-oncogene SPECC1, regulator of apoptosis ADAMTSL4, as well as with proteins involved in different signalling cascades: NF-κB, JAK2/STAT4, Ras and Hedgehog signal transduction pathways. Interestingly, hyperacetylated histone H4 in control NB4 cells was found to be associated with Nucleophosmin (NPM), a protein which regulates tumour suppressors TP53/p53 and ARF and is shown to be overexpressed in actively proliferating cells, such as various cancer and stem cells 33. It is worth mentioning that NPM was found in complexes with hyperacetylated histone H4 after treatment with 2 μM Bel as well, but to a much lesser extent.
Figure 6.

Proteins identified in association with hyperacetylated histone H4 in control NB4 cells. Untreated NB4 cells were subjected to ChIP - MS analysis. Association network of identified proteins was studied and represented using STRING database (http://string.embl.de).
After 6 hrs treatment with 2 μM Bel (Table2, Fig.7) hyperacetylated histone H4 was identified to be associated with proteins that are pro-apoptotic and necessary for apoptotic response (S100A9, S100A8, LGALS7, GOLGA3, PPT1). Tumour suppressor APC was found in immunoprecipitated complexes as well. It is important to underline that hyperacetylated histone H4 has been also found to be associated with proteins that are involved in the defence against oxidative stress (TXNRD2) and access of RA to the nuclear retinoic acid receptors regulation (CRABP1).
Figure 7.

Proteins identified in association with hyperacetylated histone H4 in Bel treated NB4 cells. 2 μM Bel treated NB4 cells were subjected to ChIP - MS analysis. Association network of identified proteins was studied and represented using STRING database (http://string.embl.de).
Discussion
It is widely accepted that epigenetic modifiers and changes in the epigenetic landscape play a very significant role in APL pathogenesis. The onset of this haematological malignancy, at least in part, was shown to be induced by abnormal HDACs recruitment to RA target genes 3,34. Therefore, activation of the RA signalling pathway via HDAC activity inhibition may serve as a promising strategy in APL differentiation therapy.
In this study, we investigated the effect of a novel HDACi belinostat, alone or in combination with differentiation inducer RA, on human promyelocytic leukaemia NB4 and HL-60 cells growth and granulocytic differentiation. Notably, belinostat in combination with RA indeed showed a valuable activity towards APL cells. Combined treatment of 0.2 μM Bel + 1 μM RA inhibited NB4 and HL-60 cells proliferation and arrested cell cycle in G0/G1 phase. In addition, combined treatment was shown to accelerate and induce higher percent of granulocytic differentiation, compared to treatment with RA alone. These results are similar to the effect of some other well-known HDACi inhibitors used for leukaemia granulocytic differentiation therapy. Our group previously demonstrated a comparable effect on NB4 and HL-60 cells granulocytic differentiation upon combined treatment with HDACi Phenyl butyrate or BML-210 with RA 35,36. Also it was demonstrated that HDACi FK228 in HL-60 cells increases RA-induced granulocytic differentiation more than 1.8-fold, whereas this combination has only a minor pro-differentiational effect on NB4 cells 37.
Considering molecular mechanisms, associated with APL cell growth suppression and increased granulocytic differentiation in more detail, the effect of Bel, RA and Bel + RA treatments on NB4 cells certain epigenetic modifiers (HDAC1, HDAC2, PCAF) and cell cycle regulators (p27) gene and protein expression were evaluated. Our group have showed numerous times earlier 22,23 that although NB4 and HL-60 cells differ in the way that NB4 is a typical APL cell line, possessing t(15;17), whereas HL-60 does not have PML-RARα, the differences in their responses to various HDACi treatments are minimal. The manner of the effect is similar or identical, only kinetics can sometimes be found to be different. Therefore, further investigations were restricted to NB4 cells only.
We showed, that treatment with belinostat sharply down-regulated NB4 cells HDAC1 and HDAC2 mRNA levels. Reduction in HDAC1 protein level was also observed, together with an increase in histone H4 hyperacetylation. In addition, our group previously demonstrated 22 that belinostat induces accumulation of H4K16Ac mark, which in turn is associated with the transcriptional activation. It was also shown that belinostat up-regulates H3K9 acetylation level depending on treatment duration and used dose 22.
As it has been described earlier 38–40, in AML cells HDACs, predominantly HDAC1, are overexpressed, which leads to histone hypoacetylation, whereas during granulocytic differentiation HDAC1 protein expression diminishes and acetylation patterns are restored. Therefore, the results we observed in NB4 cells using belinostat alone and in combination with RA are promising and in line with published data.
We also were interested in belinostat’s effect not only on HDACs but on HATs as well, in this case particularly on PCAF. This protein has shown to function as an acetylase, as it directly modifies histones and other proteins. Furthermore, it associates with additional activators, such as CBP/p300, ACTR 41 and is able to increase histone acetylation at transcriptional sites, targeting predominantly histones H3 and H4 42. It has been demonstrated earlier 43 that RA induces PCAF expression and accumulation in P19 carcinoma cells’ nucleus. It also has been postulated that increase in PCAF mRNA levels potentiates retinoid-dependent gene expression 44. In accordance to this data, we showed that in NB4 cells, RA indeed induced PCAF gene expression. However, in this study we also revealed that 6 hrs exposure with belinostat reduced PCAF gene expression more than twofold, in comparison with control cells, whereas later PCAF gene expression was restored. Furthermore, in a previous research 22 the dose-dependent down-regulation in PCAF protein level upon treatment with belinostat has been demonstrated. Therefore, it seems that belinostat, indeed, perturbs histone acetylating enzymes machinery in APL cells, but the precise mechanism still remains elusive and needs further investigation. It can only be speculated on the basis of Hirano et al. research 45, that down-regulation of PCAF gene and protein expression may be associated with belinostat’s pro-apoptotic effect, as it was demonstrated that down-regulation of PCAF sensitized human prostate cancer PC3 cells to chemotherapeutic therapy, induced G1 arrest and apoptosis.
Because of HDACs association with histone methyl transferases and DNA methyl transferases (DNMTs) belinostat exerts an impact not only on HDACs or HATs but on other chromatin remodelling enzymes as well. Our previous research 22 demonstrated that treatment with Bel depletes Polycomb repressive complex 2 subunits’ EZH2 and SUZ12 proteins, however, not bearing any significant effect on H3K27 trimethylation. In addition, in this study the activity of Bel and combined treatment Bel + RA on global DNA methylation level was also evaluated. Upon 72 hrs treatment with Bel + RA global NB4 cells DNA methylation level was down-regulated more than 14%. This is in agreement with Arzenani et al. 46, who showed that HDACi Trichostatin A down-regulates DNMT1 gene and protein expression and reduces global DNA methylation in the hepatoma cells.
In addition, our group previously indicated 22 that, although belinostat promotes APL granulocytic differentiation, as a single agent it has no effect on NB4 cells C/EBPα and C/EBPε genes expression or genes coding for transcription factors that drive granulocytic differentiation 47,48. In this study, using ChIP, we aimed to determine histone H4 hyperacetylation level in C/EBPα and C/EBPε promoter regions. We found that upon treatment with belinostat alone, basal histone H4 hyperacetylation level was up-regulated, but no increase in histone H4 hyperacetylation levels in C/EBPα and C/EBPε promoter regions were detected. Therefore, this suggests that other mechanisms than direct activation of C/EBPα and C/EBPε are responsible for ability of belinostat to enhance RA-induced granulocytic differentiation. We suppose that belinostat may mainly accomplish its action via effects on the cell cycle. RT-qPCR analysis revealed that upon NB4 cells treatment with 0.2 μM belinostat, p27 gene expression is firmly up-regulated (more than fourfold after 72 hrs treatment). In addition, ChIP results indicated the obvious increase in hyperacetylated histone H4 association with p27 promoter region. It was demonstrated earlier 22, that belinostat up-regulates NB4 cells p27 protein levels as well. Taken this data together, it is plausible not to underestimate the role of p27 activation in belinostat-mediated antileukaemic effect.
Very interesting results, regarding belinostat’s activity on APL cells, were revealed by MS analysis. We found that upon 6 hrs treatment with 2 μM belinostat hyperacetylated histone H4 was no longer associated with proteins involved in gene transcription or translation, as it was the case in untreated NB4 cells, but was found to associate with proteins, that are usually detected in cytosolic fraction as components of neutrophil extracellular traps (NETs). To express in more detail, NETs were shown to be a composition of DNA, histones and antimicrobial proteins, that form an extracellular mesh able to trap and kill pathogens 49 and they are released during a cell death that depends on reactive oxygen species (ROS) produced by the NADPH oxidase complex 50.
Regarding composition of NETs, it has been indicated that NETs contain calprotectin (S100A8/S100A9) 51 a protein complex composed of two calcium-binding proteins which are abundantly found in neutrophils cytosolic fraction and have shown to have apoptosis inducing activity 52. Upon NB4 cells treatment with belinostat MS analysis results indeed revealed the association of hyperacetylted histone H4 with calprotectin (both S100A8 and S100A9). Calprotectin is essential for the neutrophilic NADPH oxidase activation 53. The importance of S100A8/A9 complex in NB4 cells NADPH oxidase activation has been demonstrated by the S100A9 expression blockage, after which NADPH oxidase activity has been impaired 54. In addition, S100A8/A9-dependent NADPH stimulation has shown to increase ROS levels in keratinocytes 55. Notably, in PANC-1 cells belinostat promoted ROS production 56. It has been shown for multiple myeloma cells as well 57.
Furthermore, besides the calprotectin, we also found a probable serine protease TMPRSS11A associated with hyperacetylated histone H4, which is in agreement with data, showing that NETs contain serine proteases, as they may execute antimicrobial functions in those structures 58. Taking all together, we assume that belinostat’s cell death-inducing activity in some manner may relate to NETs formation. Although, it is already known that belinostat triggers apoptosis in myeloid cells 22,20,21, not NETosis (the different mechanism of cell death when NETs are released, named by Steinberg and Grinstein 59), the possibility that belinostat intervenes in NETs formation may not be rejected completely.
It is evident, that APL cells, despite of their differentiation state, manage to generate very few NETs, which was demonstrated for HL-60 cells 60. This might be explained by their inability to induce autophagy, as not only NADPH activity, but also autophagy has shown to be crucial for NETosis 61. In addition, recently it was demonstrated, that NETs formation does not necessarily require cell death 62,63 and that cells, which have formed NETs, may retain the capacity to die via apoptosis 64 However, possible role of belinostat in triggering NETs formation in APL cells is only an educated guess, at least at this stage. Therefore, further investigations are needed to confirm or reject this hypothesis.
Summarizing, our findings, regarding belinostat’s effect on cell growth, differentiation, gene and protein expression, as well as on epigenetic modifications, confirmed potential value belinostat has in APL therapy. In this study some new insights in possible molecular mechanisms of belinostat were also revealed.
Acknowledgments
This research was funded by a grant from the Research Council of Lithuania (no. LIG-06/2012).
Disclosure
The authors declare that there are no conflicts of interest.
References
- Nasr R, Lallemand-Breitenbach V, Zhu J, et al. Therapy-induced PML/RARA proteolysis and acute promyelocytic leukemia cure. Clin Cancer Res. 2009;15:6321–6. doi: 10.1158/1078-0432.CCR-09-0209. [DOI] [PubMed] [Google Scholar]
- Piazza F, Gurrieri C, Pandolfi PP. The theory of APL. Oncogene. 2001;20:7216–22. doi: 10.1038/sj.onc.1204855. [DOI] [PubMed] [Google Scholar]
- Grignani F, De Matteis S, Nervi C, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391:815–8. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- Lin RJ, Nagy L, Inoue S, et al. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998;391:811–4. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- Yang L, Zhao H, Li SW, et al. Gene expression profiling during all-trans retinoic acid-induced cell differentiation of acute promyelocytic leukemia cells. J Mol Diagn. 2003;5:212–21. doi: 10.1016/S1525-1578(10)60476-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muindi J, Frankel SR, Miller WH, Jr, et al. Continuous treatment with all-trans retinoic acid causes a progressive reduction in plasma drug concentrations: implications for relapse and retinoid “resistance” in patients with acute promyelocytic leukemia. Blood. 1992;79:299–303. [PubMed] [Google Scholar]
- Ferrara FF, Fazi F, Bianchini A, et al. Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res. 2001;61:2–7. [PubMed] [Google Scholar]
- Bruserud Ø, Stapnes C, Tronstad KJ, et al. Protein lysine acetylation in normal and leukaemic haematopoiesis: HDACs as possible therapeutic targets in adult AML. Expert Opin Ther Targets. 2006;10:51–68. doi: 10.1517/14728222.10.1.51. [DOI] [PubMed] [Google Scholar]
- Khan N, Jeffers M, Kumar S, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–9. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Witter DJ, Belvedere S, Chen L, et al. Benzo[b]thiophene-based histone deacetylase inhibitors. Bioorg Med Chem Lett. 2007;17:4562–7. doi: 10.1016/j.bmcl.2007.05.091. [DOI] [PubMed] [Google Scholar]
- Gravina GL, Marampon F, Giusti I, et al. Differential effects of PXD101 (belinostat) on androgen-dependent and androgen-independent prostate cancer models. Int J Oncol. 2012;40:711–20. doi: 10.3892/ijo.2011.1270. [DOI] [PubMed] [Google Scholar]
- Qian X, LaRochelle WJ, Ara G, et al. Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol Cancer Ther. 2006;5:2086–95. doi: 10.1158/1535-7163.MCT-06-0111. [DOI] [PubMed] [Google Scholar]
- Ramalingam SS, Belani CP, Ruel C, et al. Phase II study of belinostat (pxd101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J Thorac Oncol. 2009;4:97–101. doi: 10.1097/JTO.0b013e318191520c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccone G, Rajan A, Berman A, et al. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J Clin Oncol. 2011;29:2052–9. doi: 10.1200/JCO.2010.32.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo W, Chung HC, Chan SL, et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: a multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the mayo phase II consortium and the cancer therapeutics research group. J Clin Oncol. 2012;30:3361–7. doi: 10.1200/JCO.2011.41.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizon DS, Blessing JA, Penson RT, et al. A phase ii evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: a gynecologic oncology group study. Gynecol Oncol. 2012;125:367–71. doi: 10.1016/j.ygyno.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizon DS, Damstrup L, Finkler NJ, et al. Phase II activity of belinostat (pxd-101), carboplatin, and paclitaxel in women with previously treated ovarian cancer. Int J Gynecol Cancer. 2012;22:979–86. doi: 10.1097/IGC.0b013e31825736fd. [DOI] [PubMed] [Google Scholar]
- Grassadonia A, Cioffi P, Simiele F, et al. Role of hydroxamate-based histone deacetylase inhibitors (Hb-HDACIs) in the treatment of solid malignancies. Cancers. 2013;5:919–42. doi: 10.3390/cancers5030919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschbaum MH, Foon KA, Frankel P, et al. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: a California Cancer Consortium Study. Cancers. 2013;5:919–42. doi: 10.3109/10428194.2013.877134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Chen S, Wang L, et al. Bortezomib interacts synergistically with belinostat in human acute myeloid leukaemia and acute lymphoblastic leukaemia cells in association with perturbations in NF-κB and Bim. Br J Haematol. 2011;153:222–35. doi: 10.1111/j.1365-2141.2011.08591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapnes C, Ryningen A, Hatfield K, et al. Functional characteristics and gene expression profiles of primary acute myeloid leukaemia cells identify patient subgroups that differ in subgroups that differ in susceptibility to histone deacetylase inhibitors. Int J Oncol. 2007;31:1529–38. [PubMed] [Google Scholar]
- Savickiene J, Treigyte G, Valiuliene G, et al. Epigenetic and molecular mechanisms underlying the antileukemic activity of the histone deacetylase inhibitor belinostat in human acute promyelocytic leukemia cells. Anticancer Drugs. 2014;25:937–49. doi: 10.1097/CAD.0000000000000122. [DOI] [PubMed] [Google Scholar]
- Savickiene J, Treigyte G, Vistartaite G, et al. C/EBPα and PU.1 are involved in distinct differentiation responses of acute promyelocytic leukemia HL-60 and NB4 cells via chromatin remodeling. Differentiation. 2011;81:57–67. doi: 10.1016/j.diff.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, et al. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–62. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Simoliūnas E, Kaliniene L, Truncait≐ L, et al. Klebsiella phage vB_KleM-RaK2 - a giant singleton virus of the family Myoviridae. PLoS ONE. 2013;8:e60717. doi: 10.1371/journal.pone.0060717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunaitis V, Sadzeviciene I, Suriaikaite K, et al. Expression and subcellular localization of histone deacetylases in mesenchymal stem-like cells from exfoliated deciduous teeth. Biologija. 2008;54:306–11. [Google Scholar]
- Jeon ES, Moon HJ, Lee MJ, et al. Sphingosylphosphorylcholine induces differentiation of human mesenchymal stem cells into smooth-muscle-like cells through a TGF-beta-dependent mechanism. J Cell Sci. 2006;119:4994–5005. doi: 10.1242/jcs.03281. [DOI] [PubMed] [Google Scholar]
- Iwanaga R, Komori H, Ishida S, et al. Identification of novel E2F1 target genes regulated in cell cycle-dependent and independent manners. Oncogene. 2006;25:1786–98. doi: 10.1038/sj.onc.1209210. [DOI] [PubMed] [Google Scholar]
- Timchenko NA, Harris TE, Wilde M, et al. CCAAT/enhancer binding protein alpha regulates p21 protein and hepatocyte proliferation in newborn mice. Mol Cell Biol. 1997;17:7353–61. doi: 10.1128/mcb.17.12.7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna-Gupta A, Zibello T, Sun H, et al. Chromatin immunoprecipitation (ChIP) studies indicate a role for CCAAT enhancer binding proteins alpha and epsilon (C/EBP alpha and C/EBP epsilon) and CDP/cut in myeloid maturation-induced lactoferrin gene expression. Blood. 2003;101:3460–8. doi: 10.1182/blood-2002-09-2767. [DOI] [PubMed] [Google Scholar]
- Morosetti R, Park DJ, Chumakov AM, et al. A novel, myeloid transcription factor, C/EBP epsilon, is upregulated during granulocytic, but not monocytic, differentiation. Blood. 1997;90:2591–600. [PubMed] [Google Scholar]
- Radomska HS, Huettner CS, Zhang P, et al. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. 1998;18:4301–14. doi: 10.1128/mcb.18.7.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MJ, Wang XW. Nucleophosmin and human cancer. Cancer Detect Prev. 2006;30:481–90. doi: 10.1016/j.cdp.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidez F, Ivins S, Zhu J, et al. Reduced retinoic acid-sensitivities of nuclear receptor corepressor binding to PML- and PLZF-RARalpha underlie molecular pathogenesis and treatment of acute promyelocytic leukemia. Blood. 1998;91:2634–42. [PubMed] [Google Scholar]
- Savickiene J, Borutinskaite VV, Treigyte G, et al. The novel histone deacetylase inhibitor BML-210 exerts growth inhibitory, proapoptotic and differentiation stimulating effects on the human leukemia cell lines. Eur J Pharmacol. 2006;549:9–18. doi: 10.1016/j.ejphar.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Savickiene J, Treigyte G, Borutinskaite VV, et al. Antileukemic activity of combined epigenetic agents, DNMT inhibitors zebularine and RG108 with HDAC inhibitors, against promyelocytic leukemia HL-60 cells. Cell Mol Biol Lett. 2012;17:501–25. doi: 10.2478/s11658-012-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savickiene J, Treigyte G, Borutinskaite V, et al. The histone deacetylase inhibitor FK228 distinctly sensitizes the human leukemia cells to retinoic acid-induced differentiation. Ann N Y Acad Sci. 2006;1091:368–84. doi: 10.1196/annals.1378.081. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- Shimizu FR, Kitamura T, Wada YT, et al. Progenitors determine the cell fate of hematopoietic expression. J Biol Chem. 2009;284:30673–83. doi: 10.1074/jbc.M109.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Kikuchi J, Nishimura N, et al. Expression levels of histone deacetylases determine the cell fate of hematopoietic progenitors. J Biol Chem. 2009;284:30673–83. doi: 10.1074/jbc.M109.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Schiltz RL, et al. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–80. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–3. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yan L, Zhang Y, et al. Role of acetylated p53 in regulating the expression of map2 in retinoic acid-induced P19 cells. Chin Med Sci J. 2010;25:71–5. doi: 10.1016/s1001-9294(10)60025-9. [DOI] [PubMed] [Google Scholar]
- Blanco JCG, Minucci S, Lu J, et al. The histone acetylase PCAF is a nuclear receptor coactivator. Genes Dev. 1998;12:1638–51. doi: 10.1101/gad.12.11.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano G, Izumi H, Kidani A, et al. Enhanced expression of PCAF endows apoptosis resistance in cisplatin-resistant cells. Mol Cancer Res. 2010;8:864–72. doi: 10.1158/1541-7786.MCR-09-0458. [DOI] [PubMed] [Google Scholar]
- Arzenani MK, Zade AE, Ming Y, et al. Genomic DNA hypomethylation by HDAC inhibition implicates DNMT1 nuclear dynamics. Mol Cell Biol. 2011;31:4119–28. doi: 10.1128/MCB.01304-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeshan K, Santilli G, Corradini F, et al. Transcription activation function of C/EBPalpha is required for induction of granulocytic differentiation. Blood. 2003;102:1267–75. doi: 10.1182/blood-2003-02-0477. [DOI] [PubMed] [Google Scholar]
- Khanna-Gupta A, Zibello T, Sun H, et al. C/EBP epsilon mediates myeloid differentiation and is regulated by the CCAAT displacement protein (CDP/cut) Proc Natl Acad Sci USA. 2001;98:8000–5. doi: 10.1073/pnas.141229598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartha F, Beiter K, Normark S, et al. Neutrophil extracellular traps: casting the NET over pathogenesis. Curr Opin Microbiol. 2007;10:52–6. doi: 10.1016/j.mib.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–41. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban CF, Ermert D, Schmid M, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639. doi: 10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui S, Nakatani Y, Mikami M. Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol Pharm Bull. 2003;26:753–60. doi: 10.1248/bpb.26.753. [DOI] [PubMed] [Google Scholar]
- Brechard S, Plancon S, Tschirhart EJ. New insights into the regulation of neutrophil NADPH oxidase activity in the phagosome: a focus on the role of lipid and Ca2+ signaling. Antioxid Redox Signal. 2013;18:661–76. doi: 10.1089/ars.2012.4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkhoff C, Nacken W, Benedyk M, et al. The arachidonic acid-binding protein S100A8/A9 promotes NADPH oxidase activation by interaction with p67phox and Rac-2. FASEB J. 2005;19:467–9. doi: 10.1096/fj.04-2377fje. [DOI] [PubMed] [Google Scholar]
- Voss A, Bode G, Sopalla C, et al. Expression of S100A8/A9 in HaCaT keratinocytes alters the rate of cell proliferation and differentiation. FEBS Lett. 2011;585:440–6. doi: 10.1016/j.febslet.2010.12.037. [DOI] [PubMed] [Google Scholar]
- Wang B, Wang XB, Chen LY, et al. Belinostat-induced apoptosis and growth inhibition in pancreatic cancer cells involve activation of TAK1-AMPK signaling axis. Biochem Biophys Res Commun. 2013;437:1–6. doi: 10.1016/j.bbrc.2013.05.090. [DOI] [PubMed] [Google Scholar]
- Feng R, Oton A, Mapara MY, et al. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br J Haematol. 2007;139:385–97. doi: 10.1111/j.1365-2141.2007.06772.x. [DOI] [PubMed] [Google Scholar]
- Papayannopoulos V, Metzler KD, Hakkim A, et al. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–91. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE. 2007;2007:pe11. doi: 10.1126/stke.3792007pe11. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li M, Stadler S, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–13. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remijsen Q, Kuijpers TW, Wirawan E, et al. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011;18:581–8. doi: 10.1038/cdd.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi S, Mihalache C, Kozlowski E, et al. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–44. doi: 10.1038/cdd.2009.96. [DOI] [PubMed] [Google Scholar]
- Yipp BG, Petri B, Salina D, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386–93. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah E, Andrade F. NETs: the missing link between cell death and systemic autoimmune diseases. Front Immunol. 2013;3:428. doi: 10.3389/fimmu.2012.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]