

Abstract
Background
Clinical studies implying the sunitinib multi-kinase inhibitor have led to disappointing results for breast cancer care but mostly focused on HER2-negative subtypes. Preclinical researches involving this drug mostly concern Triple Negative Breast Cancer (TNBC) murine models. Here, we explored the therapeutic efficacy of sunitinib on a PyMT-derived transplanted model classified as luminal B (HER2-positive) and monitored the response to treatment using both in vivo and ex vivo approaches.
Methods
Tumour-induced animals were treated for 9 (n = 7) or 14 (n = 8) days with sunitinib at 40 mg/kg or with vehicle only. Response to therapy was assessed in vivo by monitoring glucose tumour metabolism and hypoxia using 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) and [18F]fluoromisonidazole ([18F]FMISO) Positron Emission Tomography (PET). After primary tumour excision, ex vivo digital microscopy was performed on treated and control samples to estimate vascular density (CD31), apoptosis (Tunel), proliferation (Ki-67), Tumour-Associated Macrophage (TAM) infiltration (F4/80), metabolism (GLUT1) and cellular response to hypoxia (HIF1 alpha). The drug impact on the metastasis rate was evaluated by monitoring the PyMT gene expression in the lungs of the treated and control groups.
Results
Concomitant with sunitinib-induced tumour size regression, [18F]FDG PET imaging showed a stable glycolysis-related metabolism inside tumours undergoing treatment compared to an increased metabolism in untreated tumours, resulting at treatment end in 1.5 less [18F]FDG uptake in treated (n = 4) vs control (n = 3) tumours (p < 0.05). With this small sample, [18F]FMISO PET showed a non-significant decrease of hypoxia in treated vs control tumours. The drug triggered a 4.9 fold vascular volume regression (p < 0.05), as well as a 17.7 fold induction of tumour cell apoptosis (p < 0.001). The hypoxia induced factor 1 alpha (HIF1 alpha) expression was twice lower in the treated group than in the control group (p < 0.05). Moreover, the occurrence of lung metastases was not reduced by the drug.
Conclusions
[18F]FDG and [18F]FMISO PET were relevant approaches to study the response to sunitinib in this luminal B (HER2-positive) model. The sunitinib-induced vascular network shrinkage did not significantly increase tumour hypoxia, suggesting that tumour regression was mainly due to the pro-apoptotic properties of the drug. Sunitinib did not inhibit the metastatic process in this PyMT transplanted model.
Electronic supplementary material
The online version of this article (doi:10.1186/s12885-015-1540-2) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, PyMT, Sunitinib, PET, Digital microscopy
Background
Based on encouraging preclinical data, the sunitinib drug, a multi-kinase inhibitor, has been investigated in several clinical studies in association with various cytotoxic drugs but led to disappointing results in breast cancer patients. Classically, breast cancers are classified according to the expression levels of the estrogen (ER) and progesterone (PR) receptors, and of the human epidermal growth factor receptor 2 (HER2) oncogene. The large majority of studies with sunitinib involved advanced and heavily treated breast cancer focusing on the HER2-negative (or trastuzumab (TZM) insensitive) subtypes (phase I [1], phases II [2, 3], phases III [4–6]). Interestingly, Burstein et al. administered sunitinib alone and detected superior overall response rate (ORR) for the HER2-positive subtype (25 % vs 11 % in the whole population) [7]. Moreover, two recent reports focusing on HER2-positive breast cancers found an improved ORR in adding sunitinib to regimens based on TZM with or without docetaxel administration [8, 9].
The breast cancer classification is now reconsidered in the light of global gene expression analyses of human biopsies leading to six identified subtypes: luminal A and B, basal-like, claudin-low, HER2-enriched and normal breast like [10, 11]. A detailed panorama of the relationships between the histological- and the transcriptomic-based classifications has been recently published [12]. In this context, the breast cancer patient care is evolving as it is expected that the efficacies of chemotherapeutic regimens should depend on the considered subtype.
In the case of sunitinib, many preclinical studies were performed using various Triple Negative Breast Cancer (TNBC) mouse models, and all found that the drug delayed the tumour growth at doses ranging between 20 to 60 mg/kg/day. Interestingly, sunitinib treatment induced tumour regression in a MCF7 xenograft model [13], which is a typical luminal A cancer [14], as well as on a MMTV-v-Ha-Ras transgenic model [15], which has been classified as luminal B [16]. Among the breast cancer diversity, the luminal subset represents mainly the ER+ group, for which an endocrine therapy is recommended. The luminal A cancers are defined as ER+ PR+ HER2- and low Ki-67 whereas luminal B carcinomas are ER+ HER2+ or ER+ PR+/− HER2- and high Ki-67 [17, 18]. The luminal A cancers present a relatively good outcome, but the luminal B tumours, which represent 10 to 20 % of all breast cancers, are associated with a poor prognosis and identification of new therapeutic options for this subtype is still very challenging. Thus, as most anterior preclinical studies with sunitinib focused on TNBC models, we investigated here its efficacy in a luminal B-type breast cancer model combining in vivo PET and ex vivo histochemical analyses of tumours.
For this purpose, we used the MMTV-PyMT murine model whose oncogenesis is induced by expression of the polyoma virus middle T oncoprotein under control of the Mouse Mammary Tumour Virus (MMTV) promoter (PMID: 1312220). Following the recommendations of Varticovski et al. [19] about the limitations of using genetically engineered mouse models in preclinical studies, we generated a transplanted orthotopic and syngeneic model from the original transgenic mice. In order to characterize the therapy response to sunitinib in the PyMT model, we then performed in vivo Positron Emission Tomography (PET) with 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) and [18F]fluoromisonidazole ([18F]FMISO) radiotracers, which allow to monitor tumour glucose metabolism and hypoxia respectively. Furthermore, in vitro analyses were used to quantify the chemotherapy impact on several cancer-associated parameters, namely vascularization (CD31), apoptosis (TUNEL), proliferation (Ki-67), hypoxia (HIF1 alpha), TAM infiltration (F4/80), metabolic activity (GLUT1) and metastasis.
Methods
Animal studies were approved by the animal ethics committee “Comité d'EThique en Expérimentation Animale” (CETEA DSV n°44) under reference 12–036 and conducted in accordance with the Directives of the European Union.
Tumour removal and preparation of cell suspensions
FVB/N-Tg (MMTV-PyMT)634Mul/J (PyMT) 12-weeks-old mice were used as tumour donor. Aseptically collected mammary tumours from PyMT mice were minced and immersed in cold Dulbecco's Modified Eagle's Medium (Sigma, USA). Mechanical cell dissociation was performed using Medicon disposable chambers (BD bioscience, USA). The cell suspension was then progressively filtered using Filcon filters with pore sizes of 500 μm, 200 μm and 70 μm (BD bioscience). Finally, cells were aliquoted in freezing medium (Life Technologies, USA) and stored in liquid nitrogen.
Tumour implantation and monitoring
After freezing medium removal and enumeration, the tumour cells were directly inoculated, without any in vitro culture step, in the mammary fat pad of the posterior nipple in FVB mice. The tumour volumes were calculated using calliper measurements and the approximated formula for a prolate ellipsoid, given by:
To evaluate drug toxicity, body animal weights were also monitored.
Chemotherapy
Two sets of mice were used in this study. For the main set A, 7 animals were implanted with 3 million viable cells. PyMT tumours were allowed to grow for 21 days. The mice were randomized into treated (n = 4) and control (n = 3) groups. The treated one received per os a daily dose of sunitinib at 40 mg/kg in 20 mM dimethyl sulfoxyde (DMSO, Sigma). The control group received only the DMSO solution. Drug administration was performed during 9 consecutive days.
To further explore the neoadjuvant therapy effects on the metastatic incidence, we extended primary tumour growth and treatment times before mammary tumour surgical resection. Thus, a secondary set B of treated (n = 4) and control (n = 4) mice was obtained implanting 400 000 viable cells. Treatment began at day 25 post implantation. It continued for 14 days until resection at day 39. For both sets A and B, the primary tumours were surgically removed after treatment and the mice were kept alive for 60 supplementary days before euthanasia to analyse the lungs for metastasis content.
[18F]FDG and [18F]FMISO positron emission tomography
[18F]FDG and [18F]FMISO PET scans were performed on mice from set A at days 0 and −1 respectively prior to treatment and at days 5 and 6 of treatment. 15 min long PET acquisitions were performed 60 min after [18F]FDG injection and 90 min after [18F]FMISO injection. PET data were corrected for attenuation, scatter and radioactive decay and reconstructed using a two dimensional ordered-subset expectation maximization (2D-OSEM) algorithm after Fourier rebinning, with a voxel size of 0.5 × 0.5 × 0.8 mm3 (sofware ASIPro VM™, CTI Concorde Microsystems). Radioactivity uptake in regions of interest (ROIs) was measured using BrainVISA 4.0 and Anatomist 4.0.2 (CEA/Neurospin/SHFJ, France) and expressed in Standardized Uptake Value (SUV) calculated using:
Histochemistry
Primary tumours from set A of animals were fixed in zinc solution (BD bioscience) and included in paraffin. Series of tissue sections were sequentially cut. For blood vessels, macrophages and cellular hypoxia sensor labelling, the slides were immersed in toluene and progressively rehydrated. Endogenous peroxidases and biotin were blocked with 3 % hydrogen peroxide solution (Sigma) and biotin blocking kit (Life technologies) respectively. Rat anti CD31 (Pharmingen, USA), rat anti F4/80 (Caltag, UK) and rabbit anti Hypoxia Inducible Factor 1 alpha (HIF1 alpha, LSBio, USA) were used as primary antibodies for each labelling respectively. Biotin-goat anti rat IgG (Life technologies) was used as secondary antibody for vascular and macrophage staining. The tyramide signal amplification (TSA) system (Perkin Elmer, USA) was then used following manufacturer’s instructions. For HIF1 alpha labelling, HRP-goat anti rabbit IgG (Life technologies) was incubated as secondary antibody. For cellular proliferation and Glucose transporter 1 (GLUT1) expression labelling, paraffin removal was performed using heated PT module buffer pH8 (Fischer Scientific, USA). As above, after the blocking steps, the slides were incubated with goat anti Ki-67 (Santa Cruz, USA) or rabbit anti GLUT1 (Neomarker, USA) for each labelling respectively. Secondary detection reagents were biotin-rabbit anti goat IgG (Life technologies) followed by TSA system for Ki-67 or HRP-goat anti rabbit IgG (Life technologies) for GLUT1. After 3-3'–diamino-benzidine (DAB, Sigma) revelation, counterstaining was performed with hematoxylin (Sigma) and slides were mounted with Eukitt (Sigma). For late apoptosis staining, terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL, Promega, USA) was used according to the manufacturer’s protocol. Slides were then mounted with ProLong Gold Antifade Reagent containing 4',6'-diamidino-2-phenylindole (DAPI, Life technologies).
Microscopy image acquisition and analysis methods
The set of tissue sections uniformly sampling the whole volume of each tumour was entirely scanned at high resolution (0.37 μm per pixel) using an AxiObserver Z1 (Zeiss, Germany). The resulting brightfield image series were analysed using the CellProfiler software [20]. After a colour deconvolution step, the segmentation of each structure of interest was based on a constant labelling-dependent threshold. A filtering step was added for size-based vessel clustering. Logic diagrams of the processing pipelines are available as supplementary data (see Additional files 1 and 2). The DAB-labelled surface areas and whole hematoxylin areas were measured by the software. The consistency of the automatic segmentation was controlled visually on the original images supplemented with the outlines of identified objects. Whole tissue sections fluorescently labelled with the TUNEL method were acquired using two excitation/emission filter sets: 365/445 nm for DAPI and 470/525 nm for TUNEL staining. TIF-format images were processed using the ImageJ software [21], yielding the total area corresponding to fluorescent pixels above a given constant threshold. The measured areas were multiplied by the distance between each tissue slide to get volume estimates.
Quantitative real time polymerase chain reaction (qRT-PCR)
The whole-lung tissue ribonucleic acids (RNA) were extracted using the total RNA isolation kit (Macherey-Nagel, Germany) following manufacturer’s instructions. RNA was reverse transcribed using SuperScript II (Life technologies) with random primer hexamers. On a LightCycler 1.5 (Roche, Switzerland), a subsequence of the PyMT cDNA was amplified in Master SYBR Green I mix (Roche) using the previously described primers [22]. The housekeeping myelin protein zero (P0, MPZ) gene was used as an internal control. A relative quantification analysis was performed applying the delta-delta Ct method.
Statistical analyses
For statistical analysis, unpaired Student t-tests were performed using GraphPad Prism software. A p-value of 0.05 or less was interpreted as statistically significant. In all graphs, values are reported as mean ± one standard deviation (SD).
Results
Sunitinib-induced mammary tumour regression on the PyMT model
In set A of mice, the mean tumour volume measured by calliper was 209 ± 38 mm3 (n = 7) just before treatment (day 21). During the treatment phase until day 30, the tumours of the control group continued to grow up to 418 ± 62 mm3 (n = 3), while the size of the treated tumours decreased down to 109 ± 24 mm3 (n = 4) (Fig. 1a, p < 0.001). In set B of mice, the mean tumour volume measured by calliper was 115 ± 9 mm3 when treatment started (day 25, n = 8). At resection (day 39), tumour volumes were 282 ± 43 mm3 in the control group (n = 4) and 57 ± 11 mm3 in the treated group (n = 4) (Fig. 1b, p < 0.0001). The mouse weights corrected for their tumour weight (Fig. 1c-d) were not significantly different between the treated and control arms.
Fig. 1.
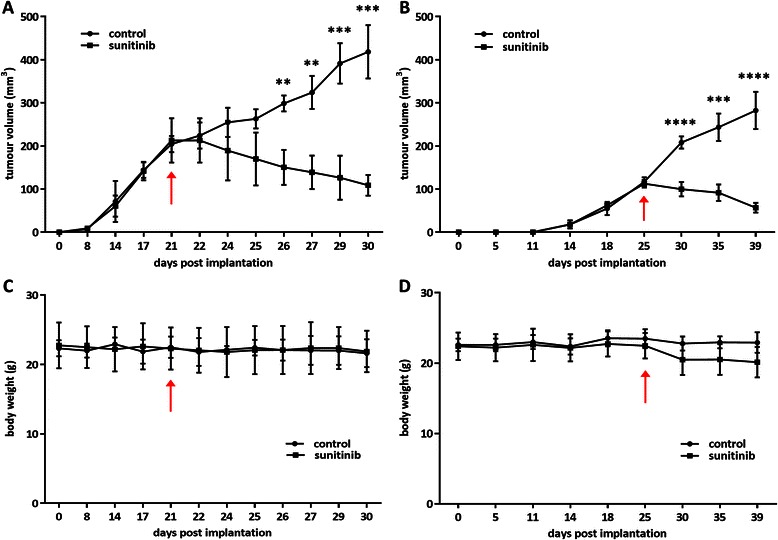
In vivo therapy model follow-up. a-b Tumour volume evolution, as measured by calliper, for sets A and B of mice respectively (set A: n = 3 for control, n = 4 for treated/set B: n = 4 for each group). c-d Mice body weight evolution for sets A and B respectively. In all graphs, arrows indicate the first day of sunitinib treatment
Effect of sunitinib administration on [18F]FDG and [18F]FMISO in vivo uptakes
PET monitoring was only performed for set A of mice. In the reconstructed images, the signal appeared more prominent in the tumour, bladder and heart compared to the rest of the body (Fig. 2a). At randomization, [18F]FDG uptakes expressed in SUV were similar in both groups. In the control group, the tumour [18F]FDG uptake was 1.4 greater after 5 days than at randomization, from 1.2 ± 0.1 to 1.6 ± 0.3 (n = 3) in SUV, although the difference was not statistically significant (NS). In the treated group, the [18F]FDG uptake remained stable during sunitinib administration, from 1.1 ± 0.2 to 1.1 ± 0.2 (n = 4) in SUV (NS). As a result, after 5 days of treatment, the [18F]FDG uptake was significantly lower, by a factor of 1.5 (p < 0.05), in the treated versus the control tumours (Fig. 2b).
Fig. 2.
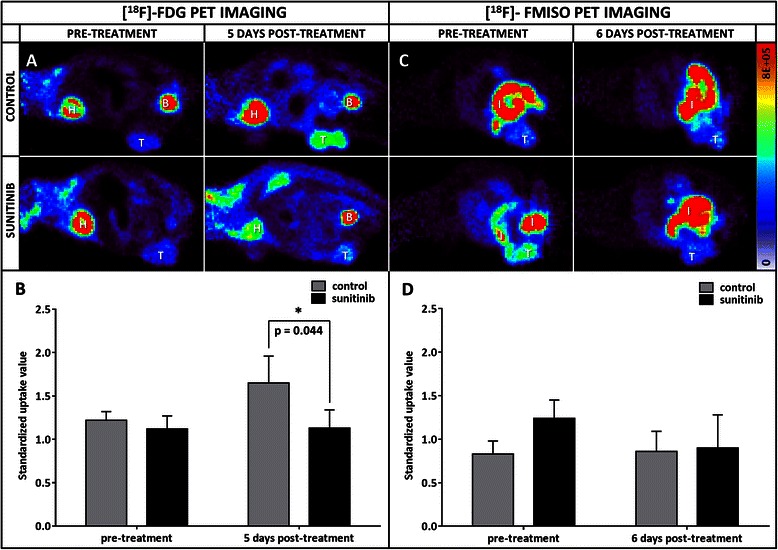
Evolution of PET radiotracer uptakes. a Representative images of a tumour-bearing mouse injected with [18F]FDG prior to treatment (left) and after 5 days (right) of sunitinib (lower part) or DMSO (upper part) administration (B: bladder, H: heart, T: tumour). b Tumour [18F]FDG SUV evolution for both 5 day-treated (n = 4) and control (n = 3) groups. c PET longitudinal images of a grafted mice injected with [18F]FMISO before treatment (left) and after 6 days (right) of treatment with sunitinib (bottom) or DMSO only (top) (I: intestine, T: tumour). d Tumour [18F]FMISO uptake evolution for both 6 day-treated (n = 3) and control (n = 3) groups
The [18F]FMISO PET images exhibited an enhanced contrast in the tumour and intestine regions (Fig. 2c). At randomization, [18F]FMISO tumour uptake was not significantly different between the group to be treated (1.2 ± 0.2 in SUV, n = 3) and the control group (0.8 ± 0.1 in SUV, n = 3), although lower in the control group. After 6 days of sunitinib or vehicle only administration, the tracer uptake remained stable in the control group (0.9 ± 0.2 in SUV, n = 3) and decreased in the treated one (0.9 ± 0.4 in SUV, n = 3), thus reducing the initial differences between the two groups (Fig. 2d).
Ex vivo evaluation of sunitinib incidence on PyMT tumour hallmarks
Digital microscopy analysis was exclusively performed on set A of mice. In the control and treated groups, the mean hematoxylin volumes estimated using digital microscopy were 126 ± 6 (n = 3) and 45 ± 20 mm3 (n = 4) respectively (p < 0.01). The nine day sunitinib treatment induced a 4.9 fold regression of the vascular volume reported to the hematoxylin volume, with values of 3.6 ± 1.8 % (n = 3) and 0.7 ± 0.05 % (n = 4) in the control and treated groups respectively (p < 0.05) (Fig. 3a). The sunitinib treatment induced a reduction of the large vessel proportion (from 31.3 ± 15.6 % in the control group to 8.3 ± 4.0 % in the treated group, p < 0.05), an increase of the small size vessels (from 33.4 ± 16.5 % in the control group to 61.0 ± 11.1 % in the treated group, p < 0.05) and no evolution for medium size vessels (Fig. 4). Interestingly, the vascular volume decrease did not induce a global increase in HIF1 alpha protein expression (Figs. 3b and 5a-b). On the contrary, sunitinib therapy led to a twofold reduction of HIF1 alpha labelling, from 11.5 ± 1.0 % (n = 3) in control tumours to 5.7 ± 3.5 % in treated ones (n = 4, p < 0.05). TUNEL labelling revealed a high induction of apoptosis by a factor of 17.7. Indeed, in nutrient supplied expanding tumours, very low programmed cell death was observed, representing 4.1 ± 0.3 % in volume (n = 3), while this value was 72.6 ± 10.7 % in treated tumours (n = 4, p < 0.001) (Figs. 3c and 5c-d). A mean pool of 1339 ± 248 cells per mm3 of tumour (n = 3) were over-expressing Ki-67 in non-treated tumours and the therapeutic agent reduced this population to 730 ± 334 proliferating cells per mm3 (n = 4, p < 0.05). This represents a 1.8 fold reduction of the tumour proliferation process (Figs. 3d and 5e-f). Tumours grown in the control conditions presented a mean density of 933 ± 212 macrophage cells (F4/80 positive) per mm3 of viable tumour tissue (n = 3). In the sunitinib treated mice, this value was at 546 ± 169 macrophages per mm3 (n = 4) (Figs. 3e and 5g-h) (p < 0.05 compared to the control mice). Finally, GLUT1 whole tumour expression was enhanced by a factor of 2.57 in the sunitinib treated group when compared to control (Figs. 3f and 5i-j). Indeed, transporter labelling represented 15.5 ± 2.6 % of control hematoxylin volume (n = 3), and reached 40.0 ± 12.3 % after the 9 days-long treatment (n = 4, p < 0.05).
Fig. 3.
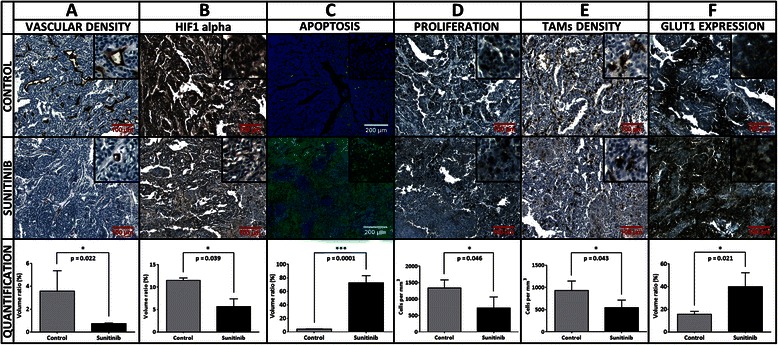
Biomarker quantification by digital microscopy. Each column corresponds to a labelling: a CD31, b HIF1 alpha, c TUNEL, d Ki-67, e F4/80, f GLUT1. Representative control and 9 day-treated tissues are displayed on first and second rows respectively. The third row presents the associated values. In bright field images, the biomarker of interest is labelled in brown and nuclei are counterstained in blue. In fluorescence images, TUNEL labelling is represented in green and nuclei are counterstained in blue
Fig. 4.
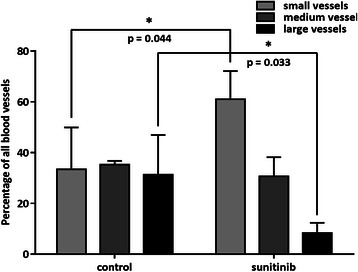
Sunitinib effect on blood vessel size. Comparison of the proportion of small, medium and large vessels between the control and treated groups (set A of mice)
Fig. 5.
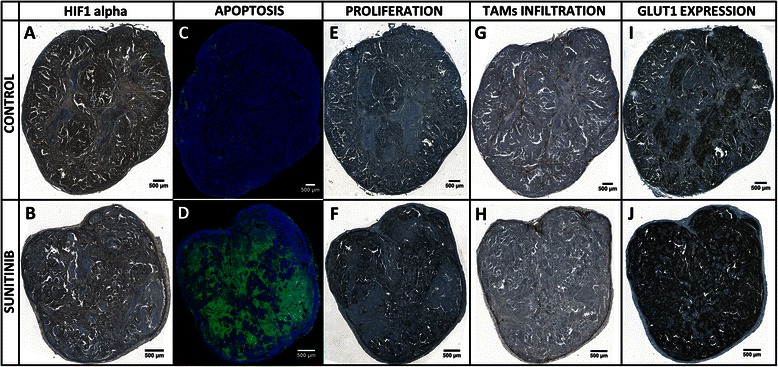
Whole tumour slide imaging. HIF1 alpha: (a) in control tumours, the HIF1alpha expression is mainly located inside heaps of high cellularity (b) in treated ones, the labelling is weaker, globally as necrotic regions get larger, and even locally inside living cell islets. TUNEL: (c) control section with low level of apoptosis (d) highly apoptotic sunitinib-treated tumour. Ki-67: (e) proliferating cell density remains at a relatively low level in control tumours, whereas (f) in treated ones, necropsied areas get larger but the density of Ki-67 positive cells increases in the remaining living cell islets. F4/80: (g) in controls, the highest TAMs density is encountered at the interface of tumour and necrotic regions; (h) in treated tumours, TAMs tend to relocate at the tumour external edges. GLUT1: (i) in control conditions, necrotic areas are the place of high GLUT1 expression; (j) under sunitinib treatment, necropsied areas are larger and GLUT1 expression changed accordingly
Impact of sunitinib administration on the metastatic dissemination in lungs
In set A, with a primary tumour growth duration of 30 days, comprising a 9-day sunitinib or DMSO administration, no lung metastasis was detected 60 days after tumour resection in the treated (n = 4) and control (n = 3) groups. In set B with a 39 days tumour growth duration, including 14 days of sunitinib or DMSO treatment, the incidence of lung metastasis was of 50 % in the treated (n = 4) and control (n = 4) groups (Fig. 6).
Fig. 6.
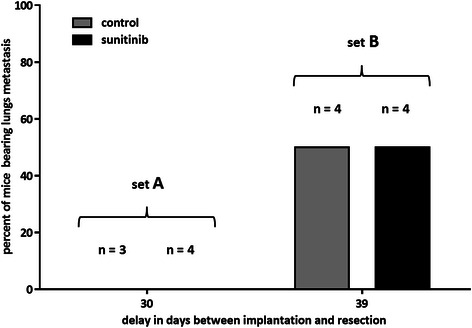
Lung metastasis incidence according to the primary tumour growth duration in control and treated groups. The percentage of lungs bearing metastasis is plotted against the delay between tumour implantation and resection. At 30 days, no lung metastasis is present in treated (n = 4) and control (n = 3) groups (set A). At 39 days, metastases are detected in half the lungs in both the 14 day-sunitinib treated group (n = 4) and the corresponding control group (n = 4) (set B)
Discussion
With an ER+/− PR+/− HER2+ status and a luminal transcriptomic signature, the PyMT model is considered to mimic human luminal B (HER2-positive) breast cancers [23–25]. The monitoring of mouse body weights during the sunitinib administration phases revealed no significant variation by comparison to control groups, which suggests that the drug, when administered at 40 mg/kg per os, had acceptable toxicity on this model. The 9-day sunitinib treatment induced a significant regression of the tumour volume and a 14 day-treatment duration further reduced the tumour volumes when compared to their respective controls. These results are consistent with those reported by Bousquet [13] and Abrams [15] regarding the luminal mammary cancer responsiveness to sunitinib.
The high efficiency of sunitinib on the MMTV-PyMT model by comparison to TNBC models might be partly explained by its dependency to particular pathways activated by the middle-T oncogene. Indeed, the middle-T antigen was shown to act through co-optation of several transduction pathways including: i) the protein phosphatase 2A (PP2A) activating the cytosolic tyrosine kinases (PTK) of the Src family (Src, Fyn, Yes), ii) the phospholinositide 3-kinase (PI3K) activating the Akt/mTOR cell-survival pathway, iii) the mitogen-activated protein kinases (MAPK/ERK) pathway through recruitment of the Shc adapter protein and iv) the phospholipase Cγ1 (PLC-γ) pathway inducing the protein kinase C (PKC) activation and a cytosolic Ca2+ concentration increase [26, 27]. As highlighted in Additional file 3, sunitinib is known to interact with the Src family cytosolic tyrosine kinases and with more than 10 tyrosine kinase membrane receptors that participate in the regulation of the MAPK/ERK and PI3 kinase pathways. As recently demonstrated on a medulloblastoma model, this drug might also repress these two signalling cascades through the induction of PTEN expression [28].
In addition to the decrease in tumour size, the vascular network evaluation demonstrated that sunitinib impacts vascular density and maturity. Moreover, the treated tumours were characterised by a higher level of apoptosis by comparison to controls. Previously, this multi-kinase inhibitor has already been shown to present in vitro and in vivo anti-angiogenic effects as well as direct pro-apoptotic properties [29, 30]. Regarding TAMs, their density was slightly reduced under sunitinib treatment versus control. As recently reviewed, TAMs roles are many and include the promotion of neo-angiogenesis, tumour immune evasion and metastatic behaviour [31]. To our knowledge our study is the first to evaluate the therapy response to sunitinib of primary tumours in a mammary cancer model using [18F]FDG or [18F]FMISO PET. The closest related work describes a [18F]FDG PET monitoring of the sunitinib response on lung metastases in a 4T1 intravenously induced metastatic model [32] and showed an increased [18F]FDG signal in the lungs of the sunitinib-treated mice compared to the control mice, which correlated with an enhanced seeding of lung metastases associated with sunitinib administration. In our [18F]FDG PET data, the mean SUV increased during the 5 day-tumour growth in the control group, whereas it remained stable in the treated tumours. We checked that the stable [18F]FDG uptake in treated tumours that were concomitantly decreasing in size was not due to partial volume effect (PVE) [33] and found that PVE alone could not explain our observations. The uptake mechanism of [18F]FDG has been previously studied emphasizing the role of the GLUT protein family [34]. In our work, we only measured GLUT1 expression and showed that sunitinib increased the presence of this transporter. The associated lack of increase in apparent [18F]FDG uptake in sunitinib-treated tumours might be at least partly explained by the lower levels in vascularisation, TAM infiltration and cell viability in sunitinib treated by comparison to control tumours. Yet, the overall conclusion is therefore that [18F]FDG PET evidenced the response to sunitinib treatment in this tumour model.
In our [18F]FMISO PET scans, randomization did not yield two perfectly equivalent groups regarding the hypoxia levels as expressed in SUV. Nevertheless, the untreated tumours remained stable in hypoxia over the treatment course, whereas the sunitinib administration tended to reduce hypoxia, although the difference was not significant in our small sample. Therefore, despite the reduction in blood supply, the treated tumours did not become more hypoxic than before the sunitinib administration. This might seem paradoxical as the sunitinib-induced anti-angiogenic effects are often associated with an increase in hypoxia due to the tumour starvation in nutrients. This enhanced hypoxia phenomenon has for instance been described by Welti et al. [32] and contributes to explain the sunitinib efficacy on the preclinical models. In our case, even in absence of enhanced hypoxia, we observed a huge increase of the apoptotic level in the treated tumours compared to the control ones. As explained above, the sunitinib is known to repress many cell survival pathways that are over-activated by the middle-T oncoprotein, and to present pro-apoptotic properties on tumour cells [30]. This supports the idea that the tumour cell apoptosis observed in our model might be mainly induced by the direct pro-apoptotic properties of sunitinib, owing to its multi-kinase inhibitor activity. Indeed, since more apoptosis occurred in the sunitinib-treated tumours compared to the control ones, the drug induced tumour regression, which finally could explain the absence of enhanced hypoxia even in a reduced angiogenesis context. The HIF1 alpha protein has a central role in the cellular adaptation process under a stressful hypoxic environment. Its regulation has been extensively reviewed [35]. Here the mild, but not statistically significant, decrease of tumour hypoxia observed in the sunitinib group was concomitant with a reduced level of HIF1 alpha expression in sunitinib tumours compared to control ones. [18F]FMISO PET therefore appeared useful to characterise the hypoxia level inside the tumours, and also to unveil the preferential way of action of the drug on this model.
Interestingly, under sunitinib treatment, apoptosis was highly increased by a factor of 17.7 whereas Ki-67-positive cell number decreased only by a factor of 1.8 when compared with the control group. Areas of proliferating cells were reduced but the Ki-67 marker was denser in the remaining living cell islets. Thus, we hypothesize that a resistance mechanism of a few cancer cells to sunitinib might act through an induction of their cell-division cycle. Moreover, two recent publications proved that one of the effects of this drug on TNBC xenograft models is to increase the cancer stem cells (CSCs) population [36] by generating intra-tumoral hypoxia [37]. Further investigations might tell whether with no increased hypoxic level, as observed in our model, the proliferating cell pool still displays a few typical CSCs markers. Those cells are indeed of major importance as they present enhanced epithelial-mesenchymal transition properties and thus high metastatic potential [38]. Their promotion under sunitinib treatment might at least partly explain its disappointing efficiency on several models of metastasis [32, 39]. In our work, the 9 day-treated set A of mice did not allow us to study the effect of the drug on the metastatic process. Comparing the 14 day-treated group against controls (set B) implanted for 39 days, the treatment did not appear to impact the incidence of lung metastases. Further molecular characterisation of this sunitinib-resistant cellular pool is required to specifically target them, for instance by combining sunitinib treatment with a c-Met inhibition strategy using crizotinib [40].
Conclusion
We showed that the luminal B (HER2-positive) type PyMT model was particularly sensitive to sunitinib compared to other preclinical breast cancer models, such as TNBC models that have been extensively used to study the effects of this drug. Our histology, [18F]FDG PET and [18F]FMISO PET imaging results suggest that in addition to its anti-angiogenic effects, the sunitinib efficacy on this model is mostly due to its direct pro-apoptotic properties.
Acknowledgements
This research was funded by the “Institut National du Cancer (INCA)” under grant agreement n° PL 051/RPT06018LLP.
Abbreviations
- [18F]FDG
2-deoxy-2-[18F]fluoro-D-glucose
- [18F]FMISO
[18F]fluoromisonidazole
- %ID/g
Percent of injected dose per gram
- 2D-OSEM
Two dimensional ordered-subset expectation maximization
- CSCs
Cancer stem cells
- DAB
3-3'–diamino-benzidine
- DAPI
4',6'-diamidino-2-phenylindole
- DMSO
Dimethyl sulfoxyde
- ER
Estrogen receptor
- GLUT1
Glucose transporter 1
- HER2
Human epidermal growth factor receptor 2
- HIF1 alpha
Hypoxia induced factor 1 alpha
- MMTV
Mouse mammary tumour virus
- NS
Not significant
- ORR
Overall response rate
- PET
Positron emission tomography
- PMID
Pubmed-indexed for MEDLINE
- PR
Progesterone receptor
- PVE
Partial volume effect
- PyMT
Polyoma virus middle T
- qRT-PCR
Quantitative real time polymerase chain reaction
- RNA
Ribonucleic acid
- ROIs
Regions of interest
- SD
Standard deviation
- SUV
Standardized uptake value
- TAMs
Tumour-associated macrophages
- TNBC
Triple negative breast cancer
- TSA
Tyramide signal amplification
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labelling
- TZM
Trastuzumab
Additional files
General diagram for the CellProfiler pipeline dedicated to image segmentation of GLUT1, HIF1 alpha, KI67 and F4/80 labelled tissue slides. This example displays the step-by-step image processing of a tumour tissue labelled for the F4/80 antigen. The modules used in the pipeline are noted in bold. The image names appear in italic by the image side.
CellProfiler pipeline for vessel segmentation and clustering in CD31 labelled tissue slides. The above example displays the step-by-step image processing of a tumour tissue labelled for the CD31 antigen. The modules used in the pipeline are noted in bold. The image names appear in italic by the image side.
List of the main known high affinity targets for sunitinib. Sunitinib interaction partners were determined using a semi-quantitative affinity chromatography method followed by LC/MS analysis. Data collected from Bairlein et al. [29].
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BThézé contributed to design the study, carried out the animal model set up, the therapy model monitoring, the histochemistry experiments, and the analyses led by digital microscopy and qRT-PCR. He performed the statistical analyses and drafted the manuscript. NB participated in the therapy model monitoring, and carried out the PET experiments and related image analyses. AB participated in model monitoring and PET experiments. SB participated in tissue sample preparation for histochemistry. BK carried out [18F]FMISO radiosynthesis and its quality control. IB participated to data analyses and interpretation, particularly those obtained by PET imaging, and helped to draft and to correct the manuscript. BTavitian contributed to design the study, provided funding and helped to draft the manuscript. RB contributed to design the study, participated in its coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Benoît Thézé, Phone: +33-169867706, Email: benoit.theze@cea.fr.
Nicholas Bernards, nicholas.bernards@googlemail.com.
Audrey Beynel, Email: audrey.beynel@gmail.com.
Stephan Bouet, Email: bouet.stephan@sfr.fr.
Bertrand Kuhnast, Email: bertrand.kuhnast@cea.fr.
Irène Buvat, Email: irene.buvat@cea.fr.
Bertrand Tavitian, Email: bertrand.tavitian@inserm.fr.
Raphaël Boisgard, Email: raphael.boisgard@cea.fr.
References
- 1.Robert F, Sandler A, Schiller JH, Liu G, Harper K, Verkh L, Huang X, Ilagan J, Tye L, Chao R, Traynor AM. Sunitinib in combination with docetaxel in patients with advanced solid tumors: a phase I dose-escalation study. Cancer Chemother Pharmacol. 2010;66:669–680. doi: 10.1007/s00280-009-1209-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wildiers H, Fontaine C, Vuylsteke P, Martens M, Canon JL, Wynendaele W, Focan C, Greve JD, Squifflet P, Paridaens R. Multicenter phase II randomized trial evaluating antiangiogenic therapy with sunitinib as consolidation after objective response to taxane chemotherapy in women with HER2-negative metastatic breast cancer. Breast Cancer Res Treat. 2010;123:463–469. doi: 10.1007/s10549-010-1066-x. [DOI] [PubMed] [Google Scholar]
- 3.Mayer EL, Dhakil S, Patel T, Sundaram S, Fabian C, Kozloff M, Qamar R, Volterra F, Parmar H, Samant M, Burstein HJ. SABRE-B: an evaluation of paclitaxel and bevacizumab with or without sunitinib as first-line treatment of metastatic breast cancer. Ann Oncol. 2010;21:2370–2376. doi: 10.1093/annonc/mdq260. [DOI] [PubMed] [Google Scholar]
- 4.Robert NJ, Saleh MN, Paul D, Generali D, Gressot L, Copur MS, Brufsky AM, Minton SE, Giguere JK, Smith JW, II, Richards PD, Gernhardt D, Huang X, Liau KF, Kern KA, Davis J. Sunitinib plus paclitaxel versus bevacizumab plus paclitaxel for first-line treatment of patients with advanced breast cancer: a phase iii, randomized, open-label trial. Clinical Breast Cancer. 2011;11:82–92. doi: 10.1016/j.clbc.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergh J, Bondarenko IM, Lichinitser MR, Liljegren A, Greil R, Voytko NL, Makhson AN, Cortes J, Lortholary A, Bischoff J, Chan A, Delaloge S, Huang X, Kern KA, Giorgetti C. First-line treatment of advanced breast cancer with sunitinib in combination with docetaxel versus docetaxel alone: results of a prospective, randomized phase iii study. JCO. 2012;30:921–929. doi: 10.1200/JCO.2011.35.7376. [DOI] [PubMed] [Google Scholar]
- 6.Barrios CH, Liu M-C, Lee SC, Vanlemmens L, Ferrero J-M, Tabei T, Pivot X, Iwata H, Aogi K, Lugo-Quintana R, Harbeck N, Brickman MJ, Zhang K, Kern KA, Martin M. Phase III randomized trial of sunitinib versus capecitabine in patients with previously treated HER2-negative advanced breast cancer. Breast Cancer Res Treat. 2010;121:121–131. doi: 10.1007/s10549-010-0788-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, Lehman M, Adams BJ, Bello CL, DePrimo SE, Baum CM, Miller KD. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. JCO. 2008;26:1810–1816. doi: 10.1200/JCO.2007.14.5375. [DOI] [PubMed] [Google Scholar]
- 8.Cardoso F, Canon J-L, Amadori D, Aldrighetti D, Machiels J-P, Bouko Y, Verkh L, Usari T, Kern KA, Giorgetti C, Dirix L. An exploratory study of sunitinib in combination with docetaxel and trastuzumab as first-line therapy for HER2-positive metastatic breast cancer. The Breast. 2012;21:716–723. doi: 10.1016/j.breast.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Bachelot T, Garcia-Saenz JA, Verma S, Gutierrez M, Pivot X, Kozloff MF, Prady C, Huang X, Khosravan R, Wang Z, Cesari R, Tassell V, Kern KA, Blay J-Y, Lluch A. Sunitinib in combination with trastuzumab for the treatment of advanced breast cancer: activity and safety results from a phase II study. BMC Cancer. 2014;14:166. doi: 10.1186/1471-2407-14-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parker JS, Mullins M, Cheang MCU, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5:5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eroles P, Bosch A, Alejandro Pérez-Fidalgo J, Lluch A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat Rev. 2012;38:698–707. doi: 10.1016/j.ctrv.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Bousquet G, Varna M, Ferreira I, Wang L, Mongiat-Artus P, Leboeuf C, de Bazelaire C, Faivre S, Bertheau P, Raymond E, Germain S, Janin A. Differential regulation of sunitinib targets predicts its tumor-type-specific effect on endothelial and/or tumor cell apoptosis. Cancer Chemother Pharmacol. 2013;72:1183–1193. doi: 10.1007/s00280-013-2300-0. [DOI] [PubMed] [Google Scholar]
- 14.Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13:215. doi: 10.1186/bcr2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abrams TJ, Murray LJ, Pesenti E, Holway VW, Colombo T, Lee LB, Cherrington JM, Pryer NK. Preclinical evaluation of the tyrosine kinase inhibitor SU11248 as a single agent and in combination with “standard of care” therapeutic agents for the treatment of breast cancer. Mol Cancer Ther. 2003;2:1011–1021. [PubMed] [Google Scholar]
- 16.Eades G. Breast cancer MicroRNAs: signaling networks and clinical applications. In: López-Camarillo C, Marchat LA, editors. MicroRNAs in Cancer. Boca Raton, FL: CRC Press; 2013. pp. 1–42. [Google Scholar]
- 17.Creighton CJ. The molecular profile of luminal B breast cancer. Biologics. 2012;6:289–297. doi: 10.2147/BTT.S29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheang MCU, Chia SK, Voduc D, Gao D, Leung S, Snider J, Watson M, Davies S, Bernard PS, Parker JS, Perou CM, Ellis MJ, Nielsen TO. Ki67 Index, HER2 Status, and Prognosis of Patients With Luminal B Breast Cancer. J Natl Cancer Inst. 2009;101:736–750. doi: 10.1093/jnci/djp082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varticovski L, Hollingshead MG, Robles AI, Wu X, Cherry J, Munroe DJ, Lukes L, Anver MR, Carter JP, Borgel SD, Stotler H, Bonomi CA, Nunez NP, Hursting SD, Qiao W, Deng CX, Green JE, Hunter KW, Merlino G, Steeg PS, Wakefield LM, Barrett JC. Accelerated preclinical testing using transplanted tumors from genetically engineered mouse breast cancer models. Clin Cancer Res. 2007;13:2168–2177. doi: 10.1158/1078-0432.CCR-06-0918. [DOI] [PubMed] [Google Scholar]
- 20.Jones TR, Kang IH, Wheeler DB, Lindquist RA, Papallo A, Sabatini DM, Golland P, Carpenter AE. Cell profiler analyst: data exploration and analysis software for complex image-based screens. BMC Bioinformatics. 2008;9:482. doi: 10.1186/1471-2105-9-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Meth. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cuevas BD, Winter-Vann AM, Johnson NL, Johnson GL. MEKK1 controls matrix degradation and tumor cell dissemination during metastasis of polyoma middle-T driven mammary cancer. Oncogene. 2006;25:4998–5010. doi: 10.1038/sj.onc.1209507. [DOI] [PubMed] [Google Scholar]
- 23.Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, Backlund MG, Yin Y, Khramtsov AI, Bastein R, Quackenbush J, Glazer RI, Brown PH, Green JE, Kopelovich L, Furth PA, Palazzo JP, Olopade OI, Bernard PS, Churchill GA, Van Dyke T, Perou CM. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakasone ES, Askautrud HA, Kees T, Park J-H, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan Y-X, Qiu J, Park J, Sinha P, Bissell MJ, Frengen E, Werb Z, Egeblad M. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fluck MM, Schaffhausen BS. Lessons in signaling and tumorigenesis from polyomavirus middle T antigen. Microbiol Mol Biol Rev. 2009;73:542–563. doi: 10.1128/MMBR.00009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiu TH, Chandramouli GVR, Hunter KW, Alkharouf NW, Green JE, Liu ET. Global expression profiling identifies signatures of tumor virulence in MMTV-PyMT-transgenic mice correlation to human disease. Cancer Res. 2004;64:5973–5981. doi: 10.1158/0008-5472.CAN-04-0242. [DOI] [PubMed] [Google Scholar]
- 27.Schaffhausen BS, Roberts TM. Lessons from polyoma middle T antigen on signaling and transformation: A DNA tumor virus contribution to the war on cancer. Virology. 2009;384:304–316. doi: 10.1016/j.virol.2008.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abouantoun TJ, Castellino RC, MacDonald TJ. Sunitinib induces PTEN expression and inhibits PDGFR signaling and migration of medulloblastoma cells. J Neuro Oncol. 2010;101:215–226. doi: 10.1007/s11060-010-0259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bairlein M. PhD thesis. Munich: Technical University of Munich, Department of Genetics; 2010. Characterization of the small molecule kinase inhibitor SU11248 (Sunitinib, SUTENT) in vitro and in vivo: towards response prediction in cancer therapy with kinase inhibitors. [Google Scholar]
- 30.Young E, Miele L, Tucker KB, Huang M, Wells J, Gu J-W. SU11248, a selective tyrosine kinases inhibitor suppresses breast tumor angiogenesis and growth via targeting both tumor vasculature and breast cancer cells. Cancer Biol Ther. 2010;10:703–711. doi: 10.4161/cbt.10.7.12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olopade O. The role of tumor-associated macrophages in breast cancer progression (Review) Int J Oncol. 2013;43:5–12. doi: 10.3892/ijo.2013.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welti J, Powles T, Foo S, Gourlaouen M, Preece N, Foster J, Frentzas S, Bird D, Sharpe K, van Weverwijk A, Robertson D, Soffe J, Erler J, Pili R, Springer C, Mather S, Reynolds A. Contrasting effects of sunitinib within in vivo models of metastasis. Angiogenesis. 2012;15:623–641. doi: 10.1007/s10456-012-9291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soret M, Bacharach SL, Buvat I. Partial-Volume Effect in PET Tumor Imaging. J Nucl Med. 2007;48:932–945. doi: 10.2967/jnumed.106.035774. [DOI] [PubMed] [Google Scholar]
- 34.Krzeslak A, Wojcik-Krowiranda K, Forma E, Jozwiak P, Romanowicz H, Bienkiewicz A, Brys M. Expression of GLUT1 and GLUT3 Glucose Transporters in Endometrial and Breast Cancers. Pathol Oncol Res. 2012;18:721–728. doi: 10.1007/s12253-012-9500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koh MY, Spivak-Kroizman TR, Powis G. HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci. 2008;33:526–534. doi: 10.1016/j.tibs.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Chinchar E, Makey KL, Gibson J, Chen F, Cole SA, Megason GC, Vijayakumar S, Miele L, Gu J-W. Sunitinib significantly suppresses the proliferation, migration, apoptosis resistance, tumor angiogenesis and growth of triple-negative breast cancers but increases breast cancer stem cells. Vasc Cell. 2014;6:12. doi: 10.1186/2045-824X-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Nat Acad Sci. 2012;109:2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur M-H, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D, Wicha MS. Breast Cancer Cell Lines Contain Functional Cancer Stem Cells with Metastatic Capacity and a Distinct Molecular Signature. Cancer Res. 2009;69:1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ebos JML, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated Metastasis after Short-Term Treatment with a Potent Inhibitor of Tumor Angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shojaei F, Simmons BH, Lee JH, Lappin PB, Christensen JG. HGF/c-Met pathway is one of the mediators of sunitinib-induced tumor cell type-dependent metastasis. Canc Lett. 2012;320:48–55. doi: 10.1016/j.canlet.2012.01.026. [DOI] [PubMed] [Google Scholar]