

Abstract
Glutamate transporters catalyze the concentrative uptake of glutamate from synapses and are essential for normal synaptic function. Despite extensive investigations of glutamate transporters, the mechanisms underlying substrate recognition, ion selectivity and the coupling of substrate and ion transport are not well understood. Deciphering these mechanisms requires the ability to precisely engineer the transporter. In this study, we describe the semisynthesis of GltPh, an archaeal homolog of glutamate transporters. Semisynthesis enables the precise engineering of GltPh through the incorporation of unnatural amino acids and peptide backbone modifications. In the semisynthesis, the GltPh polypeptide is initially assembled from a recombinantly expressed thioester peptide and a chemically synthesized peptide using the native chemical ligation reaction followed by in vitro folding to the native state. We have developed a robust procedure for the in vitro folding of GltPh. Biochemical characterization of the semisynthetic GltPh indicates that it is similar to the native transporter. We used semisynthesis to substitute Arg397, a highly conserved residue in the substrate binding site with the unnatural analog, citrulline. Our studies demonstrate that Arg397 is required for high affinity substrate binding and based on our results we propose that Arg397 is involved in a Na+- dependent remodeling of the substrate binding site required for high affinity Asp binding. We anticipate that the semisynthetic approach developed in this study will be extremely useful in investigating functional mechanisms in GltPh. Further, the approach developed in this study should also be applicable to other membrane transport proteins.
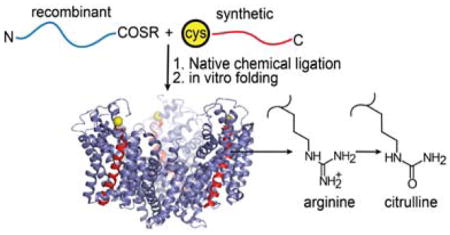
Glutamate is the major excitatory neurotransmitter in the central nervous system.1 Following release into the synaptic cleft during neurotransmission, glutamate is cleared by the actions of glutamate transporters that are also referred to as excitatory amino acid transporters or EAATs.2, 3 Glutamate transporters are present in plasma membranes of neuronal and glial cells and carry out the concentrative uptake of glutamate by coupling the transmembrane movement of glutamate to the co-transport of three Na+ ions, one H+, and the counter transport of one K+ ion.4, 5 Normal function of glutamate transporters is essential for maintaining the low extracellular concentration of glutamate that is important for efficient synaptic transmission and for preventing glutamate-induced neurotoxicity.1, 6
Glutamate transporters are members of the solute carrier 1 or SLC1 family of secondary solute transporters which also includes a large number of prokaryotic and archaeal amino acid transporters.1, 7 Structural information on glutamate transporters is available from studies on the archaeal homolog GltPh from Pyrococcus horikoshii (and the closely related GltTk from Thermococcus kodakarensis).8–12 GltPh is a Na+ coupled - aspartate transporter.9, 13 Structural analysis has revealed that GltPh is a homotrimer (Fig. 1A). Each subunit consists of eight transmembrane helices (TM) and two re-entrant hairpin loops (HP) that are arranged into two distinct domains: a central trimerization domain and a peripheral transport domain (Fig. 1A, B). The binding sites for Asp and the sodium ions are contained within the transport domain.9 The crystal structure shows an intricate network of interactions between the bound substrate and residues positioned at the tips of the hairpin loops, the highly conserved NMDGT sequence located in the unwound region of TM7, and polar residues on TM8 (Fig. 1C). Transport of Asp in GltPh is coupled to 3 Na+ ions 14, 15 and the binding sites for 2 of the 3 Na+ ions were visualized in the crystal structure (Fig. 1C).9 As observed in other secondary active transporters, the Na+ binding sites in GltPh show a heavy involvement of the backbone carbonyl oxygen atoms in coordinating the bound ions.9
Figure 1. Structure of GltPh and semisynthesis using native chemical ligation.
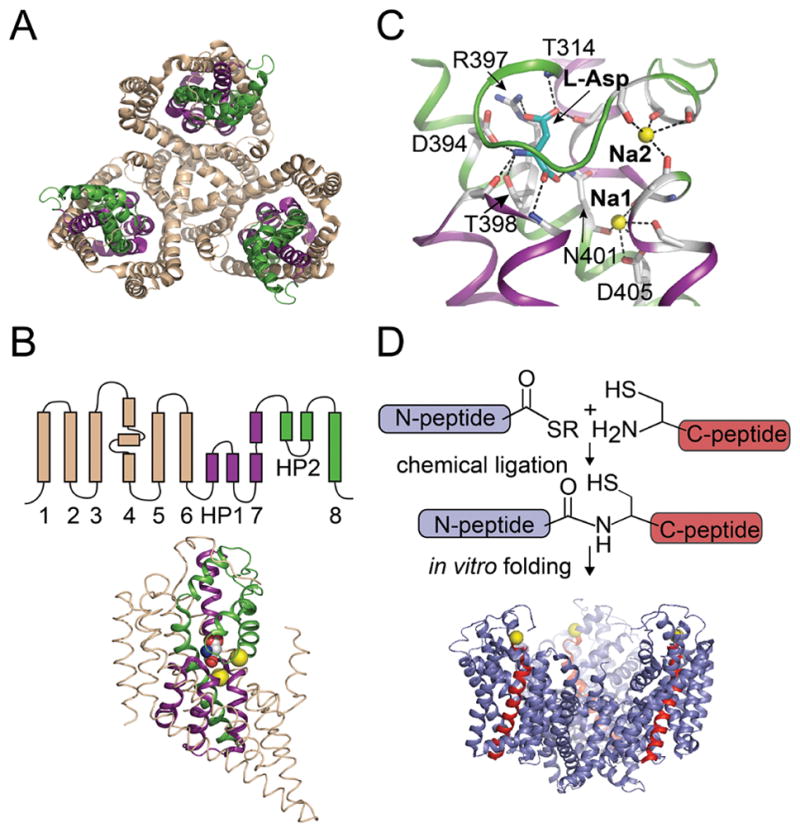
A) Top view of the trimeric GltPh transporter (pdb: 2nwx) shows the central trimerization domain (colored wheat) and the peripheral transport domain (colored green and purple). B) Topology and structure of a single subunit of GltPh. The regions of the subunit that contribute to the trimerization and the transport domain are colored as in panel A. Asp (space fill) and Na+ ions (yellow spheres) bound to the transport domain are shown. C) Close-up view of the Asp and Na+ binding sites. Asp is shown in stick representation and Na+ ions are shown as yellow spheres. Interactions between the bound ligands and GltPh are indicated by dashed lines. D) Semisynthesis of GltPh using native chemical ligation. The GltPh polypeptide is assembled by the ligation reaction of a recombinantly expressed thioester peptide (N-peptide: residues 1-384, blue) and a synthetic peptide with an N-terminal Cys (C-peptide: residues 385-418, red). The ligation product is folded in vitro to the native state. The Cys residue at the ligation site, M385C is represented as a yellow sphere.
The structural information presently available has set the stage for investigating the mechanisms underlying substrate recognition, ion selectivity, and the coupling of substrate and ion transport. These investigations require the ability to precisely manipulate the interactions observed in the crystal structures between the protein and the bound substrate and ions. Conventional site directed mutagenesis is of limited utility in this endeavor as it does not allow precise changes in the amino acid side chain or modifications of the protein backbone. Strategies for incorporating unnatural amino acids and protein backbone modifications will therefore be of great utility in these investigations.
Unnatural modifications of membrane proteins can be carried out by using nonsense suppression approaches.16 There are two approaches presently being used. The first approach, which has been popularized by the Dougherty group, uses a suppressor t-RNA chemically acylated with the unnatural amino acid in conjunction with expression in Xenopus oocytes.17 This approach is not suitable for GltPh as it requires protein expression in oocytes. The other approach, pioneered by the Schultz group, uses an orthogonal suppressor t-RNA and synthetase pair that has been evolved for the unnatural amino acid.18 This approach is presently limited in the types of unnatural amino acids that can be introduced. An alternate to these nonsense suppression approaches for incorporating unnatural modifications is to use chemical synthesis. Chemical synthesis is a very powerful method for protein modification as it enables the incorporation of a wide variety of unnatural amino acids and also allows modification of the protein backbone.19 A key advantage of chemical synthesis over the nonsense suppression approaches is that it is not dependent on the ability of the ribosome to incorporate the modification and therefore provides greater freedom in the variety of modifications that can be introduced. With the goal of using chemical synthesis to modify GltPh, we set out to develop the synthetic methodology required for GltPh.
Chemical synthesis of a protein consists of peptide synthesis followed by in vitro folding to the native state. Peptide synthesis is carried out using solid phase peptide synthesis (SPPS) and is only efficient for peptides ~50–60 amino acids in length.19 As the GltPh polypeptide is over 400 amino acids in length, we used a semisynthetic approach in which SPPS is used for the region of interest, such as the Asp binding site, while the rest of the protein is obtained using recombinant means.20 The synthetic peptide and the recombinant segment(s) are coupled using native chemical ligation (NCL) to form the GltPh polypeptide (Fig. 1D). In NCL, a peptide with a C-terminal thioester reacts with a peptide with an N-terminal Cys, linking the peptides with a native peptide bond at the ligation site.20, 21 A critical step in the synthesis is the in vitro folding process required to convert the synthetic polypeptide to the native state. GltPh presents a substantial challenge for in vitro folding as it is a multimeric, multidomain membrane protein.22
In the present study we describe the semisynthesis of GltPh. As required for the semisynthesis, we develop an efficient procedure for the in vitro folding of GltPh. We use semisynthesis to investigate the role of Arg397, a conserved Arg in the substrate binding site. Our results demonstrate that the Arg397 residue is required for high affinity substrate binding and based on our results we propose that Arg397 is involved in a Na+- dependent remodeling of the substrate binding site required for high affinity Asp binding.
EXPERIMENTAL PROCEDURES
Native expression and purification of GltPh
GltPh constructs used in this study carried an N-terminal His6 tag and a C-terminal Strep tag (WSHPQFEK). The GltPh constructs were sub-cloned into the pBCH/G4 vector (kindly provided by Dr. Eric Gouaux) and expressed in Escherichia coli TOP10 cells (Life Technologies). Protein expression was induced by 0.1% (w/v) arabinose at an optical density of 0.8 – 1.0 for 4 hours at 37 °C. Following expression, cells were pelleted, suspended in 20 mM HEPES-NaOH pH 7.5, 200 mM NaCl and membranes were prepared as described.23 Membranes were solubilized in dodecyl-β-D-maltopyranoside (DDM, 2% w/v) and the GltPh protein was purified using Ni-NTA resin (Qiagen), followed by size exclusion chromatography (SEC). SEC was carried out using a Superdex S200 column (GE Biosciences) using 20 mM HEPES-NaOH pH 7.5, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.5 mM glutamate and 0.1% (w/v) DDM as the column buffer.
Unfolding and refolding of the GltPh transporter
Unfolding of GltPh was carried out as described for the KvAP channel.23 Briefly, Triton X-100 (Tx-100) was added to the GltPh solution to a concentration of 2% (v/v) and the protein was precipitated by the addition of 15% trichloroacetic acid (TCA, w/v) and incubation at 4 °C for 30 min. The protein precipitate was collected by centrifugation, washed twice with acetone + 0.1 % TFA (trifluoroacetic acid) and then solubilized in 50% TFE (trifluoroethanol) + 0.1% TFA. The TFE solution was lyophilized to provide the unfolded GltPh polypeptide that was used for the refolding studies.
The lipid vesicles used for refolding GltPh were prepared from Soy Total Lipid Extract (Asolectin, Avanti Polar Lipids). For vesicle formation, lipids were dissolved in cyclohexane and lyophilized. The lyophilized lipids were hydrated for 1 hour at a concentration of 20 mg/mL in 20 mM HEPES-NaOH, pH 7.5, 200 mM NaCl, 10 mM DTT, 1 mM glutamate and then sonicated to yield the lipid vesicles.
For refolding, the unfolded polypeptide was dissolved in 20 mM HEPES-NaOH, pH 7.5, 200 mM NaCl, 1% SDS, 10 mM DTT, 1 mM glutamate and then diluted 10 - fold into the lipid vesicle solution and briefly sonicated. Refolding was allowed to proceed for 3 – 4 hours at room temperature and then dialyzed against 20 mM HEPES-NaOH, pH 7.5, 200 mM NaCl, 0.5 mM DTT, 1 mM glutamate. Regenerated cellulose membranes with a 6,000–8,000 molecular weight cut-off was used for dialysis. Refolded proteins were solubilized and purified as described for the native GltPh.
Crosslinking of GltPh
Assessment of the multimeric nature of GltPh was carried out by chemical crosslinking using 0.25% (w/v) glutaraldehyde for 10 min at room temperature. The crosslinking reaction was quenched by the addition of 100 mM Tris. The cross-linked products were separated by SDS-PAGE and visualized by staining with Coomassie Blue.
Aspartate binding assay
Binding assays were conducted on GltPh transporters with the L130W substitution as previously described.9 Briefly, purified GltPh was dialyzed (3X) against 200 volumes of the assay buffer (20 mM HEPES/Tris buffer, pH 7.5, containing 200 mM choline chloride, 1 mM NaCl, and 0.1% DDM) to ensure that the transporters were devoid of Asp. The binding assays were carried out using ~100 nM GltPh and binding of Asp was monitored by the change in Trp fluorescence with excitation at 295 nm and emission monitored at 334 nm. The changes in fluorescence were normalized to the initial fluorescence, and ligand binding curves were fit to the equation, to determine Kd for Asp. When the Kd for Asp was comparable to the protein concentration, the following equation: was used.9
Aspartate transport assays
The GltPh transporter was reconstituted into liposomes as previously described and the proteoliposomes obtained were snap-frozen in liquid N2 and stored at −80 °C.9, 13 Previously frozen proteoliposomes were thawed, and centrifuged at (265,000 g) for 70 min. Pelleted proteoliposomes were resuspended in 100K buffer (20 mM HEPES-KOH, pH 7.5, 100 mM KCl) at 5 mg/mL of lipid, subjected to two freeze/thaw cycles with liquid N2, and extruded through 400 nm filters. Extruded proteoliposome were centrifuged and resuspended in 100K buffer at 333 mg/mL of lipid. The uptake reaction was initiated by diluting the proteoliposomes 133-fold into the reaction buffer (20 mM HEPES-NaOH, pH 7.5, 200 mM NaCl, and 100 nM 14C-Asp at room temperature. For each time point, a 250 μl aliquot was removed and diluted 10 - fold into ice-cold quench buffer (20 mM HEPES - KOH, pH 7.5, 100 mM KCl) followed by filtration over nitrocellulose filters (0.22 μm, Millipore). Filters were washed twice with 2 mL of ice-cold quench buffer and assayed for radioactivity. Background levels of 14C-Asp uptake were determined in the absence of sodium (100K buffer on both sides). The inhibition experiments were performed by first incubating proteoliposomes in buffer (20 mM HEPES-NaOH, pH 7.5, 200mM NaCl) containing 10 μM TBOA (Tocris Bioscience) for 5 min, following addition of 100 nM 14C-Asp. Uptake data are fit to single exponentials for presentation.
Recombinant expression of GltPh (1-384) thioester
A sandwich fusion strategy was used for expression of the GltPh 1-384 (N-peptide) thioester.24 The fusion protein consisted of GltPh residues 1-384 sandwiched between Glutathione-S-transferase (GST) at the N-terminus and the gyrA intein-chitin binding domain at the C-terminus. A thrombin site, a His6 tag and a factor Xa site were present between GST and the GltPh sequence. Expression of the sandwich fusion in inclusion bodies was carried out in Escherichia coli Rosetta2 (DE3) cells (Merck) using the auto-induction protocol.23, 25 For isolation of the inclusion bodies, cells were pelleted and resuspended in 20 mM Tris-HCl pH 7.5, 0.2 M NaCl, 1 mM MgCl2, DNAse (5 μg/mL), lysozyme (0.1 mg/mL), and 1 mM phenylmethanesulfonyl fluoride. The cells were incubated at room temperature with gentle stirring for 30 min and then lysed by sonication. Tx-100 was added (1%) and the cell lysate was stirred at room temperature for 30 min. The soluble and insoluble fractions were separated by centrifugation at 12000g for 10 min. The insoluble fraction, which contains the inclusion bodies, was washed 2 X with 20 mM Tris-HCl pH 7.5, 200 mM NaCl, 1% Tx-100. The inclusion bodies were solubilized in 20 mM Tris-HCl pH 7.5, 200 mM NaCl, 1% N-Lauryl Sarcosine (NLS, w/v) and digested with thrombin (Roche,1U/L of culture) overnight, to cleave the GltPh-intein fusion from GST.
For purification of the GltPh-intein fusion, the thrombin cleavage mixture was diluted with an equal volume of 20 mM Tris-HCl pH 7.5, 200 mM NaCl. Tx-100 was added to 2% and the GltPh-intein fusion was purified using metal affinity chromatography (Talon, Clontech). Following purification, the GltPh-Intein fusion was dialyzed overnight against 20 mM Tris-HCl (pH 7.5), 200 mM NaCl, and 1% Tx-100 and then cleaved by the addition of 2-mercaptoethanesulfonic acid (MESNA, 0.15 M) to generate the GltPh thioester peptide. Typically ~50% cleavage of the intein fusion was observed following 1–2 days of incubation with MESNA at room temperature. The thiolysis mixture was precipitated using TCA and lyophilized similar to the procedure used for unfolding of GltPh.
Chemical synthesis of the GltPh C terminal peptides
The C-peptide, GltPh residues 385-418 with a C-terminal Strep tag (in italics):
CILGIDAILDMGRTMVNVTGDLTGTAIVAKTEGTGSGWSHPQFEK was synthesized on a PAM (phenylacetamidomethyl) resin using a slightly modified version of the in situ neutralization/2-(1H-benzo-triazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) activation protocol for Boc-solid phase peptide synthesis.26 The β-branched amino acids in the sequence were double-coupled using HBTU in DMSO to ensure complete coupling.27 Following chain assembly, global de-protection and cleavage from the solid phase was carried out using anhydrous hydrofluoric acid (HF). The crude material obtained after HF cleavage was solubilized in 50% buffer B (9: 1 acetonitrile: H2O + 0.1% TFA), purified by RP-HPLC on a preparative C4 column using a 30 – 70% gradient of buffer B and confirmed by electrospray-mass spectrometry [observed mass for wild-type peptide = 4691.6 ± 0.1 Da (SD), calculated = 4692.3 Da, Supplementary Fig. 1A, B]. To generate the R397Citrulline (Cit) mutant, a C-peptide with the Arg underlined substituted with Cit was synthesized. The R397Cit peptide was synthesized and purified as described for the wild type peptide (observed mass for the R397Cit peptide = 4693 ± 0.8 Da, calculated = 4693.3 Da, Supplementary Fig. 1C, D).
Assembly of the Semisynthetic GltPh transporters
The ligation reaction between the recombinant GltPh N- peptide thioester and the synthetic C peptide was carried out in 0.1 M phosphate buffer (pH 8.0), 1% dodecylphosphocholine (Fos-12, wt/vol). A ~5- to 10- fold molar excess of the C peptide was used to drive the ligation reaction to completion. The reaction was initiated by the addition of thiophenol (2%, vol/vol) and carried out at room temperature with gentle stirring. The reaction was monitored by SDS-PAGE and was typically complete after 12 – 24 hours. The ligation reaction was terminated by the addition of DTT (0.1 M, 30 min at room temperature), diluted 10 fold with 0.1 M phosphate buffer (pH 8.0), 300 mM NaCl, 0.1% DDM, and purified using Streptactin resin (IBA Lifesciences). The purified ligation product was TCA-precipitated, lyophilized, and folded in vitro as described above for the native GltPh.
RESULTS
In vitro folding of GltPh
A key requirement for our synthetic strategy is the ability to carry out in vitro folding of GltPh. As in vitro folding of GltPh has not been previously demonstrated, we initially investigated the feasibility of this step. For these studies, we used the GltPh polypeptide obtained by unfolding the native protein. To ensure extensive unfolding, GltPh was precipitated with TCA/acetone, dissolved in TFE/Buffer A [H2O + 0.1% TFA, (v/v)], lyophilized and then solubilized in 1% SDS. Unfolding of GltPh using this protocol was confirmed by glutaraldehyde cross-linking. Native GltPh is a trimer and glutaraldehyde cross-linking of the native transporter gives a protein band that migrates corresponding to a trimer on SDS-PAGE (Fig. 2A). Unfolding of GltPh results in a loss of the trimer and glutaraldehdye cross-linking of the unfolded protein only gives a protein band corresponding to the monomer on SDS-PAGE (Fig. 2A).
Figure 2. In vitro folding of GltPh.
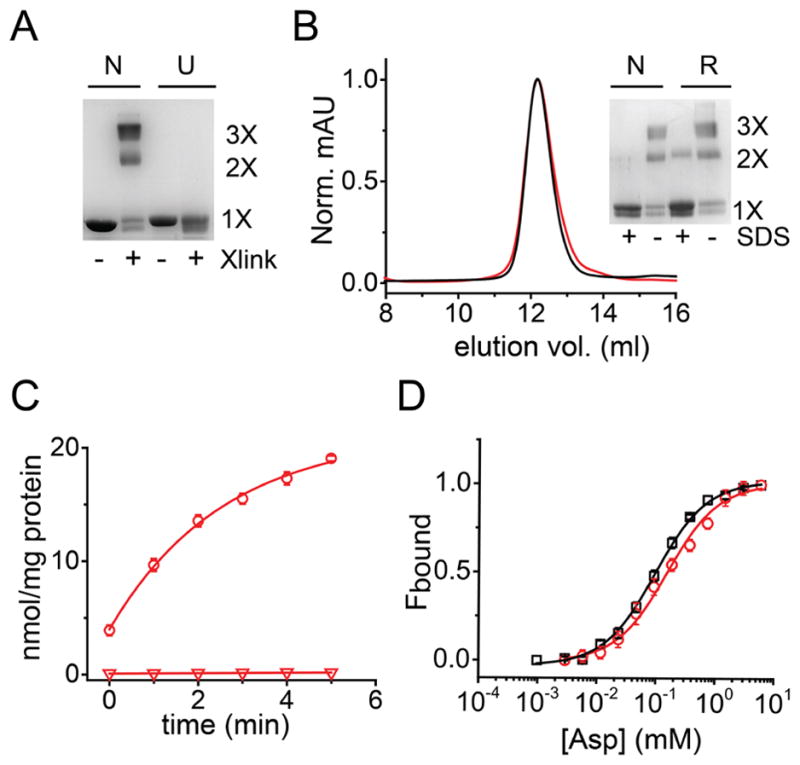
A) SDS PAGE gel showing the native (N) and unfolded (U) GltPh with (+) and without (−) glutaraldehyde cross-linking. Native GltPh cross-links to a trimer while the unfolded protein is monomeric. B) Size exclusion chromatography showing a similar elution profile for the native (black) and the refolded GltPh (red). Inset shows glutaraldehyde cross-linking of the peak fraction for native and refolded GltPh before (−) and after (+) treatment with 1 % SDS. In panels A and B, the oligomeric nature of the cross-linked band (1X, 2X, 3X) is indicated. C) Asp uptake by the refolded GltPh. Time course of 14C-Asp uptake into vesicles containing the refolded GltPh in the presence of a Na+ gradient (circles, n= 3). 14C-Asp uptake is not observed in the absence of a Na+ gradient (100 mM K+ on both sides of the membrane, triangles). D) Asp binding by native and refolded GltPh. The fraction of the protein bound (Fbound) was determined by dividing the fluorescence change upon addition of Asp to the total change at the end of the titration. Solid lines are fits to the data using the equation described in methods with a Kd value of 97 ± 4.8 μM (n = 7) for the native (black) and 154 ± 19 μM (n = 6) for refolded (red) GltPh. The binding assays were carried out in 1 mM Na+. Error bars correspond to SEM.
Guided by our previous success in using lipid vesicles for in vitro folding of ion channels,23, 28 we investigated whether a similar approach could be used for refolding GltPh. We used glutaraldehyde cross-linking to check for refolding. We observed that on dilution of the unfolded GltPh into lipid vesicles, glutaraldehyde cross-linking gave a protein band corresponding to a trimer suggesting refolding of GltPh. We purified the refolded GltPh by using metal affinity chromatography followed by size exclusion chromatography (SEC). The SEC elution profile for the refolded protein was similar to native GltPh and glutaraldehyde cross-linking confirmed the trimeric nature of refolded GltPh (Fig. 2B).
We used substrate binding and Asp transport assays to examine the functionality of refolded GltPh. We reconstituted the purified refolded GltPh into lipid vesicles for measurement of transport activity. We observed robust uptake of 14C- Asp in the presence of an inwardly directed Na+ gradient in proteoliposomes containing the refolded transporter (Fig. 2C). No uptake activity was observed in the absence of a Na+ gradient (100 mM K+ on both sides of the membrane). The specific activity of transport measured for the refolded protein was comparable to specific activity values reported for native GltPh.9, 13, 29, 30. Uptake of Asp in GltPh is blocked by TBOA (DL-threo-β-benzyloxyaspartic acid)9, 13 and we observed a similar extent of inhibition by TBOA for the native and the refolded GltPh (31 ± 10% for native compared to 40 ± 1% for refolded GltPh, n= 3). To investigate substrate binding, we used a fluorescence based assay that relies on a Trp substitution at position 130 (L130W) and reports on the coupled binding of Asp and Na+. 9 Using this assay, we measured similar Kd values for Asp binding to the native and refolded L130W GltPh (Fig. 2D). These similar biochemical and functional properties indicate that the refolded GltPh is similar to native transporter.
Semisynthesis of GltPh
In the semisynthesis of GltPh we focused on TM8, which contains residues implicated in both substrate and cation binding.8 We selected Met385, a residue upstream of TM8 as the ligation site because Asp transport assays indicated that the Cys substitution at this site, which is required for the ligation chemistry, is well tolerated. The semisynthesis therefore calls for the recombinant generation of a thioester polypeptide corresponding to residues 1-384 of GltPh (N-peptide) and a synthetic fragment corresponding to residues 385-418 with an N-terminal Cys (C-peptide) (Fig. 3A, see also Fig. 1D). To test the synthetic strategy, we initially assembled semisynthetic GltPh with the wild type sequence. The WT synthetic C-peptide was obtained by SPPS and purified using RP-HPLC in good yields (Supplementary Fig. 1A, B). We introduced a Strep tag at the C-terminus of the synthetic peptide for ease of purification of the ligation product.31 To generate the recombinant N-peptide thioester, we used the sandwich fusion approach that we have previously described.24 In this approach, the gyrA intein is fused to residues 1-384 of GltPh for introduction of the C-terminal thioester while GST is appended to N-terminus. The N-terminal GST directs expression of the fusion to inclusion bodies thereby avoiding cell lethality caused by expression of the gyrA intein fused to a transmembrane segment.27 Following expression, the inclusion bodies were isolated, the GST tag was removed by proteolysis and the N-peptide intein fusion was purified. The GltPh N-peptide thioester was then generated by thiolysis of the intein fusion (Fig. 3A, B).
Figure 3. Semisynthesis of GltPh.
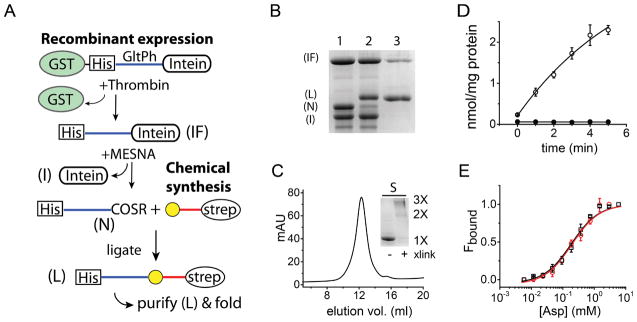
A) Strategy for the semisynthesis of GltPh. GltPh residues 1-384 are sandwiched between glutathione-S-transferase (GST) and the gyrA intein (I). A thrombin cleavage site and His6 tag are present between GST and the GltPh sequence. Proteolysis with thrombin releases the GltPh-intein fusion (IF), which is purified using the His6 tag and cleaved with MESNA to provide the N-peptide thioester (N). The N-peptide thioester is ligated to a synthetic C-peptide (residues 385- 418) with an N-terminal Cys (yellow sphere) and a C-terminal Strep tag. The ligation reaction yields the full length GltPh polypeptide (L), which is purified using the Strep tag and then folded in vitro to the native state. B) SDS-PAGE gel detailing the assembly and the purification of the semisynthetic GltPh polypeptide. Lane 1: Treatment of the GltPh-intein fusion (IF) with MESNA cleaves the N-peptide thioester (N) from the intein (I); Lane 2: NCL of the N-peptide thioester with the synthetic C-peptide yields the semisynthetic GltPh polypeptide (L); Lane 3: The semisynthetic GltPh polypeptide following purification using the C-terminal Strep tag. C) Size exclusion chromatography of semsiysnthetic GltPh. Inset: SDS-PAGE gel of the semisynthetic GltPh with (+) and without (−) glutaraldehdye cross-linking. D) Asp uptake by the semisynthetic GltPh. Time course of 14C-Asp uptake into vesicles containing the semisynthetic GltPh in the presence (open circles, n = 3) and in the absence of a Na+ gradient (filled circles, n = 3). E) Asp binding by semisynthetic GltPh. Asp binding assays as described in Fig. 2D for the semisynthetic (red circles, Kd = 229 ± 28 μM, n=3) and the native control, M385C-GltPh (black squares, Kd = 210 ± 21 μM, n=3).
Our attempts at purifying the thioester polypeptide using RP-HPLC were not successful likely due to the size and the hydrophobic nature of the polypeptide and so we carried out the ligation reaction with the C-peptide without purification. Prior to ligation, the thiolysis mixture was TCA/acetone precipitated and co-lyophilized with the C-peptide. The ligation reaction was carried out in the presence of dodecylphosphocholine (Fos-12) to keep the reactants soluble during the course of the reaction. We observed that the ligation reaction proceeded to ~90 % completion after a 12 – 24 hour incubation at room temperature (Fig. 3B).
Following ligation, the semisynthetic GltPh polypeptide (and the unreacted C-peptide) was separated from the unreacted N-peptide thioester and the un-cleaved N-peptide intein fusion by using the Strep tag present at the C-terminus (Fig. 3A, B). Following Strep tag purification, further separation of the GltPh polypeptide from the unreacted C-peptide can be carried out using the His6 tag present at the N-terminus of the ligation product. However, the C-peptide did not interfere with the folding of GltPh and so the folding reaction was carried out without further purification. In vitro folding of the semisynthetic GltPh was carried out using the lipid based protocol that we identified using the unfolded GltPh.
The semisynthetic GltPh transporter after in vitro folding was purified similarly to the refolded GltPh. The semisynthetic GltPh had a retention time on SEC similar to that of native GltPh, and glutaraldehyde cross-linking confirmed the trimeric nature of the semisynthetic transporter (Fig. 3C). We reconstituted the purified semisynthetic GltPh transporter into lipid vesicles for measurement of Asp uptake. Transport assays showed uptake of 14C - Asp in the presence of an inwardly directed Na+ gradient while no uptake activity was observed with 100 mM K+ on both sides of the membrane indicating the semisynthetic GltPh is functional (Fig. 3D). The specific activity of transport measured for semisynthetic GltPh was lower than native GltPh but was still within the range of values reported in the literature for the native transporter.9, 13, 29, 30 Further, Asp uptake observed for the semisynthetic GltPh was inhibited by TBOA to a similar extent as observed for native GltPh (42 ± 4% for semisynthetic compared to 31 ± 10% for native GltPh, n =3). We also assembled a semisynthetic GltPh with the L130W substitution to assay for Asp binding. These assays indicated that the Kd values for Asp binding to the semisynthetic GltPh was similar to the native control (M385C-GltPh, Fig. 3E). Taken together, these assays indicate the functional similarity of semisynthetic GltPh to the native GltPh. The limiting component in the semisynthesis of GltPh is the N-peptide thioester. Starting with the thioester obtained from 8 L of culture, we were able to obtain ~0.2 –0.3 mg of the purified semisynthetic GltPh.
Arg397 in the Asp binding site
The substrate binding site in GltPh contains an Arg residue, Arg397 that forms a salt bridge with the β–carboxyl group of the bound Asp (Fig. 4A). Arg397 is proposed to play a key role in determining amino acid selectivity as it is highly conserved in EAATs and the prokaryotic acidic amino acid transporters but is substituted with a Thr or Cys in the neutral amino acid transporters.9, 32 Further, substitution of the equivalent Arg in mammalian glutamate transporters with a neutral or a negatively charged amino acid abolished glutamate uptake but did not affect the interaction of neutral amino acids with the transporter.33–35 To further investigate the role of Arg397 in GltPh, we used semisynthesis to replace Arg397 with the unnatural amino acid, Cit. Cit is isosteric to Arg but lacks a positive charge on the side chain (Fig. 4B). The Arg to Cit substitution is therefore minimally perturbing to the substrate binding site and specifically tests the role of a positively charged side chain in substrate binding and transport. To substitute Arg397 with Cit, we synthesized a C-peptide with the R397Cit substitution and used this peptide to assemble a semisynthetic R397Cit GltPh (Supplementary Fig. 1C, D).
Figure 4. Arg397 in the substrate binding site of GltPh.

A) Close up view of the Asp binding site highlighting the interaction of Arg397 with the β-carboxyl group of the bound Asp. B) Structures of the side chains of Arg and the unnatural amino acid Citrulline (Cit). C) Time course for 14C-Asp uptake into vesicles containing the R397Cit GltPh in the presence 0.1 μM 14C-Asp (open diamonds, n = 2) and 1.0 μM 14C-Asp (filled diamonds, n = 3). For comparison, the time course for 14C-Asp uptake into vesicles containing the semisynthetic WT GltPh in the presence of 0.1 μM 14C-Asp (open circles) is also shown (data from Fig. 3D). For clarity, data presented has been corrected for background uptake determined in the absence of a Na+ gradient (100 mM K+ on both sides of the membrane). D and E) Asp binding to R397Cit (D) and the WT (E) at 1 mM (filled symbols) and 100 mM Na+ (open symbols) carried out as described in Fig. 2D. A shift in Na+ from 1 mM to 100 mM Na+ decreases the Kd for Asp binding to the native control (M385C-GltPh) from 210 ± 21 μM (n= 3) to 3.01 ± 0.6 nM (n= 4) while only a modest decrease [961 ± 102 μM (n = 3) to 204 ± 15 μM (n =4)] is observed for the R397Cit-GltPh. F) Logarithmic plots of Asp Kd (μM) values against log [Na+] (mM) are shown for the native control (black squares) and R397Cit GltPh (blue diamonds).
We purified the semisynthetic R397Cit GltPh and reconstituted it into lipid vesicles to measure Asp uptake. We did not observe Asp uptake in the Cit mutant under the standard assay conditions (0.1 μM Asp) or on using a 10-fold higher concentration of Asp (Fig. 4C). The lack of uptake at the Asp concentrations tested led us to investigate whether the compromised uptake in the Cit mutant was due to perturbed binding of Asp. We used semisynthesis to generate R397Cit GltPh with the L130W substitution for binding assays. We carried out Asp binding assays over a range of Na+ concentrations from 1 to 100 mM. Surprisingly, we observed that the effect of the Cit substitution on Asp binding was dependent on the Na+ concentration. At 1 mM Na+, the Kd for Asp binding to the Cit mutant (Kd = 961 ± 102 μM) was only slightly altered compared to the native control, M385C GltPh (Kd = 210 ± 21 μM, Fig. 4D, E). However at 100 mM Na+, there was a ~50000 fold difference in the Kd for the Cit mutant (Kd = 204 ± 15 μM) compared to M385C GltPh (Kd =3.01 ± 0.6 nM). The Asp binding assays at different Na+ concentrations show that the removal of the positive charge has a minimal effect on substrate binding at low Na+ but a substantial effect at high Na+. In wild type GltPh, the binding of Asp is coupled to the binding of Na+. A plot of the Log Kd for Asp against Log [Na+] yields a straight line with a slope of −2.48 indicating that the binding of Asp is coupled to the binding of at least 2 Na+ ions (Fig 4F).9 In the Cit mutant, the plot had a slope of −0.30 indicating a lack of coupling between Asp and Na+ binding in this mutant (Fig. 4F).
DISCUSSION
In this study we developed a semisynthesis for GltPh. GltPh, is to the best of our knowledge, the largest membrane protein to be obtained using synthetic or semisynthetic means. A critical step in the semisynthesis was the in vitro folding and we have developed a robust procedure using lipid vesicles for the in vitro folding of GltPh. The successful in vitro folding of GltPh demonstrates that integral membrane proteins with complex subunit topology can be folded in vitro.
The semisynthetic strategy that we developed is for TM8 but can be easily extended to other functionally important regions such as TM7 or the hairpin regions by selecting appropriate ligation sites. The yields of the semisynthesis presently are sufficient for functional/biochemical studies but are not yet sufficient for structural studies. The limiting step in the current strategy is the generation of the N-peptide thioester. We envision that by exploring various inteins, we will be able to identify an intein that is more efficient than the gyrA intein presently used, to improve the yields of the thioester peptide and thereby the yields of semisynthetic GltPh.
We used semisynthesis to replace Arg397 in the substrate binding site with Cit. Cit is isosteric to Arg but neutral and thereby provides us with a precise means to evaluate the role of the positively charged residue in the substrate binding site. Comparison of Asp binding to the wild type and the Cit mutant showed at low Na+ concentration, substituting the Arg side chain had only a small (4.5 fold) effect on Asp binding compared to a substantial (~50000 fold) effect at high Na+. This difference in the effect of substituting the Arg side chain (on Asp binding) with Na+ concentration indicates that R397 is critical for high affinity Na+ coupled Asp binding. The coupling between Na+ and Asp binding in GltPh has been proposed to involve conformational changes in the substrate binding site.12 As Na+ coupling to Asp binding is lost on substitution of Arg397, we speculate that the conformational change in the substrate binding site involves Arg397. A precedent for a conformational change in Arg397 on ion binding is observed on comparing the inward-facing structure of GltPh in the apo-state to the inward-facing structure in the Tl+-bound state (Fig. 5A, B).12 A similar repositioning of the equivalent Arg side chain is observed on comparing the outward facing apo structure of GltTk to the outward facing Na+ and Asp bound structure of GltPh.9, 10.
Figure 5. Ion-induced conformational change in substrate binding site of GltPh.
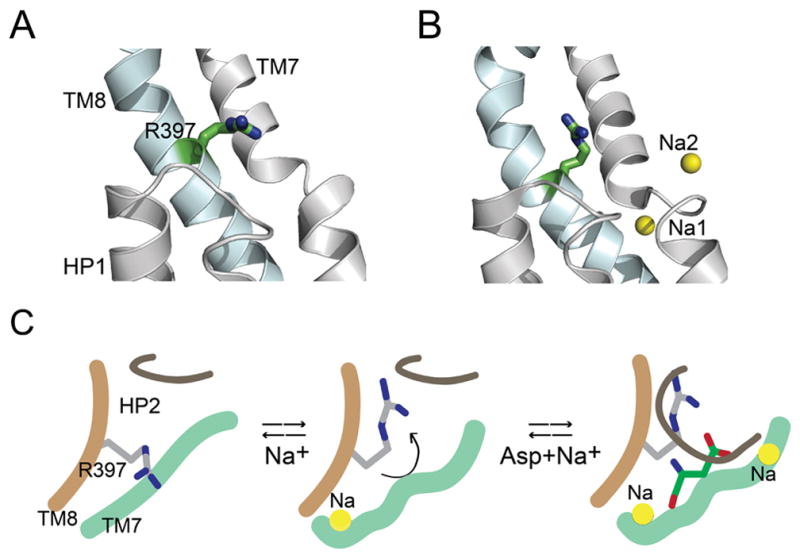
A and B) Crystal structures of the inward-facing conformation of GltPh in the apo-state (A, pdb: 4p19) and in the presence of Tl+ (B, pdb: 4p6h). Close-up view of the substrate binding site showing HP1 and TM7 (colored grey) and TM8 (blue). Arg397 is shown in stick representation. The cation-binding sites, Na1 and Na2 are represented as yellow spheres. C) Model depicting a proposed role for Arg397. Binding of a Na+ ion causes a conformational change in the substrate binding site that reorients Arg397 to create a high affinity Asp binding site. Binding of Asp is followed by the binding of another Na+ ion that is concomitant with the closure of HP2 leading to transport. Only the sodium sites visualized in the GltPh crystal structures 9 are shown in the model.
Studies on EAATs indicate an ordered binding process in which two Na+ ions bind prior to glutamate, followed by the binding of the third Na+ ion.2, 3, 36 Recent studies suggest a similar binding order for Na+ and Asp in GltPh.9, 12, 29 We propose that at low Na+, the Arg397 is positioned unable to interact with the β-carboxyl group of Asp while Na+ binding causes a conformational change in the substrate binding site resulting in a repositioning of the Arg side chain for optimal coordination of substrate (Fig. 5C).
In conclusion, we have developed a semisynthesis of GltPh. The semisynthesis enables the precise engineering of GltPh through the use of chemical synthesis for the incorporation of unnatural amino acids and modifications of the peptide backbone. We anticipate that these unnatural modifications introduced using semisynthesis will be extremely useful in investigating functional mechanisms in GltPh. Recent years have seen a rapid increase in the structural information available on transporters. We anticipate that the semisynthetic approaches that we developed for GltPh will be applicable to these membrane transport proteins and will permit a detailed analysis of the transport mechanisms in these transporters.
Supplementary Material
Acknowledgments
Funding Source Statement: This research was supported by grants to FIV from the NIH (GM087546) and a Pew Scholar Award. PJF was supported by a postdoctoral fellowship from the American Heart Association (12POST11910068).
We thank Dr. Scott Landfear and Dr. Marco Sanchez for assistance with transport assays. We thank Dr. H. Peter Larsson and Dr. Olga Boudker for helpful discussions.
Abbreviations used are
- TM
transmembrane helices
- HP
hairpin loops
- SPPS
solid phase peptide synthesis
- NCL
native chemical ligation
- DDM
dodecyl-β-D-maltopyranoside
- Tx-100
Triton X-100
- NLS
N-Lauryl Sarcosine
- MESNA
2-mercaptoethanesulfonic acid
- PAM
phenylacetamidomethyl
- HBTU
2-(1H-benzo-triazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HF
hydrofluoric acid
- Cit
Citrulline
- Fos-12
dodecylphosphocholine
- GST
Glutathione-S-transferase
- SEC
size exclusion chromatography
- TBOA
DL-threo-β-benzyloxyaspartic acid
Footnotes
Author Contributions
PF and FV designed experiments, PF and AA performed experiments, PF and FV analyzed data, and wrote the manuscript.
References
- 1.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 2.Jiang J, Amara SG. New views of glutamate transporter structure and function: advances and challenges. Neuropharmacology. 2010;60:172–181. doi: 10.1016/j.neuropharm.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vandenberg RJ, Ryan RM. Mechanisms of glutamate transport. Physiol Rev. 2013;93:1621–1657. doi: 10.1152/physrev.00007.2013. [DOI] [PubMed] [Google Scholar]
- 4.Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- 5.Levy LM, Warr O, Attwell D. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J Neurosci. 1998;18:9620–9628. doi: 10.1523/JNEUROSCI.18-23-09620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8:935–947. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- 7.Slotboom DJ, Konings WN, Lolkema JS. Structural features of the glutamate transporter family. Microbiol Mol Biol Rev. 1999;63:293–307. doi: 10.1128/mmbr.63.2.293-307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- 9.Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 10.Jensen S, Guskov A, Rempel S, Hanelt I, Slotboom DJ. Crystal structure of a substrate-free aspartate transporter. Nat Struct Mol Biol. 2013;20:1224–1226. doi: 10.1038/nsmb.2663. [DOI] [PubMed] [Google Scholar]
- 11.Reyes N, Ginter C, Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462:880–885. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verdon G, Oh S, Serio RN, Boudker O. Coupled ion binding and structural transitions along the transport cycle of glutamate transporters. Elife. 2014;3:e02283. doi: 10.7554/eLife.02283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan RM, Compton EL, Mindell JA. Functional characterization of a Na+-dependent aspartate transporter from Pyrococcus horikoshii. J Biol Chem. 2009;284:17540–17548. doi: 10.1074/jbc.M109.005926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groeneveld M, Slotboom DJ. Na(+):aspartate coupling stoichiometry in the glutamate transporter homologue Glt(Ph) Biochemistry. 2010;49:3511–3513. doi: 10.1021/bi100430s. [DOI] [PubMed] [Google Scholar]
- 15.Reyes N, Oh S, Boudker O. Binding thermodynamics of a glutamate transporter homolog. Nat Struct Mol Biol. 2013;20:634–640. doi: 10.1038/nsmb.2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pless SA, Ahern CA. Unnatural amino acids as probes of ligand-receptor interactions and their conformational consequences. Annu Rev Pharmacol Toxicol. 2013;53:211–229. doi: 10.1146/annurev-pharmtox-011112-140343. [DOI] [PubMed] [Google Scholar]
- 17.Dougherty DA, Van Arnam EB. In vivo incorporation of non-canonical amino acids by using the chemical aminoacylation strategy: a broadly applicable mechanistic tool. ChemBioChem. 2014;15:1710–1720. doi: 10.1002/cbic.201402080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 19.Kent SB. Chemical synthesis of peptides and proteins. Annu Rev Biochem. 1988;57:957–989. doi: 10.1146/annurev.bi.57.070188.004521. [DOI] [PubMed] [Google Scholar]
- 20.Muir TW. Semisynthesis of proteins by expressed protein ligation. Annu Rev Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 21.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 22.Harris NJ, Booth PJ. Folding and stability of membrane transport proteins in vitro. Biochim Biophys Acta. 2012;1818:1055–1066. doi: 10.1016/j.bbamem.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Devaraneni PK, Devereaux JJ, Valiyaveetil FI. In vitro folding of KvAP, a voltage-gated K+ channel. Biochemistry. 2011;50:10442–10450. doi: 10.1021/bi2012965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Komarov AG, Linn KM, Devereaux JJ, Valiyaveetil FI. Semisynthesis of K+ channels. Methods Enzymol. 2009;462:135–150. doi: 10.1016/S0076-6879(09)62007-3. [DOI] [PubMed] [Google Scholar]
- 25.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 26.Schnolzer M, Alewood P, Jones A, Alewood D, Kent SB. In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int J Pept Protein Res. 1992;40:180–193. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 27.Valiyaveetil FI, MacKinnon R, Muir TW. Semisynthesis and folding of the potassium channel KcsA. J Am Chem Soc. 2002;124:9113–9120. doi: 10.1021/ja0266722. [DOI] [PubMed] [Google Scholar]
- 28.Valiyaveetil FI, Zhou Y, MacKinnon R. Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry. 2002;41:10771–10777. doi: 10.1021/bi026215y. [DOI] [PubMed] [Google Scholar]
- 29.Bastug T, Heinzelmann G, Kuyucak S, Salim M, Vandenberg RJ, Ryan RM. Position of the third Na+ site in the aspartate transporter GltPh and the human glutamate transporter, EAAT1. PLoS One. 2012;7:e33058. doi: 10.1371/journal.pone.0033058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulligan C, Mindell JA. Mechanism of transport modulation by an extracellular loop in an archaeal Excitatory Amino Acid Transporter (EAAT) homolog. J Biol Chem. 2013 doi: 10.1074/jbc.M113.508408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt TG, Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat Protoc. 2007;2:1528–1535. doi: 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- 32.Scopelliti AJ, Ryan RM, Vandenberg RJ. Molecular determinants for functional differences between alanine-serine-cysteine transporter 1 and other glutamate transporter family members. J Biol Chem. 2013;288:8250–8257. doi: 10.1074/jbc.M112.441022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bendahan A, Armon A, Madani N, Kavanaugh MP, Kanner BI. Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J Biol Chem. 2000;275:37436–37442. doi: 10.1074/jbc.M006536200. [DOI] [PubMed] [Google Scholar]
- 34.Seal RP, Leighton BH, Amara SG. A model for the topology of excitatory amino acid transporters determined by the extracellular accessibility of substituted cysteines. Neuron. 2000;25:695–706. doi: 10.1016/s0896-6273(00)81071-5. [DOI] [PubMed] [Google Scholar]
- 35.Slotboom DJ, Konings WN, Lolkema JS. Cysteine-scanning mutagenesis reveals a highly amphipathic, pore-lining membrane-spanning helix in the glutamate transporter GltT. J Biol Chem. 2001;276:10775–10781. doi: 10.1074/jbc.M011064200. [DOI] [PubMed] [Google Scholar]
- 36.Grewer C, Gameiro A, Rauen T. SLC1 glutamate transporters. Pflugers Arch. 2014;466:3–24. doi: 10.1007/s00424-013-1397-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.