

Abstract
Transcription factors of the forkhead box, class O (FoxO) family are important regulators of the cellular stress response and promote the cellular antioxidant defense. On one hand, FoxOs stimulate the transcription of genes coding for antioxidant proteins located in different subcellular compartments, such as in mitochondria (i.e. superoxide dismutase-2, peroxiredoxins 3 and 5) and peroxisomes (catalase), as well as for antioxidant proteins found extracellularly in plasma (e.g., selenoprotein P and ceruloplasmin). On the other hand, reactive oxygen species (ROS) as well as other stressful stimuli that elicit the formation of ROS, may modulate FoxO activity at multiple levels, including posttranslational modifications of FoxOs (such as phosphorylation and acetylation), interaction with coregulators, alterations in FoxO subcellular localization, protein synthesis and stability. Moreover, transcriptional and posttranscriptional control of the expression of genes coding for FoxOs is sensitive to ROS. Here, we review these aspects of FoxO biology focusing on redox regulation of FoxO signaling, and with emphasis on the interplay between ROS and FoxOs under various physiological and pathophysiological conditions. Of particular interest are the dual role played by FoxOs in cancer development and their key role in whole body nutrient homeostasis, modulating metabolic adaptations and/or disturbances in response to low vs. high nutrient intake. Examples discussed here include calorie restriction and starvation as well as adipogenesis, obesity and type 2 diabetes.
Keywords: Forkhead box proteins, Oxidative stress, Stress signaling, Antioxidant proteins, DAF-16, C. elegans, Insulin signaling, Akt
Graphical abstract
Highlights
-
•
FoxO transcription factors are regulators of metabolism and antioxidant defense.
-
•
Stressful stimuli, including oxidative stress, modulate FoxO activity.
-
•
FoxO activities are regulated by posttranslational modifications.
-
•
FoxO levels are controlled transcriptionally and post-transcriptionally.
-
•
Redox dysregulation of FoxOs contributes to the development of metabolic diseases.
1. Introduction
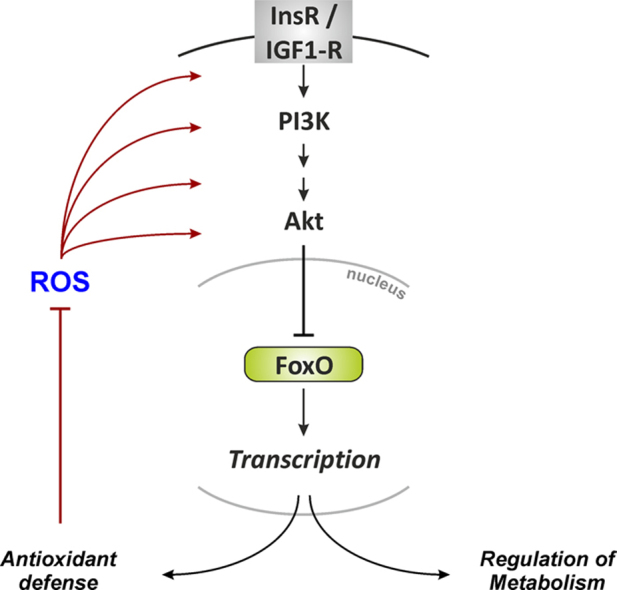
“Fork head” was first identified in Drosophila as a potential transcriptional regulator [1], and demonstrated to harbor a so-called winged-helix DNA binding domain that was then recognized to be present in other transcriptional regulators, including the mammalian hepatocyte-enriched nuclear factor (HNF)-3A (now FoxA1) [2]. This domain was christened the fork head domain [2], and later the dozens of proteins with such a winged helix/fork head domain identified by then were categorized into different classes of forkhead box (Fox) proteins [3]. Fox proteins – specifically, the forkhead box, class O, proteins (FoxO) – were first linked to stress resistance when long-lived mutants of Caenorhabditis elegans were analyzed with respect to genetic traits contributing to their longevity. It was found that insulin-like signaling along the cascade orthologous to the mammalian insulin receptor/phosphoinositide 3′-kinase/Akt (InsR/PI3K/Akt) cascade was involved in that mutants with impaired signaling along this cascade had extended life spans [4] (Fig. 1). It was then demonstrated that the daf-16 gene conferred this life span extension [5] and that DAF-16 (DAF, dauer formation) is a transcription factor of the Fox family (specifically, a FoxO orthologue) essential to this process [6,7].
Fig. 1.

Insulin signaling in mammalian cells and in C. elegans. See text for further details. Right panel: C. elegans transgenic strain TJ356 stably expresses a DAF-16::GFP fusion protein. DAF-16::GFP accumulates in nuclei upon exposure of worms to an oxidative stress (induced by diamide, a thiol oxidizing agent). Speckles (arrow) represent nuclei with DAF-16::GFP.
Mutants with deficient daf-2, coding for a C. elegans InsR orthologue [8], were then shown to not only display a long-lived phenotype but also a phenotype (“Oxr”) characterized by oxidative stress resistance: A daf-2-inactive mutant had an enhanced resistance towards redox cycling compounds such as paraquat or menadione [9]. Like the longevity phenotype, this Oxr phenotype was prevented by mutations in daf-16, suggesting that transcriptional targets of DAF-16 might be involved in conferring stress resistance. In fact, the expression of sod-3, the gene for one of the two manganese-containing superoxide dismutases of C. elegans (but neither sod-1 nor sod-2, coding for Cu, Zn-dependent SOD and a second Mn-dependent SOD, respectively [10]), was upregulated in long-lived daf-2 mutants, which was prevented by an additional daf-16 mutation [9].
These findings suggested that the expression of genes coding for antioxidant enzymes such as superoxide dismutases might be under the control of forkhead-type transcription factors. In fact, expression of the human Mn-SOD (mitochondrial SOD-2 in humans) was demonstrated to be transcriptionally controlled by the human DAF-16 orthologue, the forkhead box transcription factor, FoxO3a [11]. Despite the fact that it was later demonstrated that SOD-3 is not essential to the longevity phenotype in daf-2 mutants [12], a link was established between forkhead box transcription factors and cellular antioxidant defense.
The purpose of this review is to provide an overview on the role of FoxO transcription factors in the cellular response to (oxidative) stress – including antioxidant defense – and on the contribution of redox signaling to the biological activity of FoxOs.
Four FoxO proteins are present in humans (Fig. 2), FoxO1a, FoxO3a, FoxO4 and FoxO6 (for an overview on FoxO nomenclature, see [13]). All are widely expressed in diverse tissues [14] – including FoxO6, which has frequently been said to be primarily found in brain, but has now been shown to be ubiquitously expressed as well [15]. In terms of a functional classification of FoxO target genes, three major categories are “stress response and antioxidative defense”, “metabolism”, and “cell death and proliferation” [16].
Fig. 2.

Domain organization of human FoxO proteins. Positions of the most conserved domains and of some functionally characterized sequence motifs in human FoxO proteins are depicted. The numbers next to the domain or motif indicate its beginning and end within the sequence. Total length (in aa, amino acids) of each FOXO protein is indicated to the right of its schematic depiction. CR1 and CR3, conserved regions 1 and 3; CR3 represents a conserved C-terminal transactivation domain [326,327]. FH/DBD, forkhead box/DNA-binding domain [129,309,328]; NLS, nuclear localization signal; NES, nuclear export sequence. The amino acid sequence range of FoxO4 NLS is according to Obsilova et al. [329]. The corresponding homologous regions are depicted for FoxO1a, FoxO3a, and FoxO6 NLS. Whereas NES regions were defined for FoxO1a, 3a and 4 [330–332], the presence of a NES in FoxO6 is being debated [312,313]. The scheme and numbers depicted are based on the following NCBI RefSeq (National Center for Biotechnology Information Reference Sequence Database; [333]) entries: FoxO1a – NP_002006.2 (GI:9257222); FoxO3a – NP_001446.1 (GI:4503739); FoxO4 – NP_005929.2 (GI:103472003); FoxO6 – NP_001278210.2 (GI:849540648).
2. FoxO target genes: antioxidant defense
FoxO targets include genes coding for both intra- and extracellular antioxidant proteins interfering with all levels of oxygen reduction that would otherwise generate diverse ROS and cause oxidative damage to biomolecules. In humans, these FoxO-regulated antioxidants include Mn-SOD (SOD-2) [11], catalyzing disproportionation (dismutation) of the first oxygen reduction product, superoxide, to generate oxygen and hydrogen peroxide (H2O2). Moreover, there are indications of a regulation of cytoplasmic Cu,Zn-SOD (SOD-1) in murine erythroblasts by FoxOs, further supporting a role of these transcription factors in the cellular defense against superoxide [17]. H2O2 is further dismutated to water and oxygen in a reaction catalyzed by catalase, a peroxisomal heme peroxidase whose generation was found to be regulated by FoxO3a [18]. Alternatively, H2O2 may be reduced to water at the expense of reducing equivalents provided by NADPH via thioredoxin or glutathione (GSH), and the formation of two enzymes catalyzing this reduction was demonstrated to be regulated by FoxOs (peroxiredoxin-3, Prx3, and Prx5) [19,20] or to be likely regulated by FoxOs (glutathione peroxidase-1, GPx-1) [17], respectively. The interesting aspect here is that in addition to peroxisomal catalase, the mitochondrial Prx3 and Prx5, and the cytoplasmic GPx-1 appear to establish a FoxO-regulated battery of enzymes ascertaining that H2O2 may be reduced in multiple cellular compartments in parallel. Moreover, the expression of mitochondrial thioredoxin (Trx2) and mitochondrial thioredoxin reductase (TrxR2) were demonstrated to be regulated by FoxO3a in bovine aortic endothelial cells [20] and can be anticipated to contribute to the reduction of mitochondrial Prx3.
In the presence of redox-active metal ions, e.g. Fe2+ or Cu+, H2O2 may be reduced in a Fenton-type reaction to generate the hydroxyl radical •OH, a most aggressive oxidant. Chelating such metal ions would prevent hydroxyl radical formation and its initiating lipid peroxidation and oxidation of other biomolecules. In cells, chelation of copper ions is achieved by metallothioneins (MT), and one cellular iron sink is ferritin. Expression of C. elegans metallothionein-1 and a resulting resistance of the worms towards copper stress appears to be supported by DAF-16 [21], and expression of a ferritin ortholog, ftn-1, is regulated by DAF-16 [22]. Similarly, metallothionein mRNA levels were shown in mammalian cells to be increased upon stimulation of FoxO3a, particularly so following FoxO3a phosphorylation by AMP-activated kinase (AMPK) [23]. Interestingly, hepatic expression of the major plasma copper protein in mammals, ceruloplasmin (Cp), is controlled by FoxOs [24,25]. Cp harbors several copper ions per molecule and has antioxidant activity in that it acts as ferroxidase to oxidize Fe2+ released from cells to Fe3+. Not only does this prevent Fenton-type reactions but it also allows for transport of iron as Fe3+ by transferrin [26,27].
Another aspect of FoxO proteins regulating antioxidant networks was established when expression of the SEPP1 gene, coding for the major plasma selenoprotein, selenoprotein P (SelP), was found to be regulated by FoxO1a [28,29]. SelP has some hydroperoxidase activity per se [30,31], protecting LDL against oxidation [32]. However, its major physiological function is the transport of selenium from the liver to extrahepatic tissues via blood [33], providing selenium for the synthesis of cellular antioxidant selenoenzymes, such as glutathione peroxidases, including GPx-1 and GPx-4 [34], or thioredoxin reductases, and thus rendering cells more resistant against oxidative stress [35,36].
In summary, FoxO transcription factors regulate the expression of genes coding for intra- and extracellular antioxidant proteins, with different intracellular compartments as well as the major required steps in antioxidant defense covered (Fig. 3).
Fig. 3.

FoxO target genes coding for antioxidant proteins: subcellular localization and functional significance of gene products. See text for further details. Abbreviations: CP, ceruloplasmin; GPx, glutathione peroxidase; GSH, glutathione; GSSG, glutathione disulfide; LPO, lipid peroxidation; MT, metallothionein; Prx, peroxiredoxin; SelP, selenoprotein P; SOD, superoxide dismutase; Trx, thioredoxin; TrxR, thioredoxin reductase. Inset: color code to indicate subcellular localization of proteins. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Activity of FoxO transcriptional regulators is modulated at several levels, including (i) posttranslational modifications, (ii) subcellular localization, (iii) interaction with coregulators and (iv) FOXO gene expression and FoxO formation and stability. Subcellular localization and interaction with coregulators are governed by posttranslational modifications (PTM), some of which will be discussed in the following section.
3. Redox regulation of FoxO activity
FoxO PTMs that respond to changes in ROS levels and/or regulate FoxO antioxidant activity include phosphorylation, acetylation and ubiquitination. These modifications affect FoxO subcellular localization, activity as a transcriptional regulator, and stability. More recently, methylation and O-glycosylation were added to the list of FoxO modifications. For a compilation of FoxO posttranslational modifications and the respective FoxO sites of modification, see [16,37].
3.1. Endogenous and exogenous sources of ROS and their effect on FoxO phosphorylation
Stimulation of receptor tyrosine kinases (RTK) by their natural ligands frequently comes with a transient generation of ROS – this is true for stimulation of the insulin receptor with insulin [38], the epidermal growth factor (EGF) receptor with EGF [39,40] or the platelet-derived growth factor (PDGF) receptor with PDGF [41]. NADPH oxidases are the source of these ROS, whose formation results in modulation of downstream signaling. These signaling events occur through transient oxidative inhibition of protein tyrosine phosphatases (PTP) that are associated with the respective RTK. This appears to be required for a significant ligand-induced increase in RTK phosphorylation and therefore a significant intracellular signal to be initiated. For example, NOX4 generates H2O2, which was shown to attenuate dephosphorylation of EGFR [42]. Similarly, insulin-dependent signaling is modulated by NOX-derived reactive oxygen species. Not only does insulin stimulation of cells cause the generation of H2O2, but this peroxide formation is also essential to insulin signaling: by reversible oxidation of a redox-sensitive cysteine residue, H2O2 transiently inhibits PTP1B, a PTP that controls insulin receptor tyrosine phosphorylation [43–45]. A major H2O2 source in insulin-exposed adipocytes was then identified as NOX4, whose activity controls PTP1B activity and insulin receptor-dependent signaling [46].
NADPH oxidase complexes were originally identified as membrane-bound flavoenzymes responsible for the generation of superoxide in phagocytes upon stimulation. Five different isoforms of the catalytic subunit, NOX 1 through 5, have been identified, and NADPH oxidases are now known to be present in many non-phagocytes, to be activated by numerous stimuli, and to be crucial mediators in cellular signaling processes [47,48].
The exact mode of coupling the insulin receptor to NOX4 for an acute increase in ROS generation is unclear at present, particularly considering the current view of NOX4 as largely constitutively active (in contrast to NOX1 or 2) and regulated mainly at the level of expression [49]. However, a link between insulin exposure and a prolonged increase in generation of NOX4-derived ROS was established in 3T3-L1 fibroblasts [50]; insulin-induced signaling results in an enhanced expression of NOX4. Interestingly, insulin-induced NOX4 expression entailed the enhanced formation of MAPK-phosphatase-1 (MKP1) in that study, a dual-specificity phosphatase regulating MAPK phosphorylation that is also known as an immediate-early gene expressed under stress [51].
Stimulation of RTK-dependent signaling, including insulin signaling, may result in modulation of cascades that ultimately affect FoxO transcription factors. Two classical RTK-dependent signaling cascades result in activation of the Ser/Thr kinases Akt (protein kinase B) and the extracellular signal-regulated kinases (ERK) -1 and -2. Both are stimulated in cells exposed to various stressful stimuli, including ROS such as hydrogen peroxide, singlet oxygen or peroxynitrite [52–56]. Moreover, both modulate FoxO activities (see below). Further stress-responsive kinases include the other major mitogen-activated protein kinase (MAPK) family members in addition to ERK-1/ERK-2, p38MAPK isoforms and c-Jun-N-terminal kinases (JNKs). Again, p38MAPK and JNKs are stimulated by ROS and stimuli acting via the formation of ROS, such as ultraviolet radiation [57–59], singlet oxygen [60,61], peroxynitrite [55] or hydrogen peroxide [62]. Akt-induced FoxO phosphorylation (at three sites in FoxO1a, 3a and 4, at two in FoxO6), as elicited upon stimulation of cells with insulin, usually results in FoxO inactivation and nuclear exclusion, resulting in an attenuation of FoxO-dependent expression of genes like those coding for glucose 6-phosphatase or phosphoenolpyruvate carboxykinase [63, 64]. Similarly, FoxO phosphorylation by ERK-1/-2 causes modulation of its activity: ERK-dependent phosphorylation of FoxO3a triggers its poly-ubiquitination by murine double minute (MDM)-2, followed by FoxO3a degradation [65]. Moreover, Asada et al. identified phosphorylation of (murine) Foxo1a by ERK (and by p38MAPK) as regulating its transcriptional coregulator activity of Ets-1 transcription factor [66].
Mitochondria are another important endogenous source of ROS. Mitochondrial dysfunction can be the result of a metabolic imbalance, such as in the case of hyperglycemia, which is also linked to glycation of proteins and the formation of advanced glycation end products (AGEs), and fosters other oxidative processes causing elevated ROS formation, including NOX activation and uncoupling of eNOS [67]. Human aortic endothelial cells held under hyperglycemic conditions had elevated iNOS levels and activities, enhancing oxidation of LDL coincubated with the cells; FoxO1a was identified as the mediator, being upregulated by hyperglycemia, inducing iNOS and causing LDL oxidation [68]. High levels of H2O2 are also generated in peroxisomal fatty acid oxidation – e.g. by fatty acyl-CoA oxidase, the first enzyme of the classical peroxisomal fatty acid β-oxidation system – and in cytochrome P450-dependent xenobiotic metabolism [69–71].
Certain xenobiotics generate reactive oxygen species in cells by undergoing redox cycles, i.e. they are reduced by cellular enzymes at the expense of reducing equivalents such as NADH or NADPH, followed by reoxidation of the product by physically dissolved molecular oxygen, generating superoxide and reaction products thereof [72,73]. Certain quinones are examples of such redox cyclers and stimulate RTK signaling [74], in part through the generation of ROS [75], and can thus be expected to affect FoxO-dependent processes by modulating kinases known to phosphorylate FoxO proteins.
Doxorubicin is an anthraquinone derivative, DNA intercalating agent and topoisomerase inhibitor known to generate ROS (although probably not through redox cycling of the quinone moiety) in cells [76]. It is a known stimulator of ERK activation [77] and also causes p38 activation as well as p38-dependent FoxO3a phosphorylation (at Ser7), resulting in its nuclear accumulation and activation in MCF-7 breast cancer cells [78]. Doxorubicin treatment also appears to affect Foxo expression as it leads to an upregulation of Foxo1a and 3a mRNA levels in rat cardiac and skeletal muscle, but it is not known whether ROS formation is involved in this effect [79].
Redox-active metal ions can undergo redox cycling as well, and exposure to copper or iron ions will cause the generation of ROS in cells. Exposure of human hepatoma cells to copper ions strongly stimulated PI3K/Akt signaling [80] and FoxO phosphorylation and inactivation [81], which – despite formation of ROS – was independent of the generation of reactive oxygen species [82]. This explains why redox-inert metal ions like Zn2+ and Cd2+ also stimulate PI3K/Akt/FoxO signaling in a similar fashion, albeit less strongly so [82]. Different from these ions, Ni2+ did not stimulate a significant Akt-dependent FoxO phosphorylation [83]. Cu, Zn and Cd ions share an affinity for thiols, and a direct interaction with PTPase-type phosphatases such as PTEN was hypothesized as a potential mechanism of signaling initiation – which was indeed demonstrated to be the case for Zn [84].
Exposure of cells to arsenite – another molecule with high affinity towards thiols – results in oxidative damage to biomolecules [85,86], suggesting the generation of ROS. Exposure of rat pheochromocytoma cells to arsenite led to an induction of apoptosis via p38-dependent Foxo3a activation, followed by Foxo-dependent Bim-EL expression [87]. In human cells – HaCaT keratinocytes [88] and HepG2 hepatoma cells [89] – a stimulation of insulin-like signaling was observed, leading to an Akt-dependent phosphorylation of FoxOs and the inactivation of FoxO signaling.
Interestingly, even certain flavonoids, commonly categorized as antioxidants, may generate hydrogen peroxide through autoxidation in cell culture [90,91], which may cause stimulation of the PI3K/Akt cascade and phosphorylation of FoxO proteins, resulting in their inactivation and nuclear exclusion [92].
MAPK are proline (Pro)-directed kinases, and FoxO proteins contain several potential phosphorylation sites, i.e. a Ser or Thr, followed by a Pro residue. Therefore, it comes as no surprise that not only ERK, but also p38MAPK and JNK were shown to phosphorylate FoxO proteins. For example, (murine) Foxo1a – with 15 potential phosphorylation sites (14 of which are conserved in human FOXO1a) – is phosphorylated by ERK and p38MAPK, but not by JNK [66], whereas FoxO4 is phosphorylated by JNK under conditions of oxidative stress, stimulating FoxO4 nuclear accumulation [93].
Other Pro-directed kinases may be expected to phosphorylate FoxOs – such as cyclin-dependent kinases (CDKs), glycogen synthase kinase-3 (GSK3) or dual specificity tyrosine-regulated kinase (DYRK)-1a. In fact, CDK2 was shown to phosphorylate FoxO1a in response to DNA damage, inducing its nuclear exclusion and inactivation [94]. In line with a potential direct interaction with FoxO proteins, GSK-3 stimulates FoxO transcriptional activity to result in enhanced IGF1-receptor formation [95]. Nevertheless, no GSK-3-dependent FoxO phosphorylation in vivo has yet been reported. DYRK-1a is a Pro-directed kinase [96,97] that phosphorylates Ser329 in FoxO1a, which appears to slightly support nuclear exclusion and to moderately affect FoxO activity [98]. The flavonoid, epigallocatechin gallate (EGCG), while generating H2O2 in cell culture at high concentrations, is a potent inhibitor of DYRK-1a [99]. This explains why very low EGCG concentrations under conditions that do not generate any detectable H2O2 in the experimental setup stimulate nuclear accumulation of FoxO1a rather than a H2O2-induced nuclear exclusion [92]. Similar effects were seen with C. elegans: EGCG induced the nuclear accumulation of DAF-16 and enhanced expression of SOD-3 [92].
In summary, several kinases phosphorylate FoxO proteins in response to elevated levels of ROS and upon exposure of cells to stressful stimuli. Table 1 provides a summary of known FoxO phosphorylation events. Akt, ERK, p38MAPK and JNK are among the stress-responsive kinases known to target FoxOs, and to contribute to the modulation of FoxO activity and subcellular localization: while Akt usually inactivates FoxOs and causes their nuclear exclusion, JNK may phosphorylate and activate FoxO4, stimulating its nuclear accumulation (see Fig. 4 for an overview on FoxO phosphorylation events and their consequences). However, other PTMs may fine-tune the consequences of FoxO phosphorylation. Thus, the following section will focus on FoxO acetylation and ubiquitination in the cellular response to oxidative stimuli.
Table 1.
Phosphorylation sites in FoxO proteinsa.
| Kinasesb | Sites phosphorylated |
Comment | ||||
| Group [304,305] | FoxO1a | FoxO3a | FoxO4 | FoxO6 | ||
| AGC [306] | Akt | T24, S256, S319 [307,308] | T32, S253, S315 [309] | T28, S193, S258 [310,311]c | T26, S184 [312,313] | Interaction with 14-3-3 ↑; FoxO1a/3a/4: inactivation, nuclear exclusion; FoxO6: inactivation |
| SGK | T32, (S253), S315 [314] | See Akt | ||||
| PKA | T24, S256, S319 [315] | See Akt | ||||
| CMGC [316] | JNK | Not defined, but likely phosphorylated [317]; (in vitro only: S294, S425 [78]) | T447, T451 [93]c | FoxO3a: inactivation, nuclear exclusion [317] | ||
| FoxO4: activation, nuclear accumulation [93] | ||||||
| ERK | S246, S284, S295, S326 (analogous to human S329), S413, S415, S429, S467, S475 (numbers for murine FoxO1) [66] | S294, S344, S425 [65] | FoxO1a: enhanced interaction with other transcription factors suggested [66] | |||
| FoxO3a: inactivation, nuclear exclusion, Mdm2-mediated degradation↑ [65] | ||||||
| p38MAPK | S284, S295, S326, S467, S475 (numbers for murine FoxO1) [66] | S7 [78]; (in vitro only: S12, S294, S344, S425 [78]) | FoxO1a: enhanced interaction with other transcription factors hypothesized (in analogy to ERK) [66] | |||
| FoxO3a: nuclear accumulation [78] | ||||||
| CDK1 | S249 [318] | FoxO1a: Interaction with 14-3-3↓; | ||||
| activation, nuclear accumulation [318] | ||||||
| CDK2 | S249, (S298) [94] | FoxO1a: inactivation, nuclear exclusion [94]; S249 phosphorylation verified, but no nuclear exclusion in some cells [318]. | ||||
| DYRK1 | S329 [98] | FoxO1a: inactivation, nuclear exclusion [98] | ||||
| GSK3 | S325 (only in vitro) [319] | |||||
| NLK | S329 (plus up to 7 other Ser-Pro) [320] | FoxO1a: inactivation, nuclear exclusion [320] | ||||
| CK1 [321] | CK1 | S322, S325 [319] | FoxO1a: phospho-S319 (Akt/SGK) generates recognition motif for CK1 to phosphorylate S322; thereafter, S325 is phosphorylated [319]. Inactivation, nuclear exclusion. | |||
| CAMK | AMPK | T179, S399, S413, S555, S588, S626 [23] | FoxO3a: activation; no effect on subcellular localization [23] | |||
| MK5 | S215 (murine FoxO1a; analogous to S218 in hFoxO1a) [322] | S215 (S253, S551, S555) [323] | FoxO1a: activation [322] | |||
| FoxO3a: nuclear accumulation and activation [323] | ||||||
| STE | MST1 | S212 [324] | S207 (S213, S229/230, S241) [324]d | FoxO3a: interaction with 14-3-3↓; nuclear accumulation and activation [324] | ||
| Other | IKKβ | S644 [325] | FoxO3a: inactivation, nuclear exclusion, degradation↑ [325] | |||
| PERK | S298, (S301, S303) [244] | Not defined, but likely phosphorylated [244] | FoxO1a: nuclear accumulation and activation [244] | |||
Numbers refer to human FoxO proteins, unless noted otherwise (e.g., ERK, MK5).
Abbreviations: AGC – kinase group incorporating, among others, the protein kinase A, protein kinase G, protein kinase C families; AMPK – AMP-activated kinase; CAMK – kinase group incorporating calcium and calmodulin-regulated kinases and related families; CDK – cyclin-dependent kinase; CK – casein kinase; CMGC – kinase group named after some of its members, such as the CDK, MAPK, GSK3, CDK-like kinase families; DYRK – dual specificity, tyrosine phosphorylation-regulated kinase; ERK – extracellular signal-regulated kinase; GSK – glycogen synthase kinase; IKK – inhibitor of κB kinase; JNK – cJun N-terminal kinase; MAPK – mitogen-activated protein kinase; MK5 – MAPK-activated protein kinase (MAPKAPK) 5; MST – mammalian sterile 20-like; NLK – nemo-like kinase; PERK – protein kinase R-like endoplasmic reticulum kinase; PKA – protein kinase A; SGK – serum/glucocorticoid-regulated kinase; STE – kinase group incorporating several yeast sterile kinase-like kinases.
FoxO4 phosphorylation sites: the corresponding positions in the current version of the human FoxO4 sequence are T32, S197, S262, T451, and T455 [NCBI RefSeq accession number NP_005929.2 (GI:103472003)].
FoxO3a phosphorylatiuon sites listed in Ref. [324]: the corresponding positions in the current version of the human FoxO3a amino acid sequence are S209, S215, S231/232, and S243 [NCBI RefSeq accession number of the sequence: NP_001446.1 (GI:4503739)].
Fig. 4.

FoxO phosphorylation and its biological consequences. Schematic representation of FoxO phosphorylation by different kinases and the consequences with respect to activity and subcellular localization. (A) ERK-catalyzed phosphorylation of FoxOs may cause nuclear exclusion and murine double-minute (Mdm)-2-dependent proteasomal degradation [65]. Similarly, Akt (B) catalyzed FoxO phosphorylation will cause FoxO inactivation, nuclear exclusion and may trigger FoxO degradation (not shown). Interestingly, FoxO1a phosphorylation at S319 was shown to prime for a consecutive phosphorylation by casein kinase 1 (CK1), which further enhances nuclear exclusion [319]. FoxO phosphorylation may also result in nuclear accumulation and activation: (C) ER-stress may cause PERK-dependent FoxO phosphorylation and activation [244]. (D) c-Jun-N-terminal kinase (JNK)-dependent FoxO phosphorylation was described as activating (FoxO4 [93]) or inactivating (FoxO3a; dashed lines [317]). Phosphorylation is indicated by a black “P” on yellow background and stands for phosphorylations at multiple different sites (e.g. Akt: T24, S256, S319 for human FoxO1a). See Table 1 for further explanations. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. ROS control of FoxO activity by lysine modification: acetylation and ubiquitination
The ε-amino group of lysine (Lys, K) residues in proteins can be modified post-translationally through acetylation and mono- or poly-ubiquitination. Calnan and Brunet [16] listed six known acetylation sites each in human FoxO1a (K245, K248, K262, K274, K294, K559) and in human FoxO3a (K242, K245, K259, K271, K290, K569) as well as five acetylation sites each in human FoxO4 (K186, K189, K215, K237, K407) and in human FoxO6 (K173, K176, K190, K202, K229). Another previous compilation specified FoxO1a (K265) and FoxO3a (K203) as additional acetylation sites [100]. Reversible lysine acetylation is accomplished by the action of histone acetyltransferases and deacetylases: CBP (CREB-binding protein), p300 and p300/CBP-associated factor (PCAF) acetylate FoxOs, using acetyl-CoA as co-substrate, whereas enzymes of the sirtuin (Sirt, orthologues of silent information regulator) family catalyze NAD+-dependent deacetylation of FoxOs [101–107]. Most of the acetylation sites in human FoxO proteins surround a consensus site for Akt-induced serine phosphorylation (S256 in the case of human FoxO1a) within the nuclear localization signal (NLS) motif, a region that is highly conserved among the FoxO isoforms [100] (Fig. 2). This conserved localization of major acetylation sites in the FoxO proteins implies functional links between their acetylation state and metabolic regulation of reversible phosphorylation, cytoplasmic/nuclear shuttling and transcriptional activity of FoxOs, which indeed have been identified and turned out to be very complex and in part controversial. Acetylation has been shown to result in both stimulation and inhibition of the transcriptional activity of FoxOs, depending on the examined FoxO isoforms and their binding partners such as other transcription factors and transcriptional co-activators, the FoxO target genes and the cell types used in the studies [103,104,106,108,109]. The molecular mechanisms underlying those discrepancies are still not completely understood. The enzymes in charge of FoxO acetylation and deacetylation also alter the acetylation state of histones and of the FoxO coactivator PGC-1α (peroxisome proliferator-activated receptor γ-coactivator-1α) [110], which may modify the effect of a stimulus on FoxO-induced gene transcription.
The histone acetyltransferase (HAT) and transcriptional coactivator p300 was initially identified as a coactivator of FoxO proteins [111]. Early reports stressed the fact that p300-dependent acetylation of FoxO proteins resulted in enhanced FoxO transcriptional activity [105,112], and the interaction between the two proteins facilitated the recruitment of p300 to the promoter regions of target genes, where p300 further enhanced gene transcription by recruiting the basal transcriptional machinery and by facilitating chromatin remodeling through its intrinsic HAT activity [113]. It was also proposed that p300-mediated acetylation of FoxOs increase their transcriptional activity. However, it was soon noted that, at least in some contexts, p300-induced acetylation could also suppress their transcriptional activity [114]. This observation was quickly followed by the identification of the histone deacetylase Sirt as a positive regulator of FoxO activity [102].
In addition to acetylation, the above-mentioned lysine residues in FoxO proteins can also become ubiquitinated. Mono-ubiquitination has been shown to result in nuclear translocation and increased transcriptional activity of FoxO4 [115]. As the same lysine residues are shared for acetylation and ubiquitination, deacetylation may facilitate FoxO ubiquitination: deacetylation by Sirt1 or Sirt2 promoted FoxO3a poly-ubiquitination mediated by increased binding of the E3 ubiquitin ligase subunit Skp2 and subsequent FoxO3a proteasomal degradation [116]. As mentioned above for FoxO3a [65], the phosphorylation state of FoxOs may also affect their susceptibility to poly-ubiquitination and degradation: FoxO1a has been shown to be ubiquitinated by Skp2, and prior Akt-mediated phosphorylation at serine residue S256 facilitated FoxO1a poly-ubiquitination [117].
Oxidative stress mediated by increases in intracellular levels of ROS, specifically H2O2, has been identified as a key mediator of the acetylation and ubiquitination state of FoxOs [101,104,109,115,118]. Besides H2O2, the redox-cycling oxidant menadione induced transient FoxO3a acetylation, whereas UV and γ-irradiation had no effect [101]. Importantly, though, application of moderately high doses of exogenous H2O2, typically 25–500 µM, was required to trigger interaction of acetyl transferases with FoxOs [101,109]. These doses are considerably higher than the (physiological) levels of 0.1–7 µM H2O2 that stimulate cellular proliferation and mimic insulin-induced phosphorylation of FoxOs [119]. In contrast, application of exogenous H2O2 in higher micromolar concentrations initially induces growth arrest and possibly a subsequent cellular adaptation to oxidative stress [119]. The molecular mechanism of H2O2-mediated FoxO acetylation has been elucidated for FoxO4: Exogenously added H2O2 at a minimum concentration of 25 µM triggered the formation of heterodimers between p300/CBP acetylases and FoxO4 through intermolecular disulfide bridges, linking redox-sensitive cysteine residues [109]. Moreover, an increase in endogenous cellular ROS production resulting from glucose deprivation of the culture medium was also sufficient to induce hetero-dimerization of p300/CBP and FoxO4, whereas the antioxidant N-acetyl cysteine (NAC) counteracted this redox-sensitive interaction between the proteins [109]. Turnover of the p300/CBP-FoxO4 complex was regulated by the disulfide-reducing activity of thioredoxin-1 [109], a small antioxidant protein localized both in cytosol and nucleus [120]. The p300/CBP-FoxO4 interaction is primarily mediated through cysteine residue C477 in human FoxO4, and intriguingly, this cysteine residue is conserved among all human and murine FoxO isoforms [109]. This suggests the possibility that redox-sensitive hetero-dimerization with acetylases might represent the general mechanism of H2O2-induced FoxO acetylation. However, a recent screening study identified several proteins including peroxiredoxins and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) that bound after H2O2 treatment to the homologous cysteine residue in human FoxO3a (C622), but acetylases were not included in the provided list. Also, the other redox-sensitive cysteine residues in human FoxO3a did not interact with acetyltransferases [121].
Under conditions of oxidative stress, acetylated FoxO3a has been shown to translocate from the cytosol into the nucleus, where it may interact with the nuclear sirtuin Sirt1 to become deacetylated [101]. Sirt1 activity is regulated by the NAD+/NADH ratio and acts as a sensor of the cellular redox status that becomes activated when reducing equivalents are limiting [122]. Likewise, FoxO1a and FoxO4 are deacetylated by Sirt1, causing their nuclear trapping and modifying their transcriptional activity [102–104]. In most cases, transcriptional activity of FoxOs is elevated upon deacetylation. As net result of the successive acetylation and deacetylation events, deacetylated FoxOs become enriched in the nucleus and increase transcription of cell cycle arrest genes such as CDKN1B (coding for cyclin-dependent kinase inhibitor p27Kip1), DNA repair genes such as GADD45 (growth arrest and DNA damage-inducible protein 45) and genes coding for antioxidant enzymes such as SOD-2, whereas transcription of apoptotic genes is decreased [101, 107, 118]. This switch may allow cells to survive oxidative and metabolic stresses, as it has been demonstrated for FoxO3a in the landmark study by Brunet et al. [101]. This model has since been confirmed for FoxO1a and FoxO4: both successive acetylation/deacetylation of FoxO1a and FoxO4 induced the expression of CDKN1B and SOD2 [102,123]. The physiological relevance of this regulation was later demonstrated in mouse models of oxidative stress-induced heart failure [124,125]. The report by Alcendor et al. [125] also showed that Sirt1-mediated FoxO deacetylation regulates the catalase gene. A later study demonstrated regulation of catalase production by the FoxO/Sirt1 complex in renal tubular cells [126]. Recently, it was found that Sirt1-mediated FoxO deacetylation regulates FoxO-dependent expression of genes coding for additional ROS detoxification enzymes, peroxiredoxins 3 and 5 (Prx3, Prx5), thioredoxin 2 (Trx2), thioredoxin reductase 2 (TrxR2), and also uncoupling protein 2 (UCP-2), a protein that protects mitochondria from excessive superoxide generation in the electron transport chain (ETC) [20]. Importantly, FoxOs are indispensable for Sirt1-dependent cell survival under oxidative stress [127]. In another study, successive acetylation/deacetylation of FoxO1a protected against acute β-cell failure induced by H2O2, preserving insulin biosynthesis and secretion through induction of the transcription factors NeuroD and MafA [128]. As deacetylated FoxOs are then again more sensitive to poly-ubiquitination and proteasomal degradation [116,128], the (de)acetylation-mediated switch in gene expression of FoxO target genes is expected to protect cells from acute but not from chronic stress.
Several studies have analyzed the mechanistic details that underlie FoxO regulation by successive acetylation/deacetylation. It has been reported that acetylation by p300 reduces the DNA binding affinity of FoxO only marginally [129], while it significantly destabilizes FoxO binding to nucleosome-bound DNA [130]. Stable nucleosome binding is essential for efficient FoxO-dependent chromatin remodeling, because FoxOs work as pioneer transcription factors capable of binding compacted hypoacetylated chromatin [131]. Daitoku et al. proposed a model by which formation of the p300–FoxO complex causes histone acetylation and the recruitment of a preinitiation complex containing RNA polymerase II (RNAPII) to the target promoter and the induced transcription could be attenuated by the subsequent FoxO1a acetylation by CBP [132]. Olmos et al. [20] analyzed Sirt1 binding and histone acetylation in the promoter regions of Sirt1/FoxO target antioxidant genes. Their study found that Sirt1 binding to these promoters was associated with decreased nucleosome acetylation and decreased RNAPII binding. Importantly, decreased RNAPII binding was compensated by enhanced formation of elongation RNAPII complexes, resulting in a net induction of gene expression. These results may suggest that in those promoters where transcription elongation is not a kinetically limiting factor, Sirt1-dependent lowering of RNAPII binding would lead to transcriptional down-regulation, while in those where transcriptional elongation is the kinetically limiting step, Sirt1 activity would result in transcriptional activation. The higher transcriptional activity of deacetylated FoxOs has also been explained by a better binding capability to target DNA sequences due to the presence of positively charged lysine residues; conversely, lysine acetylation weakened the FoxO1a-DNA interaction and made FoxO1a more prone to Akt-induced S256 phosphorylation and in turn nuclear exclusion [108]. It should be noted, too, that the higher DNA binding capability of deacetylated FoxOs can result in trans-repression of target genes of other transcription factors. As an example, iron-induced deacetylation of FoxO1a in adipocytes has been found to decrease transcription of the PPAR-γ target gene adiponectin due to enhanced binding of FoxO1a at the PPAR-γ response element in the adiponectin promoter [133].
In addition to Sirt1, other sirtuins have been proposed to regulate FoxO activity. It has been shown that the cytosolic isoform Sirt2 is capable of deacetylating FoxO1a and FoxO3a, thereby promoting their re-localization from the cytoplasm to the nucleus and increasing FoxO-dependent transcription of antioxidant enzymes [134,135].
Sirt3 is the only sirtuin whose expression has been linked to human longevity [136,137]. Initially described as a mitochondria-specific deacetylase [138], it is in fact a nuclear protein that is translocated to the mitochondria upon oxidative stress [138,139]. It has been described that FoxO3a and Sirt3 directly interact in the mitochondria and that Sirt3 activates FoxO3a-dependent gene expression, probably by increasing the binding of FoxO3a to the promoters of its target genes [140]. In cardiomyocytes, a Sirt3-mediated increase in FoxO3a activity prevented cardiac hypertrophy through induction of SOD-2 and catalase [141] and by suppressing the calcium/calcineurin-dependent activation of NFAT [142]. Therefore, NFAT inhibition might be indirect and mediated by a reduction in ROS levels.
Also, the role of Sirt6 is increasingly appreciated. In C. elegans, it has been shown that the Sirt6 orthologue SIR-2.4 promotes nuclear localization of the FoxO orthologue DAF-16 in response to stress [143]. In mammals, studies demonstrating a functional interaction of Sirt6 with FoxO3a relate to cholesterol homeostasis in the liver. Sirt6 was shown to affect FoxO3a-dependent transcription of SREBP2 (sterol-regulatory element binding protein 2), a major transcriptional regulator of cholesterol biosynthesis, and PCSK9 (proprotein convertase subtilisin/kexin type 9), a crucial enzyme for the control of LDL receptor degradation: FoxO3a recruits Sirt6 to the promoters, where Sirt6 histone deacetylation promotes a repressive chromatin state [144, 145].
4. Redox regulation of FOXO expression
Compared to the well-studied regulation of FoxO activity by posttranslational modifications and protein-protein interactions, mechanisms regulating FoxO gene transcription and mRNA stability are much less known. Here, we provide examples of studies that describe up- or downregulation of FoxO gene expression, often in response to stressful stimuli, including DNA damage, hypoxia/reoxygenation, or oxidative stress.
4.1. Transcriptional regulation of FoxO gene expression
The first transcription factor identified as regulating FoxO genes was E2F-1 [146]. E2F-1 controls cell cycle progression and apoptosis in various cell types [147–150]. Nowak et al. [146] found several putative E2F binding sites in the promoters of the human FoxO1a and FoxO3a genes. Using a human neuroblastoma cell line stably expressing an E2F-1-ER (estrogen receptor) fusion protein, they showed that both FoxO1a and FoxO3a are direct target genes of E2F-1 and are strongly upregulated by this factor [146]. Moreover, using chromatin immunoprecipitation, the direct binding of endogenous E2F-1, as well as of E2F-2 and E2F-3, to the FoxO1a promoter was demonstrated. Of note, upregulation of FoxO by E2F-1-ER was cell type- and species-specific [146], pointing to a possible involvement of additional transcription factors/co-factors or chromatin modifications.
A role of the E2F-1/FoxO axis in regulating the apoptotic response of cardiomyocytes to ischemia/reperfusion (I/R) injury was identified in mice [151,152]. Using E2F-1 knockout mice, Angelis et al. [151] showed that both E2F-1 and FoxO1a are upregulated in the wild-type mice after I/R injury, while in E2F-1-null animals FoxO1a mRNA was not increased. In agreement with that, several pro-apoptotic FoxO1a target genes were also upregulated in the wild-type but not in the E2F-1 knockout mice, and extent of I/R injury (infarct area) was attenuated in the mutant animals. The role of FoxO1a as a critical regulator of cardiomyocyte apoptosis in response to hypoxia followed by reoxygenation was further confirmed in vitro, using primary adult mouse cardiomyocytes and neonatal rat ventricular myocytes [151].
In an apparent discrepancy to the above results, Sengupta et al. [152] showed that FoxO1a and FoxO3a are necessary and sufficient to promote cardiomyocyte cell survival upon induction of oxidative stress by acute I/R or myocardial infarction (MI). The mice with conditional, cardiomyocyte-specific, combined deletion of FoxO1a and FoxO3a exhibited significant increase in infarct area and decreased expression of anti-apoptotic molecules, antioxidant enzymes and autophagy-related proteins following I/R, as compared to controls. The same conditional FoxO knockout mice subjected to MI had increased apoptotic cell death relative to controls, among other cardiac defects [152]. The seemingly opposing results of the two reports could be explained by (i) the dual role of FoxO in combating oxidative stress – pro-survival and pro-apoptotic, where both pathways might be activated following the initial stress signals, and the final outcome would depend on the severity of the cardiomyocyte injury, and (ii) the fact that the absence of the pro-apoptotic FoxO function in FoxO knockout cardiomyocytes might be compensated for by some other apoptotic pathway. Consequently, such cells and cardiac regions would display a higher degree of injury compared to the wild-type controls, as well as to the imaginary E2F-1−/− FoxO+/+ “control”, where FoxO pro-apoptotic function could be impaired, while its pro-survival role would remain operative.
The link between E2F-1 and FoxO may be more intricate than outlined above. Shats et al. [153] found that FoxO1a and FoxO3a, with their genes being E2F-1 targets, can act in a feed-forward regulatory loop by forming a complex with E2F-1 to reinforce gene induction of multiple apoptotic genes. However, as the experiments were done in U2OS human osteosarcoma cells stably expressing an E2F-1-ER fusion, it is not clear whether the same type of regulation can take place also in cardiomyocytes and/or under conditions of severe hypoxia/reoxygenation. Indeed, at least some target genes are regulated differently, as the E2F-1/FoxO complex in U2OS cells upregulates, for example, the classic apoptotic gene APAF1 [153], whereas APAF1 is not upregulated in the myocardium after I/R injury [151]. This may reflect cell context or species-specific (human vs. mouse) differences.
In human fibroblasts, E2F-1 enhances cellular senescence, whereas FoxOs antagonize senescence by upregulating the formation of ROS scavenging proteins [154,155]. Xie et al. [156] showed that E2F-1 attenuates FoxO3a-mediated expression of MnSOD and catalase. They mapped interaction between E2F-1 and FoxO3a to a region including the DNA binding domain of E2F-1 and the C-terminal transcription activation domain of FoxO3a. They propose that E2F-1 inhibits FoxO3a function by directly binding FoxO3a in the nucleus and preventing the activation of its target genes [156]. Depending on the cellular and promoter context (and possibly also on the redox conditions in the cell), the two proteins can therefore act synergistically, or one can antagonize the activity of the other.
The real promoter scenarios are likely to be even more complicated. In their impressive study, Zheng et al. [157] uncovered the mechanism that underlies and dictates two mutually exclusive biological outcomes of E2F-1 activity. They describe the site-specific methylation of E2F-1 by the asymmetrically dimethylating protein arginine methyltransferase 1 (PRMT1) and symmetrically dimethylating PRMT5. Methylation by PRMT1 blocks methylation by PRMT5, which strengthens E2F-1-driven apoptosis in cells harboring damaged DNA. Conversely, PRMT5-catalysed methylation and cyclin A binding to E2F-1 block PRMT1 methylation and promote proliferation [157]. It will be interesting to see how the PRMT1 asymmetric methylation mark on E2F-1 is read on the promoters of its apoptotic target genes.
E2F-1 is not the only transcription factor known so far to directly regulate FoxO genes. Two recent reports reveal roles for p53 tumor suppressor protein as an upstream regulator of FoxO3a [158,159]. Kurinna et al. [158] report FoxO3a as a new p53/p73 target gene. They demonstrate that in the quiescent liver of the adult mouse, p53 and the transactivating isoform of its homolog p73 (TA-p73) reside on the FoxO3a promoter and maintain its transcription active. TA-p73 can bind the same consensus site as p53, and the authors detected binding of both proteins to a predicted p53 response element located -3.7 kb upstream of the FoxO3a transcription start site. In marked contrast to the quiescent state, transcription of the FoxO3a gene is strongly downregulated during the proliferative stage of liver regeneration following partial hepatectomy. This is apparently caused by the disruption of p53, TA-p73, and acetyltransferase p300 binding to, and loss of active chromatin structure within the FoxO3a promoter region. The factors maintaining FoxO3a expression are reestablished and FoxO3a transcription upregulated with the growth completion and recovery of liver mass [158].
Loss of both p53 function and FoxO3-mediated regulation of transcription have been linked to increased proliferation and tumorigenesis [65,160,161]. In summary, the authors [158] suggest a regulatory axis between the p53 family members and FoxO tumor suppressors that functions in the surveillance of normal hepatocytes and is temporarily turned off in the course of liver regeneration. In this context, it would be interesting to survey the FoxO3a promoter status in various liver cancers.
In the second report, Renault et al. [159] point out a number of similarities in function between FoxO3a and p53, e.g. in induction of cell cycle arrest, apoptosis and DNA repair, and to both direct and indirect interactions between the two proteins (see [159] and references therein). This raised a question as to whether one of the proteins could regulate transcription of the other. Indeed, it was found that p53 specifically upregulates the transcription of the mouse FoxO3a gene in embryonic fibroblasts (MEF) and in thymocytes in response to DNA damage [159]. Furthermore, using in silico searches, the authors found four putative p53 binding sites, three of them in the promoter and one in the second intron of the FoxO3a gene. The subsequent chromatin immunoprecipitation assays with extracts from MEFs identified p53 binding to the site in the second intron, but not to those in the promoter region. Although p53 was bound to the intronic site even in the absence of DNA damage, its recruitment to the site was slightly increased following doxorubicin treatment. Moreover, the intronic p53 binding site was proven to be necessary and sufficient for p53-specific transactivation of a luciferase reporter. Further experiments showed that FoxO3a is not required for p53-dependent cell cycle arrest, but it has a role in p53-directed apoptosis [159].
The above two reports describe p53 binding to two sites within the mouse FoxO3a gene; one located in the promoter region and the other in the second intron. Notably, the site occupancy by the p53 protein differs between the adult liver and MEFs, in agreement with different modes of transcriptional regulation in the two tissues. In quiescent liver, the p53/p73 proteins seem to maintain FoxO3a expression at a constant level, in order to prevent hepatocyte proliferation, whereas in MEFs the role of p53 is to upregulate FoxO3a following DNA damage. Possibly, different locations of the binding sites reflect involvement of different cooperating factors/cofactors in order to ensure the proper regulatory mode.
A positive feedback loop in the regulation of FoxO genes transcription has been characterized within the FoxO family itself [162]. Essaghir et al. [162] showed in human fibroblasts that FoxO3a can upregulate FoxO1a and FoxO4 genes expression. At least with the FoxO1a gene, this is achieved by direct binding of FoxO3a to the FoxO binding site, identified in the FoxO1a promoter and characterized in this study. Conversely, all three genes are repressed by growth factors, e.g. PDGF and FGF, and in case of FoxO1a and FoxO4 this may be achieved by inactivation of FoxO3a protein by phosphorylation. Understanding the downregulation of the FoxO3a gene itself by growth factors such as FGF requires further studies. The authors conclude that this new mechanism operating at the transcriptional level modulates fibroblast proliferation [162].
By contrast, Zhu et al. [163] report that FoxO3a negatively regulates autophagy by inhibiting FoxO1a transcription in prostate cancer PC3 cells. It is possible that the transactivating effect of FoxO3a on the FoxO1a promoter is reversed to a repressive one in the tumor cell line context, however, this issue requires further experiments.
Bakker et al. [164] showed that FoxO3a transcription is upregulated during hypoxia in a hypoxia-inducible factor-1α (HIF-1α)-dependent manner in MEFs and NIH3T3 fibroblasts. Under these conditions, FoxO3a in turn induces transcription of CITED2, which inhibits HIF-1α-induced apoptosis in a negative feedback loop. Thus, FoxO3a plays a pro-survival role in response to hypoxic stress [164]. The authors found nine HIF-responsive elements conserved in human and mouse FoxO3a promoters. Whether FoxO3a is a direct target of HIF-1α has yet to be documented. HIF-1α -dependent increase of FoxO3a mRNA and protein levels under hypoxic conditions or following hypoxia-mimetic dimethyloxalyl glycine treatment has more recently been confirmed by Samarin et al. for mouse glomerular microvascular endothelial cell line glEND.2 [165].
Two recent reports analyze regulation of FoxO transcription during fasting and metabolic stress. Wondisford et al. [166] report increases of FoxO1a transcript and protein levels both in the liver of mice fasted for 16 h, as well as in hepatocytes treated with dibutyryl cAMP. The authors identified and functionally characterized tandem cAMP-response elements in the FoxO1a promoter and showed that co-activator p300 regulates FoxO1a gene expression in complex with the cAMP-response element-binding protein (CREB). Lützner et al. [167] identified two functional glucocorticoid-responsive elements (GREs) in the promoter of the FoxO3a gene. FoxO3 transcription was induced by glucocorticoid receptor (GR)-binding steroids and further augmented by activation of AMP-activated protein kinase (AMPK). Moreover, FoxO3a protein upregulated its own promoter, thus acting in a positive autoregulatory feedback loop. The study shows how, under conditions of metabolic stress, GR and high levels of intracellular AMP cooperate to induce FoxO3 gene transcription and post-translationally activate FoxO3a protein [167]. Multiple functional GREs were recently detected also in the murine FoxO1 promoter [168]. These experiments, performed in the C2C12 myoblasts, suggested an additional mechanism by which GR stimulates muscle atrophy [168].
Finally, the FoxO1a gene has been shown to be a direct transcriptional target of FoxC1 in cultured human trabecular meshwork (TM) cells (cells originating from the eye) and in the zebrafish developing eye [169]. FoxC1 binding to an evolutionarily conserved element in the FoxO1a promoter was demonstrated in vivo. Furthermore, siRNA-based downregulation of FoxC1 increased cell death in response to oxidative stress in TM cells (imposed by H2O2 treatment), and in the developing zebrafish eye, indicating the role of the FoxC1–FoxO1a axis in cellular homeostasis and stress protection [169].
While the above data raise an intriguing possibility that E2F-1, p53, FoxO3a, Hif1, p300, GR, and FoxC1 interact in regulating the transcription of FoxO genes, there are further reports describing the up- or downregulation of FoxO gene expression, e.g. during differentiation processes and/or as a cellular response to certain physiological cues, with the regulating transcription factor unknown. For example, an upregulation of FoxO1a and -3a but not -4 mRNAs as well as FoxO1a protein by oxidative stress was observed in murine follicular granulosa cells, followed by FoxO nuclear accumulation and apoptosis [170].
As detailed in a later paragraph, FoxO1a is involved in the regulation of adipocyte differentiation [171,172]. FoxO levels increase during the differentiation of preadipocytes to mature adipocytes, and it has been proposed that FoxO1a-mediated upregulation of antioxidant enzymes may limit the risk of oxidative damage provoked by the generation of intracellular ROS during adipogenesis [173].
FoxO1a is also upregulated during differentiation of human endometrial stromal cells into decidual cells (endometrial decidualization), a process induced by cAMP and progesterone signaling and accompanied by elevated ROS levels and oxidative stress [174–177]. In parallel to the FoxO1a gene induction in the course of the differentiation, FoxO3a expression is downregulated. It is thought that while FoxO1a enhances resistance to oxidative damage during this process, FoxO3a downregulation prevents induction of apoptosis in differentiated, decidualized cells [176].
Expression of FoxO genes has also been shown to change in response to nutritional and hormonal factors [178], aging and caloric restriction [179], and as a result of B cell receptor signaling [180,181].
In conclusion, the transcription of FoxO genes is regulated in response to a number of physiological cues and pathological stress stimuli that are frequently associated with increased oxidative stress. The altered FoxO protein levels impact pro-survival or pro-apoptotic pathways within the cells.
4.2. Posttranscriptional regulation of FoxO levels
Posttranscriptional regulation of FoxO expression emerges as a new level of complexity in controlling FoxO functions in both normal and cancer cells. Four distinct mechanisms have been identified so far: (i) the RNA-binding protein, HuR, stabilizes FoxO-mRNA, (ii) the RNA-binding protein Quaking decreases FoxO mRNA stability, (iii) the FoxO1a 3′UTR may function as a competing endogenous RNA (ceRNA), and (iv) numerous microRNAs have been described that target FoxO transcripts.
The RNA binding protein HuR responds to stressful stimuli that cause its cytosolic accumulation. These stimuli include hydrogen peroxide [182,183], or conditions that generate ROS – including UV radiation [182,184], exposure to arsenite [185], or tert-butylhydroquinone [186]. All these stimuli activate p38MAPK, which directly or indirectly (e.g., via MAPK-activated kinase-2, MK2) stimulates phosphorylation of HuR, which in turn mediates stress-induced cytoplasmic accumulation of HuR and enhances its mRNA stabilizing activity [187–189].
Additionally, it was proposed that HuR itself could act as a redox sensor. A cysteine residue in the first of the three HuR RNA recognition motifs was identified as crucial for homodimerization. Homodimerization is required for full HuR activity, and the authors of the study suggest that this cysteine may respond to oxidative stress and affect HuR homodimer formation and activity [190].
Li et al. [191] identified HuR as interacting with the 3′-untranslated region (3′UTR) of human FoxO1a mRNA, which leads to transcript stabilization and positive regulation of FoxO1a expression. Furthermore, 5-fluorouracil (5-FU) treatment induced FoxO1a expression in a HuR-dependent manner, and that enhanced 5-FU-induced apoptosis in breast cancer cells [191].
Conversely, Yu et al. [192] showed negative regulation of FoxO1 mRNA at the posttranscriptional level by the RNA-binding protein Quaking (QKI). QKI binding to three QKI-response elements (QREs), found in the FoxO1 3′UTR, destabilizes and downregulates FoxO1 mRNA in breast cancer cell lines [192].
Yang et al. found that miR-9 binds both FoxO1a and E-cadherin 3′UTR, indicating a competition for this miRNA between the two transcripts. The results suggest that FoxO1a 3′UTR can function as a ceRNA, promoting E-cadherin expression and inhibiting epithelial-to-mesenchymal transition and metastasis of breast cancer cells [193].
Numerous reports show that upregulation of specific microRNAs leads to downregulation of FoxO1a (e.g., miR-137, miR-223, miR-370) or FoxO3a (e.g., miR-96, miR-155) transcripts in various cancer cells, thus promoting their proliferation [194–199].
4.3. FoxO transcriptional coregulators in redox regulation of FoxO activity
The transcriptional coactivator PGC-1α is an upstream regulator of carbohydrate and lipid metabolism as well as mitochondrial biogenesis and function that associates with different transcription factors to regulate target gene expression; and it has been shown to regulate FoxO activity in different systems [14]. PGC-1α is a positive regulator of fasting-induced hepatic gluconeogenesis and this is mediated through its interaction with FoxO1a [200]. Similarly, the FoxO1a-mediated stimulation of selenoprotein P (SelP) expression in hepatocytes is enhanced by interaction of PGC-1α with FoxO1a [29]. PGC-1α has also been shown to interact with FoxO3a to regulate antioxidant gene expression in endothelial cells and in the skeletal muscle where PGC-1α overexpression is sufficient to attenuate muscle atrophy induced by expression of a constitutively active FoxO transgene [201]. Importantly, the role of PGC-1α inhibition of FoxO3a to increase muscle resistance to catabolic wasting does not compromise the capacity of PGC-1α to reduce oxidative stress in cardiac muscle [202].
In the kidney, high fat diet-induced renal lipotoxicity is associated with insulin resistance and dyslipidemia, possibly due to the downregulation of FoxO3a and PGC-1α and associated with increased oxidative stress. Administration of TEMPOL, a radical scavenger [203] that was recently shown to prevent renal injury by modulating PI3K-Akt-FoxO signaling [204], ameliorated high fat diet-induced renal damage, probably due to the upregulation of FoxO3a and PGC-1α which resulted in protection against oxidative stress and lipoapoptosis [205].
Coordinated upregulation of FoxO and PGC-1α in response to manganese-induced neurotoxicity has been observed, suggesting a coordinated regulation of antioxidant gene expression [206]. Such a co-regulation of genes involved in the protection against oxidative stress has been previously described in endothelial cells [207].
The transcriptional cofactor and lysine demethylase KDM has recently been shown to induce the expression of genes involved in antioxidant defense through its interaction with FoxO. The results show that the principal role for the complex is to maintain the basal expression of oxidative stress resistance genes rather than their induction in response to exogenous oxidative stress [208]. The results further suggest that FoxO access to different chromatin contexts and nuclear microenvironments may rely on different cofactors.
In human cell lines, Ataxin-3/ATXN3 has been shown to interact with FoxO4 and to increase FoxO-dependent transcription of the gene coding for SOD-2. Upon stimulation of oxidative stress, ATXN3 and FoxO4 translocate to the nucleus and coordinately induce the expression of SOD2. Cell lines from patients with spinocerebellar ataxia type 3, deficient in ATXN3, when exposed to oxidative stress, show reduced binding of FoxO4 to the SOD2 promoter, impaired upregulation of SOD-2, and a significantly increased formation of ROS that correlates with the increase in cytotoxicity [209].
5. Physiological and pathophysiological consequences of redox (dys)regulation of FoxOs: selected examples
5.1. Metabolic adaptation to low nutrient intake
The acetylation state of FoxOs affects FoxO-controlled gene regulation in metabolic adaptation to fasting, caloric restriction and starvation. In response to fasting signals, Sirt1 deacetylates both FoxO1a and PGC-1α in hepatocytes, resulting in increased transcription of gluconeogenic genes and elevated glucose release from the liver [104,110]. Nutrient deprivation has been shown to trigger intracellular formation of H2O2 that serves as signaling molecule for the induction of autophagy [210]. Autophagy is an adaptation mechanism to support cellular survival during starvation through delivering cytoplasmic constituents to lysosomes for degradation and recycling [211]. Protein levels of both FoxO1a and Sirt1 were elevated in cultured cardiac myocytes after 2 h of glucose deprivation; Sirt1-catalyzed deacetylation of FoxO1a caused an increase in the autophagic flux through stimulating the expression of Rab7, a GTP-binding protein that mediates the fusion of autophagosomes and lysosomes [212]. In vivo, deacetylation of FoxO1a by Sirt1 has also been shown to be required for induction of autophagy and for resistance against oxidative stress in the heart [125,212]. Sirt1 expression became elevated in response to pressure overload and oxidative stress in the heart of wild-type mice, while moderate Sirt1 overexpression protected the heart of transgenic mice against paraquat-induced oxidative stress through up-regulation of the FoxO-dependent antioxidant enzyme catalase [125]. Apparently, it depends on the cell type whether autophagy is stimulated by deacetylated or by acetylated FoxO proteins: FoxO3a controls fasting-induced autophagy during muscle atrophy [213]. But in contrast to liver and heart, protein levels of Sirt1 have been reported to decrease in type II skeletal muscle of starved mice; transgenic overexpression of Sirt1 resulting in FoxO3a deacetylation prevented the up-regulation of atrophy-related genes in skeletal muscle during fasting [214]. An unexpected molecular mechanism of autophagy induction, which does not depend on the activity of FoxOs as transcription factors, has been delineated for acetylated FoxO1a located in the cytosol of human cancer cell lines: in response to oxidative stress or serum starvation, cytosolic FoxO1a became acetylated following its dissociation from Sirt2. Acetylated FoxO1a then induced the autophagic process through interaction with Atg7 (ubiquitin-like modifier-activating enzyme 7), a key regulator in the formation of the autophagosome [215].
Activity of FoxO3a under conditions of nutrient restriction has also been assessed with respect to metabolism and gene regulation in mitochondria [216]. In myotubes, glucose restriction induces the formation of a FoxO3a/Sirt3 complex that recruited mitochondrial RNA polymerase at mitochondrial DNA-regulatory regions (mtDNA-RR) to activate mitochondrial transcription, resulting in increased mitochondrial respiration capacity. The relevance of Sirt3 regulation of FoxO3a to control mitochondrial function is further supported by recent studies showing that, in response to oxidative stress, Sirt3-mediated deacetylation of FoxO3a upregulates a set of nuclear genes involved in mitochondrial homeostasis including biogenesis, fusion/fission, mitophagy and ROS control [217–219]. Accordingly, it has been reported that chronic dietary restriction increases FoxO3a and FoxO4 levels in skeletal muscle, and these changes correlate with increased expression of genes associated with stress resistance, antioxidants, DNA repair, protein turnover and cell death [220].
5.2. Adipocyte differentiation
Sirt2 is the major sirtuin in adipocytes, and deacetylation of FoxO1a by Sirt2 has been implicated in the regulation of adipogenesis [106]. Adipogenesis takes place in adipose tissue, where mesenchymal stem cells are first committed to preadipocytes, which subsequently undergo clonal expansion, growth arrest and terminal differentiation into mature fat-accumulating adipocytes [221]. Hormonal induction of adipocyte differentiation in vitro is accompanied by a transient increase in intracellular superoxide and H2O2, as described for human adipose tissue-derived stem cells [173] as well as for murine 3T3-L1 preadipocytes [222], the most widely used model cell line for the study of adipogenesis. H2O2 is thought to serve as signaling molecule to increase the stimulating action of insulin on adipogenesis and lipogenesis; exogenous application of H2O2 may both induce and augment adipocyte differentiation of preadipocytes [173,223]. On the other hand, excessive ROS generation would be detrimental and is thus counteracted through staggered induction of antioxidant enzymes such as isoforms of the glutathione peroxidase and thioredoxin reductase selenoenzymes and the FoxO target genes SOD-2 and catalase [173,222,224]. Gene expression and protein synthesis, intracellular localization and posttranslational modifications (phosphorylation, acetylation) of FoxO proteins are tightly and timely regulated in the course of adipocyte differentiation: FoxO1a, FoxO3a and FoxO4 mRNA and protein levels are very low in preadipocytes and increase during adipogenesis, with FoxO1a being the major FoxO isoform in mature adipocytes and in adipose tissue [171,173]. While the levels of FoxO1a begin to rise already in the early stage of adipogenesis, it does not become transcriptionally active before the end of the clonal expansion phase [171]. The delay in FoxO1a activation is accomplished through posttranslational modifications (phosphorylation at S253 of murine Foxo1a, and acetylation), resulting in FoxO1a exclusion from the nucleus during the clonal expansion phase [106,171]. Reversible acetylation of FoxO1a during adipogenesis is controlled through strict regulation of Sirt2 levels: Sirt2 is highly expressed in preadipocytes, strongly down-regulated immediately after the initiation of adipocyte differentiation and partly restored after the clonal expansion phase [106,135]. Both overexpression of Sirt2 and overexpression of a constitutively active FoxO1a mutant, which cannot be excluded from the nucleus, suppressed adipocyte differentiation of preadipocytes [106,135,171]. Conceivably, a strict control of FoxO expression and activity is crucial for the regulation of the intracellular redox state during adipogenesis; switching off FoxOs in early stages ensures more oxidized conditions that favor adipocyte differentiation, while switching on FoxOs in later stages counter-acts oxidative damage through induction of FoxO-dependent antioxidant enzymes (Fig. 5).
Fig. 5.

Schematic representation of the time course of Sirt2, FoxO1a, SOD-2 formation as well as of intracellular ROS levels during adipocyte differentiation in 3T3-L1 murine preadipocytes. The transient increase in intracellular ROS levels is shut down through induction of FoxO1a target genes such as SOD-2. Following a brief period of Akt-dependent phosphorylation of FoxO1a (p-FoxO1a, referring to FoxO phosphorylated at Ser253 – the equivalent of human FoxO1a Ser256), it is upregulated during adipogenesis, and it becomes transcriptionally active after the clonal expansion phase due to its deacetylation through interaction with Sirt2 [106,135,171,222].
5.3. Obesity and diabetes
Carbohydrate and lipid homeostasis is not controlled properly in patients suffering from diabetes mellitus. The metabolic disturbances in diabetes are either caused by lack of insulin due to autoimmune destruction of pancreatic β-cells (type 1 diabetes mellitus, T1DM) or by insulin resistance combined with progressive β-cell failure (type 2 diabetes mellitus, T2DM). Overweight and obesity increase the risk to develop T2DM. By 2030, 573 million adults world-wide are projected to be obese and 366 million individuals may have diabetes [225,226]. The steadily increasing prevalence of overweight, obesity and T2DM gave rise to the concept of a global epidemic of diabetes and to considerable scientific efforts to decipher molecular mechanisms underlying the pathogenesis of T2DM. Oxidative stress is considered a key factor in the development and progression of diabetes and its complications. Excess of glucose and saturated fatty acids elicits overproduction of superoxide and H2O2 through activation of NOX, elevated oxidative phosphorylation in the mitochondrial respiratory chain and uncoupled NO synthase (NOS) [227]. Other potentially harmful ROS in obesity and T2DM derive from ER stress and from chronic low-grade inflammation [228]. Oxidative stress is linked to insulin resistance of liver, skeletal muscle and adipose tissue [227,228]. The insulin-producing pancreatic β-cells are particularly susceptible to oxidative stress-induced damage due to their low expression of antioxidant enzymes [229]. In addition, the vascular endothelium is impaired in diabetes, because excessive superoxide lowers the bioavailability of nitric oxide (NO) through formation of peroxynitrite. This may result in endothelial dysfunction, a major diabetic complication [230]. Increased flux of glucose and sorbitol through the polyol pathway may contribute to cellular redox imbalance under hyperglycemic conditions and is also thought to be involved in the pathogenesis of diabetic complications such as retinopathy, neuropathy, nephropathy and cardiovascular disease [227].
FoxO proteins, in particular FoxO1, are highly expressed in the major insulin target tissues as well as in the insulin-producing β-cells. Hyper-activation of FoxOs has been reported to be associated with hallmarks of overt diabetes such as hyperglycemia, hypertriglyceridemia, insulin resistance and an impaired compensatory increase in β-cell mass as well as with diabetic complications [231–234]. Overexpression of constitutively active Foxo1a in liver and pancreatic β-cells was sufficient to induce diabetes in transgenic mice, due to increased hepatic glucose production combined with decreased β-cell compensation [231]. Conversely, haploinsufficiency of the Foxo1 gene rescued the diabetic phenotype of insulin-resistant mice through lowering hepatic expression of gluconeogenic enzymes and increasing adipocyte expression of insulin-sensitizing genes [231]. In other studies with transgenic mice, both overexpression of constitutively active Foxo1a as well as knockdown of Foxo1a and Foxo3a likewise resulted in hypertriglyceridemia [232,235]. Diabetic complications such as retinopathy and impaired fracture healing have been linked to elevated FoxO1a transcriptional activity under hyperglycemic conditions [234,236,237]. However, FoxOs also have beneficial effects with respect to diabetes, as FoxO-dependent transcription of antioxidant enzymes may counteract oxidative stress-induced cellular damage [233]. FoxO1-mediated induction of NeuroD and MafA has been shown to protect pancreatic β-cells against glucose toxicity [128]. Mice with a triple knockdown of Foxos (Foxo1a, Foxo3a and Foxo4) in pancreatic β-cells developed a maturity-onset diabetes of the young (MODY)-like phenotype characterized by an insulin secretory defect due to impaired ATP generation after glucose stimulation and preferential use of lipids as nutrient source instead of glucose [238].
Dys-regulated hepatic glucose production and release contributes to the fasting and postprandial hyperglycemia that is characteristic of overt diabetes. High glucose stimulates transcription of the FoxO target gene glucose-6-phosphatase in the diabetic liver [239], resulting in a vicious cycle of further increased hepatic glucose release despite high blood glucose levels in diabetes (Fig. 6). Activation of hepatic FoxO1a at high glucose concentrations occurs through its coactivator PGC-1α: expression of PGC-1α is increased in the diabetic liver [240]. In response to high glucose, PGC-1α binds to the enzyme O-GlcNAc transferase and targets it to FoxOs, resulting in increased FoxO GlcNAcylation and increased FoxO-dependent transcription of gluconeogenic enzymes [241]. Moreover, hepatic gene expression and secretion of the FoxO1a target gene SelP is elevated at high glucose concentrations [242,243], and this may result in higher plasma SelP and selenium levels and promote the development of insulin resistance in liver and skeletal muscle [243] (Fig. 6).
Fig. 6.

Hyperactivation of FoxO1a induced by hyperglycemia and ER stress in the diabetic liver results in permanent upregulation of FoxO1a target genes. Elevated hepatic glucose and selenoprotein P (SelP) release may further augment insulin resistance in type 2 diabetes mellitus. G6Pase, glucose 6-phosphatase; OGT, O-linked N-acetylglucosamine transferase; PEPCK, phosphoenolpyruvate carboxykinase; PERK – protein kinase R-like endoplasmic reticulum kinase.
FoxO transcriptional activity is also promoted by the protein kinase PERK (Fig. 4c): PERK has been reported to phosphorylate FoxOs in response to ER stress, thus overriding the inhibitory effect of FoxO phosphorylation via the insulin/Akt signaling pathway [244]. In addition to FoxO activation through posttranslational modifications under diabetic conditions, FoxO1a gene expression has been found to be elevated in the liver of animal models for T1DM and T2DM [232]. It is still not understood completely how the actions of FoxOs on hepatic glucose and lipid metabolism are integrated under conditions of insulin sensitivity and insulin resistance or deficiency. A recently published study proposed a model that distinguishes between early and late stages in the course of pathogenesis of T2DM [245]: in early insulin resistance, the reactive increase in β-cell mass and insulin secretion results initially in hyperinsulinemia and suppression of hepatic FoxO activity, thereby switching off gluconeogenesis and redirecting glycolysis-derived pyruvate to de novo lipogenesis. In contrast, FoxOs are strongly activated in overt T2DM, resulting in elevated gluconeogenesis, while hepatic lipogenesis is predicted to be stimulated through the transcription factors sterol regulatory element-binding protein 1c (SREBP-1c) and carbohydrate-responsive element-binding protein (ChREBP) [245].
5.4. Cancer
The role of FoxOs in tumor development and progression has been widely investigated. Initially believed to be oncogenes, based on elevated FoxO levels in tumor samples, later evidence showed that tumor development was strongly associated with constitutive activation of Akt and loss of FoxO activity. It has also been well documented that loss of FoxO activity is a poor prognosis factor [246–248]. However, the identification of tumor stem cells has further complicated that picture, since it was recognized that FoxO activity, even in the presence of constitutively active Akt, was a prevalent factor linked to drug resistance and invasiveness [234]. FoxOs have a role in chemotherapy resistance due to their regulation of the synthesis of enzymes essential for detoxification. FoxO1a is involved in drug resistance in ovarian cancer exposed to paclitaxel, and is overexpressed in paclitaxel-resistant cancer, attenuating the cytotoxic effect [249]. Importantly, controlling the levels of ROS seems to play a major role in these two aspects of FoxO activity in cancer.
Due to the high degree of homology among FoxO1a, FoxO3a and FoxO4, there is some functional redundancy, and single or double FoxO knockout mice frequently do not show clear phenotypes. Single and double FoxO knockout mice did not show increased rates of tumorigenesis, but the triple deletion of Foxo1a, Foxo3a and Foxo4 resulted in accelerated tumor progression in adult mice, together with increased intracellular ROS levels [250]. In contrast, a single knockout of the less related FoxO6 resulted in increased cellular proliferation and promoted the development of gastric cancer [251].
Cancer cells tend to have elevated intracellular ROS levels [252]. ROS can induce cell proliferation and damage DNA, thereby increasing mutation rates and promoting DNA instability. Elevated ROS and a low antioxidant capacity render cancer cells generally more sensitive towards drugs that induce the formation of intracellular ROS, and this sensitivity has been exploited in chemotherapy [253]. FoxO activity is both crucial to control ROS levels and promote cell quiescence, and to induce apoptotic cell death in response to chemotherapeutic drugs.
Inactivation of FoxOs in tumors occurs through several mechanisms; many tumors show enhanced activation of the PI3K/Akt pathway. Inactivation of the phosphatase PTEN is most frequently observed, resulting in increased FoxO phosphorylation by Akt, their nuclear exclusion and degradation (see also Fig. 1). Other growth factor signaling pathways also converge at the inactivation of FoxO. Importantly, it has been recently described that ROS-dependent oxidation of PTPN12 in breast cancer cells inhibits FoxO activity by enhancing SGK1-mediated FoxO phosphorylation [254].
As mentioned above, microRNAs modulate FoxO levels. Several miRNAs have been demonstrated to be involved in the regulation of FoxO activity in tumor cells. Decreased FoxO levels due to microRNA-induced downregulation may result in higher proliferation and tumor growth in gastric cancer [255] and in bladder cancer [256]. Inactivation of FoxO1a is related to an increase of microvascular area in cancer via a ROS dependent HIF-1α stabilization and activation of the VEGF pathway [257].
Regarding the role of PTMs, some studies have suggested a role of p300 acetylation of FoxO factors in inducing apoptotic cell death in response to a chemotherapeutic agent, particularly those whose mechanism of action depends on increased cellular oxidative stress [258].
Cancer cells make limited use of mitochondria for production of ATP, but they use mitochondria as a source of ROS to stimulate proliferative signaling and as a source of intermediary metabolites to fuel cell growth [259]. Importantly, two recent studies have shown that, in addition to controlling antioxidant gene expression in cancer cells, FoxO3a inhibits c-Myc through several mechanisms: inhibition of c-Myc resulted in downregulation of a large number of nuclear genes encoding mitochondrial proteins, in suppression of mitochondrial ROS production and in prevention of ROS-dependent stabilization of the hypoxia inducible factor HIF-1α, a key player in tumor development [260]. This pathway is likely to be relevant for normal cellular adaptation to hypoxia, preventing the excessive production of mitochondrial ROS in response to low oxygen tensions [261].
6. FoxOs and ROS in different organs
6.1. Liver
The liver as the central metabolic organ has an active metabolism that generates high levels of ROS. In fact, oxidative stress is implicated in many liver diseases [262–265]. Hepatocytes express high levels of antioxidant enzymes, but excessive damage or failure of control mechanisms are common under pathological conditions and generally result in cell death. FoxO3a has recently been shown to have a pro-apoptotic function in hepatocytes exposed to excessive oxidative stress. Importantly, knockdown of β-catenin in hepatocytes rendered mice more sensitive to hepatotoxin-induced liver injury and resulted in elevated expression of FoxO3a target genes such as the pro-apoptotic proteins Bim and p27. Conversely, β-catenin prevented oxidative stress-induced apoptosis through inhibition of FoxO3a [266]. The Wnt/β-catenin pathway had been previously implicated in hepatocyte survival and shown to contribute to the activation of cellular antioxidant defense systems, activating survival pathways and suppressing apoptotic cell death [267,268]. The functional relevance of this regulatory pathway is supported by studies carried out in the context of colon cancer [269–271].
Calorie restriction has been found to result in elevated levels of FoxO1a and FoxO1a-dependent genes coding for proteins involved in antioxidant protection, cell cycle arrest, DNA repair and apoptosis in the liver; thus, the authors suggested that FoxO1a might contribute to the anti-neoplastic effect of calorie restriction [272]. The close interplay between metabolic and oxidative control is highlighted by a recent report that identified a novel mechanism of negative regulation of FoxO activity, involving class IIa HDACs (HDAC4, 5, and 7) in glioblastoma cells. In response to stimulation, mTORC2 promotes inactivation of class IIa histone deacetylases, which leads to increased acetylation of FoxO factors, release of c-Myc inhibition and induction of glycolysis [273]. In contrast, in response to fasting, glucagon in liver induces the dephosphorylation and nuclear translocation of class IIa HDACs, where they recruit HDAC3, which in turn deacetylates FoxOs and boosts gluconeogenesis [274].
6.2. Skeletal muscle
The best characterized specific function of FoxOs in skeletal muscle is the promotion of muscle atrophy in cachexia and denervation. Studies on the role of acetylation in the control of FoxO activity in the muscle have evidenced a differential effect of acetylation on FoxO3a and FoxO1. While increased p300 activity induces FoxO3a degradation and prevented its nuclear localization, it increased FoxO1a nuclear localization [275,276]. Importantly, it has been recently shown that the deacetylation of FoxO3a by HDAC1 is the main driver of muscle atrophy [277] while Sirt1-mediated deacetylation of FoxO1a and 3a inhibits muscle atrophy [214]. In this scenario, it has been an extremely controversial issue whether or not control of ROS levels by FOXOs is relevant in aging sarcopenia, and if so, which are the relevant mechanisms involved. A recent study has now conclusively demonstrated that FoxOs do not significantly contribute to sarcopenia in aging [278]. Furthermore, another recent study strongly argues that FoxO control of ROS in muscle plays a crucial role in lifespan and healthy aging [279].
6.3. Bone
FoxOs have been shown to play an important role in bone metabolism by influencing osteoblast generation and survival via ROS-dependent and -independent mechanisms. FoxO1a inactivation in osteoblasts decreases osteoblast numbers, bone formation rate, and bone volume by 50%. Importantly, it has been shown that FoxO-mediated defense against oxidative stress in osteoblasts regulates skeletal homeostasis. In fact, the bone formation phenotype of FoxO1a osteoblast deficient mice can be attributed to decreased antioxidant defense mechanisms in the absence of FoxO1. Elevated ROS activated the p53 signaling cascade, inducing cell cycle arrest and limiting osteoblast proliferation. The antioxidant N-acetyl cysteine (NAC) normalized redox levels and restored osteoblast proliferation and bone formation [280,281].
Osteoclasts are highly specialized myeloid cells capable of dissolving and digesting the organic bone matrix. In vitro, osteoclast formation induced by parathyroid hormone and interleukin-1 was accompanied by increased superoxide levels, and superoxide dismutase inhibited osteoclastic resorption, indicating that ROS are required for osteoclast differentiation and bone resorption [282]. Both RANKL and M-CSF increase the levels of ROS in osteoclast progenitors and potentiate osteoclast formation and activation [283,284]. Oxidative stress has been also implicated in the pathologic bone resorption associated with estrogen deficiency [285]. However, the link between ROS generation and osteoclast formation was circumstantial. A recent study using murine conditional loss or gain of function FoxO mutants, or mitochondria-targeted catalase in osteoclasts has shown that FoxOs inhibit osteoclast differentiation, at least in part, by stimulating catalase production and thus downregulating H2O2 levels [286].
In cartilage, the expression of FoxO1a and FoxO3a and their nuclear localization has been found to be reduced both in mice and humans in the marginal zone of cartilage exposed to maximal weight – possibly as a consequence of increased levels of pro-inflammatory cytokines [287]. A recent study has shown that knockdown of FoxO1a and FoxO3a in human articular chondrocytes resulted in significantly decreased levels of glutathione peroxidase 1 (GPx-1), catalase, light chain 3 (LC3), beclin1, and Sirt1; following treatment with tert.-butyl hydroperoxide (tBHP), susceptibility to ROS-induced apoptotic cell death was increased [288]. This study is particularly relevant in view of the previously noted observation that aging chondrocytes show reduced levels of antioxidants and increased vulnerability to ROS-induced cell death of [289].
6.4. Central nervous system
Brain-specific ablation of FoxO3a or combined inactivation of FoxO1a/3a/4 in mice resulted in a phenotype that resembled age-dependent depletion of the neural stem cell (NSC) population [290,291]. Furthermore, an ex-vivo assay demonstrated decreased self-renewal capacity of FoxO knockout cells in an age-dependent fashion. Decreased self-renewal of FoxO-deficient NSC can be partially attributable to enhanced intracellular ROS, which leads to decreased NSC reserve and neurogenesis. Analyses of gene expression profiles showed that FoxO3a regulates genes involved in cellular quiescence, differentiation, oxygen metabolism and antioxidants [290]. FoxO3a maintains NSC quiescence preventing premature differentiation under hypoxic conditions by regulating their entry into the cell cycle and ROS levels [291,292]. These studies parallel earlier observations on the role of FoxOs in the control of long-term proliferative capacity of hematopoietic stem cells [293]. In both studies, administration of the antioxidant NAC rescued the aberrant proliferation and loss of self-renewal capacity, supporting the major role of ROS in FoxO deficient stem cell phenotypes.
The amyloid precursor protein (APP) is a transmembrane protein that has been involved in the pathogenesis of Alzheimer’s disease (AD). APP can be cleaved at multiple sites to generate a series of fragments, including the amyloid β (Aβ) peptides and APP intracellular domain (AICD). A recent study has shown that FoxO is a crucial mediator of APP-induced AICD-dependent cell death. AICD functions as a transcriptional co-activator of FoxO that, together with FoxO, translocates into the nucleus upon oxidative stress, and promotes FoxO-induced transcription of the gene encoding the pro-apoptotic protein Bim [294].
Astrocytes, in collaborating with neurons in terms of antioxidant defense, contribute to the prevention of oxidative stress-induced neuronal damage and neurodegeneration [295]. Environmental exposure to manganese and other transition metals increases cellular oxidative stress and has been associated with an increased risk of neurodegeneration. A recent report shows that FoxO levels and activity were induced in astrocytes following Mn exposure [206].
6.5. Blood
FoxOs have been identified as mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Ablation of the three major FoxO isoforms in hematopoietic stem cells (HSC) results in excessive proliferation and early exhaustion of the HSC pool, due to excessive ROS levels in mice [293]. A later study using hematopoietic progenitors from Drosophila further demonstrated that ROS control by FoxOs prevents unprimed differentiation. These results are in line with a number of studies showing that differentiation factors induce a transient increase in ROS levels in the target cells [296].
Oxidative stress has also been shown to regulate erythropoiesis; in the absence of FoxO3a, excessive ROS levels induce cell cycle arrest in precursor cells, preventing maturation, and shorten the viability of mature erythrocyctes [17]. In contrast, myeloid-specific triple FoxO knockout mice showed increased proliferation of granulocyte–monocyte progenitors, resulting in neutrophilia with monocytosis, increased inducible nitric oxide synthase (iNOS) expression and oxidative stress in macrophages. As a result, these mice developed larger atherosclerotic lesions than wild-type controls, with an increased number of intralesional macrophages, but a decreased percentage of apoptotic macrophages [297].
Finally, accumulated evidence suggests that downregulation of Sirt1 and FoxO factors in endothelial cells contributes to oxidative stress, accelerates senescence [298] and induces apoptosis in response to hypoxia [299]. Vascular endothelial senescence has been proposed as a relevant factor in atherogenesis and diabetic retinopathy.
6.6. Kidney
Diabetic nephropathy and glycogen storage disease type Ia (GSD-Ia)-linked nephropathy have been associated with reduced FoxO activity suppression of antioxidant enzymes and activation of NADPH oxidases contributing to disease progression [300,301].
6.7. Skin
FoxO1a also functions in keratinocytes to reduce oxidative stress and prevent cell death as well as to regulate TGF-β signaling. Importantly, ROS are key mediators of cell migration and TGF-β signaling. As a result, the role of FoxO in wound healing is dramatically different in normal and diabetic mice. FoxO1a plays a positive role in wound healing in normal mice [302]. It coordinates the response of keratinocytes to wound healing through upregulation of TGF-β1 and its downstream targets, which are needed for keratinocyte migration. However, in the diabetic wounds, when oxidative stress is more extreme FoxO1a has been linked to impaired wound healing. FoxO1a activity is driven by TNF-α and associated with higher levels of apoptosis and reduced proliferation of fibroblasts [303].
7. Conclusions
While oxidative stress may contribute to the pathogenesis of many diseases, low physiological concentrations of ROS, particularly of hydrogen peroxide, have emerged as indispensable signaling molecules in the hormone- and growth factor-mediated control of cellular metabolism. Apparently, this is not by chance reminiscent of the partially opposite roles of FoxO transcription factors in metabolism and stress response under physiological and pathophysiological conditions. The activity of FoxOs and the cellular redox state are intrinsically linked to each other: FoxOs act as cellular redox sensors that become modified post-translationally through phosphorylation, acetylation and ubiquitination in response to oxidative stress, affecting FoxO cytoplasmic/nuclear shuttling, FoxO stability, and eventually transcription of FoxO-controlled target genes. Moreover, FoxOs may interact with other proteins including co-regulators and antioxidant peroxiredoxins in a redox-sensitive manner, and there is some evidence for redox-sensitive regulation of FoxO gene expression itself. On the other hand, some of the FoxO target genes are antioxidant enzymes that affect the intra- and extracellular redox state by catalyzing dismutation and reduction of superoxide and hydrogen peroxide, respectively. The net result of FoxO-mediated gene expression influences the cell fate upon oxidative damage, inducing either apoptotic cell death or cell cycle arrest and subsequent repair processes. Under physiological conditions, FoxOs are involved in the regulation of cellular differentiation, energy metabolism and nutrient homeostasis. In contrast, FoxO hyper-activation under some pathological conditions associated with oxidative stress may augment metabolic disturbances in type 2 diabetes mellitus or cause muscle atrophy. Such adverse effects emphasize the need for a strict and timely control of FoxO expression and activity.
Acknowledgments
Several authors of this review were supported by the European Cooperation in Science and Technology (COST) Action BM1203/EU-ROS. We thank Dr. N. Urban for the C. elegans photograph in Fig. 1.
Contributor Information
Lars-Oliver Klotz, Email: lars-oliver.klotz@uni-jena.de.
Maria Monsalve, Email: mpmonsalve@iib.uam.es.
References
- 1.Weigel D., Jürgens G., Küttner F., Seifert E., Jäckle H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell. 1989;57:645–658. doi: 10.1016/0092-8674(89)90133-5. [DOI] [PubMed] [Google Scholar]
- 2.Weigel D., Jäckle H. The fork head domain: a novel DNA binding motif of eukaryotic transcription factors? Cell. 1990;63:455–456. doi: 10.1016/0092-8674(90)90439-l. [DOI] [PubMed] [Google Scholar]
- 3.Kaestner K.H., Knochel W., Martinez D.E. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14:142–146. [PubMed] [Google Scholar]
- 4.Kenyon C. The first long-lived mutants: discovery of the insulin/IGF-1 pathway for ageing. Philos. Trans. R. Soc. Lond. B: Biol. Sci. 2011;366:9–16. doi: 10.1098/rstb.2010.0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kenyon C., Chang J., Gensch E., Rudner A., Tabtiang R.A. C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 6.Ogg S., Paradis S., Gottlieb S., Patterson G.I., Lee L., Tissenbaum H.A., Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 7.Lin K., Dorman J.B., Rodan A., Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 8.Kimura K.D., Tissenbaum H.A., Liu Y., Ruvkun G. daf-2, An insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 9.Honda Y., Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999;13:1385–1393. [PubMed] [Google Scholar]
- 10.Honda Y., Honda S. Oxidative stress and life span determination in the nematode Caenorhabditis elegans. Ann. N.Y. Acad. Sci. 2002;959:466–474. doi: 10.1111/j.1749-6632.2002.tb02117.x. [DOI] [PubMed] [Google Scholar]
- 11.Kops G.J., Dansen T.B., Polderman P.E., Saarloos I., Wirtz K.W., Coffer P.J., Huang T.T., Bos J.L., Medema R.H., Burgering B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 12.Doonan R., McElwee J.J., Matthijssens F., Walker G.A., Houthoofd K., Back P., Matscheski A., Vanfleteren J.R., Gems D. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008;22:3236–3241. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greer E.L., Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 14.Monsalve M., Olmos Y. The complex biology of FOXO. Curr. Drug Targets. 2011;12:1322–1350. doi: 10.2174/138945011796150307. [DOI] [PubMed] [Google Scholar]
- 15.Kim D.H., Perdomo G., Zhang T., Slusher S., Lee S., Phillips B.E., Fan Y., Giannoukakis N., Gramignoli R., Strom S., Ringquist S., Dong H.H. FoxO6 integrates insulin signaling with gluconeogenesis in the liver. Diabetes. 2011;60:2763–2774. doi: 10.2337/db11-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calnan D.R., Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 17.Marinkovic D., Zhang X., Yalcin S., Luciano J.P., Brugnara C., Huber T., Ghaffari S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J. Clin. Investig. 2007;117:2133–2144. doi: 10.1172/JCI31807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nemoto S., Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 19.Chiribau C.B., Cheng L., Cucoranu I.C., Yu Y.S., Clempus R.E., Sorescu D. FOXO3A regulates peroxiredoxin III expression in human cardiac fibroblasts. J. Biol. Chem. 2008;283:8211–8217. doi: 10.1074/jbc.M710610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olmos Y., Sanchez-Gomez F.J., Wild B., Garcia-Quintans N., Cabezudo S., Lamas S., Monsalve M. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1alpha complex. Antioxid. Redox Signal. 2013;19:1507–1521. doi: 10.1089/ars.2012.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barsyte D., Lovejoy D.A., Lithgow G.J. Longevity and heavy metal resistance in daf-2 and age-1 long-lived mutants of Caenorhabditis elegans. FASEB J. 2001;15:627–634. doi: 10.1096/fj.99-0966com. [DOI] [PubMed] [Google Scholar]
- 22.Anderson C.P., Leibold E.A. Mechanisms of iron metabolism in Caenorhabditis elegans. Front. Pharmacol. 2014;5:113. doi: 10.3389/fphar.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greer E.L., Oskoui P.R., Banko M.R., Maniar J.M., Gygi M.P., Gygi S.P., Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 24.Leyendecker M., Korsten P., Reinehr R., Speckmann B., Schmoll D., Scherbaum W.A., Bornstein S.R., Barthel A., Klotz L.O. Ceruloplasmin expression in rat liver cells is attenuated by insulin: role of FoxO transcription factors. Horm. Metab. Res. 2011;43:268–274. doi: 10.1055/s-0031-1271692. [DOI] [PubMed] [Google Scholar]
- 25.Sidhu A., Miller P.J., Hollenbach A.D. FOXO1 stimulates ceruloplasmin promoter activity in human hepatoma cells treated with IL-6. Biochem. Biophys. Res. Commun. 2011;404:963–967. doi: 10.1016/j.bbrc.2010.12.089. [DOI] [PubMed] [Google Scholar]
- 26.Hellman N.E., Gitlin J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002;22:439–458. doi: 10.1146/annurev.nutr.22.012502.114457. [DOI] [PubMed] [Google Scholar]
- 27.White K.N., Conesa C., Sanchez L., Amini M., Farnaud S., Lorvoralak C., Evans R.W. The transfer of iron between ceruloplasmin and transferrins. Biochim. Biophys. Acta. 2012;1820:411–416. doi: 10.1016/j.bbagen.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Walter P.L., Steinbrenner H., Barthel A., Klotz L.O. Stimulation of selenoprotein P promoter activity in hepatoma cells by FoxO1a transcription factor. Biochem. Biophys. Res. Commun. 2008;365:316–321. doi: 10.1016/j.bbrc.2007.10.171. [DOI] [PubMed] [Google Scholar]
- 29.Speckmann B., Walter P.L., Alili L., Reinehr R., Sies H., Klotz L.O., Steinbrenner H. Selenoprotein P expression is controlled through interaction of the coactivator PGC-1alpha with FoxO1a and hepatocyte nuclear factor 4alpha transcription factors. Hepatology. 2008;48:1998–2006. doi: 10.1002/hep.22526. [DOI] [PubMed] [Google Scholar]
- 30.Saito Y., Hayashi T., Tanaka A., Watanabe Y., Suzuki M., Saito E., Takahashi K. Selenoprotein P in human plasma as an extracellular phospholipid hydroperoxide glutathione peroxidase. Isolation and enzymatic characterization of human selenoprotein p. J. Biol. Chem. 1999;274:2866–2871. doi: 10.1074/jbc.274.5.2866. [DOI] [PubMed] [Google Scholar]
- 31.Takebe G., Yarimizu J., Saito Y., Hayashi T., Nakamura H., Yodoi J., Nagasawa S., Takahashi K. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J. Biol. Chem. 2002;277:41254–41258. doi: 10.1074/jbc.M202773200. [DOI] [PubMed] [Google Scholar]
- 32.Traulsen H., Steinbrenner H., Buchczyk D.P., Klotz L.O., Sies H. Selenoprotein P protects low-density lipoprotein against oxidation. Free Radic. Res. 2004;38:123–128. doi: 10.1080/10715760320001634852. [DOI] [PubMed] [Google Scholar]
- 33.Burk R.F., Hill K.E., Selenoprotein P. An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 2005;25:215–235. doi: 10.1146/annurev.nutr.24.012003.132120. [DOI] [PubMed] [Google Scholar]
- 34.Hoffmann P.R., Hoge S.C., Li P.A., Hoffmann F.W., Hashimoto A.C., Berry M.J. The selenoproteome exhibits widely varying, tissue-specific dependence on selenoprotein P for selenium supply. Nucleic Acids Res. 2007;35:3963–3973. doi: 10.1093/nar/gkm355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinbrenner H., Alili L., Bilgic E., Sies H., Brenneisen P. Involvement of selenoprotein P in protection of human astrocytes from oxidative damage. Free Radic. Biol. Med. 2006;40:1513–1523. doi: 10.1016/j.freeradbiomed.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 36.Steinbrenner H., Bilgic E., Alili L., Sies H., Brenneisen P. Selenoprotein P protects endothelial cells from oxidative damage by stimulation of glutathione peroxidase expression and activity. Free Radic. Res. 2006;40:936–943. doi: 10.1080/10715760600806248. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y., Wang Y., Zhu W.G. Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell. Biol. 2011;3:276–282. doi: 10.1093/jmcb/mjr013. [DOI] [PubMed] [Google Scholar]
- 38.May J.M., de Haen C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J. Biol. Chem. 1979;254:2214–2220. [PubMed] [Google Scholar]
- 39.Bae Y.S., Kang S.W., Seo M.S., Baines I.C., Tekle E., Chock P.B., Rhee S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 40.DeYulia G.J., Jr., Carcamo J.M. EGF receptor–ligand interaction generates extracellular hydrogen peroxide that inhibits EGFR-associated protein tyrosine phosphatases. Biochem. Biophys. Res. Commun. 2005;334:38–42. doi: 10.1016/j.bbrc.2005.06.056. [DOI] [PubMed] [Google Scholar]
- 41.Sundaresan M., Yu Z.X., Ferrans V.J., Irani K., Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 42.Chen K., Kirber M.T., Xiao H., Yang Y., Keaney J.F., Jr. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 2008;181:1129–1139. doi: 10.1083/jcb.200709049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahadev K., Wu X., Zilbering A., Zhu L., Lawrence J.T., Goldstein B.J. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J. Biol. Chem. 2001;276:48662–48669. doi: 10.1074/jbc.M105061200. [DOI] [PubMed] [Google Scholar]
- 44.Mahadev K., Zilbering A., Zhu L., Goldstein B.J. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J. Biol. Chem. 2001;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- 45.Meng T.C., Buckley D.A., Galic S., Tiganis T., Tonks N.K. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J. Biol. Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- 46.Mahadev K., Motoshima H., Wu X., Ruddy J.M., Arnold R.S., Cheng G., Lambeth J.D., Goldstein B.J. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell. Biol. 2004;24:1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katsuyama M., Matsuno K., Yabe-Nishimura C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J. Clin. Biochem. Nutr. 2012;50:9–22. doi: 10.3164/jcbn.11-06SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schulz E., Wenzel P., Munzel T., Daiber A. Mitochondrial redox signaling: interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014;20:308–324. doi: 10.1089/ars.2012.4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lassegue B., San Martin A., Griendling K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schröder K., Wandzioch K., Helmcke I., Brandes R.P. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler. Thromb. Vasc. Biol. 2009;29:239–245. doi: 10.1161/ATVBAHA.108.174219. [DOI] [PubMed] [Google Scholar]
- 51.Wancket L.M., Frazier W.J., Liu Y. Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology, physiology, and disease. Life Sci. 2012;90:237–248. doi: 10.1016/j.lfs.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guyton K.Z., Liu Y., Gorospe M., Xu Q., Holbrook N.J. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 53.Wang X., McCullough K.D., Franke T.F., Holbrook N.J. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J. Biol. Chem. 2000;275:14624–14631. doi: 10.1074/jbc.275.19.14624. [DOI] [PubMed] [Google Scholar]
- 54.Schieke S.M., von Montfort C., Buchczyk D.P., Timmer A., Grether-Beck S., Krutmann J., Holbrook N.J., Klotz L.O. Singlet oxygen-induced attenuation of growth factor signaling: possible role of ceramides. Free Radic. Res. 2004;38:729–737. doi: 10.1080/10715760410001712764. [DOI] [PubMed] [Google Scholar]
- 55.Schieke S.M., Briviba K., Klotz L.O., Sies H. Activation pattern of mitogen-activated protein kinases elicited by peroxynitrite: attenuation by selenite supplementation. FEBS Lett. 1999;448:301–303. doi: 10.1016/s0014-5793(99)00372-5. [DOI] [PubMed] [Google Scholar]
- 56.Klotz L.O., Schieke S.M., Sies H., Holbrook N.J. Peroxynitrite activates the phosphoinositide 3-kinase/Akt pathway in human skin primary fibroblasts. Biochem. J. 2000;352(Pt 1):219–225. [PMC free article] [PubMed] [Google Scholar]
- 57.Derijard B., Hibi M., Wu I.H., Barrett T., Su B., Deng T., Karin M., Davis R.J. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 58.Sachsenmaier C., Radler-Pohl A., Zinck R., Nordheim A., Herrlich P., Rahmsdorf H.J. Involvement of growth factor receptors in the mammalian UVC response. Cell. 1994;78:963–972. doi: 10.1016/0092-8674(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 59.Klotz L.O., Holbrook N.J., Sies H. UVA and singlet oxygen as inducers of cutaneous signaling events. Curr. Probl. Dermatol. 2001;29:95–113. doi: 10.1159/000060660. [DOI] [PubMed] [Google Scholar]
- 60.Klotz L.O., Briviba K., Sies H. Singlet oxygen mediates the activation of JNK by UVA radiation in human skin fibroblasts. FEBS Lett. 1997;408:289–291. doi: 10.1016/s0014-5793(97)00440-7. [DOI] [PubMed] [Google Scholar]
- 61.Klotz L.O., Pellieux C., Briviba K., Pierlot C., Aubry J.M., Sies H. Mitogen-activated protein kinase (p38-, JNK-, ERK-) activation pattern induced by extracellular and intracellular singlet oxygen and UVA. Eur. J. Biochem. 1999;260:917–922. doi: 10.1046/j.1432-1327.1999.00255.x. [DOI] [PubMed] [Google Scholar]
- 62.Wang X., Martindale J.L., Liu Y., Holbrook N.J. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem. J. 1998;333:291–300. doi: 10.1042/bj3330291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmoll D., Walker K.S., Alessi D.R., Grempler R., Burchell A., Guo S., Walther R., Unterman T.G. Regulation of glucose-6-phosphatase gene expression by protein kinase Balpha and the forkhead transcription factor FKHR. Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity. J. Biol. Chem. 2000;275:36324–36333. doi: 10.1074/jbc.M003616200. [DOI] [PubMed] [Google Scholar]
- 64.Barthel A., Schmoll D., Unterman T.G. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 2005;16:183–189. doi: 10.1016/j.tem.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 65.Yang J.Y., Zong C.S., Xia W., Yamaguchi H., Ding Q., Xie X., Lang J.Y., Lai C.C., Chang C.J., Huang W.C., Huang H., Kuo H.P., Lee D.F., Li L.Y., Lien H.C., Cheng X., Chang K.J., Hsiao C.D., Tsai F.J., Tsai C.H., Sahin A.A., Muller W.J., Mills G.B., Yu D., Hortobagyi G.N., Hung M.C. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008;10:138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Asada S., Daitoku H., Matsuzaki H., Saito T., Sudo T., Mukai H., Iwashita S., Kako K., Kishi T., Kasuya Y., Fukamizu A. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal. 2007;19:519–527. doi: 10.1016/j.cellsig.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 67.Ott C., Jacobs K., Haucke E., Navarrete Santos A., Grune T., Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014;2:411–429. doi: 10.1016/j.redox.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tanaka J., Qiang L., Banks A.S., Welch C.L., Matsumoto M., Kitamura T., Ido-Kitamura Y., DePinho R.A., Accili D. Foxo1 links hyperglycemia to LDL oxidation and endothelial nitric oxide synthase dysfunction in vascular endothelial cells. Diabetes. 2009;58:2344–2354. doi: 10.2337/db09-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yeldandi A.V., Rao M.S., Reddy J.K. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat. Res. 2000;448:159–177. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]
- 70.Lodhi I.J., Semenkovich C.F. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. 2014;19:380–392. doi: 10.1016/j.cmet.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nordgren M., Fransen M. Peroxisomal metabolism and oxidative stress. Biochimie. 2014;98:56–62. doi: 10.1016/j.biochi.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 72.Klotz L.O. Oxidative stress, antioxidants, and chemoprevention: on the role of oxidant-induced signaling in cellular adaptation. In: Jacob C., Kirsch G., Slusarenko A.J., Winyard P.G., Burkholz T., editors. Recent Advances in Redox Active Plant and Microbial Products. Springer; Dordrecht: 2014. pp. 119–146. [Google Scholar]
- 73.Klotz L.O., Hou X., Jacob C. 1,4-Naphthoquinones: from oxidative damage to cellular and inter-cellular signaling. Molecules. 2014;19:14902–14918. doi: 10.3390/molecules190914902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beier J.I., von Montfort C., Sies H., Klotz L.O. Activation of ErbB2 by 2-methyl-1,4-naphthoquinone (menadione) in human keratinocytes: role of EGFR and protein tyrosine phosphatases. FEBS Lett. 2006;580:1859–1864. doi: 10.1016/j.febslet.2006.02.048. [DOI] [PubMed] [Google Scholar]
- 75.Klaus V., Hartmann T., Gambini J., Graf P., Stahl W., Hartwig A., Klotz L.O. 1,4-Naphthoquinones as inducers of oxidative damage and stress signaling in HaCaT human keratinocytes. Arch. Biochem. Biophys. 2010;496:93–100. doi: 10.1016/j.abb.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 76.Zhang S., Liu X., Bawa-Khalfe T., Lu L.S., Lyu Y.L., Liu L.F., Yeh E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012;18:1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 77.Abdelmohsen K., von Montfort C., Stuhlmann D., Gerber P.A., Decking U.K., Sies H., Klotz L.O. Doxorubicin induces EGF receptor-dependent downregulation of gap junctional intercellular communication in rat liver epithelial cells. Biol. Chem. 2005;386:217–223. doi: 10.1515/BC.2005.027. [DOI] [PubMed] [Google Scholar]
- 78.Ho K.K., McGuire V.A., Koo C.Y., Muir K.W., de Olano N., Maifoshie E., Kelly D.J., McGovern U.B., Monteiro L.J., Gomes A.R., Nebreda A.R., Campbell D.G., Arthur J.S., Lam E.W. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. J. Biol. Chem. 2012;287:1545–1555. doi: 10.1074/jbc.M111.284224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kavazis A.N., Smuder A.J., Powers S.K. Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J. Appl. Physiol. 2014;117:223–230. doi: 10.1152/japplphysiol.00210.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ostrakhovitch E.A., Lordnejad M.R., Schliess F., Sies H., Klotz L.O. Copper ions strongly activate the phosphoinositide-3-kinase/Akt pathway independent of the generation of reactive oxygen species. Arch. Biochem. Biophys. 2002;397:232–239. doi: 10.1006/abbi.2001.2559. [DOI] [PubMed] [Google Scholar]
- 81.Walter P.L., Kampkötter A., Eckers A., Barthel A., Schmoll D., Sies H., Klotz L.O. Modulation of FoxO signaling in human hepatoma cells by exposure to copper or zinc ions. Arch. Biochem. Biophys. 2006;454:107–113. doi: 10.1016/j.abb.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 82.Barthel A., Ostrakhovitch E.A., Walter P.L., Kampkötter A., Klotz L.O. Stimulation of phosphoinositide 3-kinase/Akt signaling by copper and zinc ions: mechanisms and consequences. Arch. Biochem. Biophys. 2007;463:175–182. doi: 10.1016/j.abb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 83.Eckers A., Reimann K., Klotz L.O. Nickel and copper ion-induced stress signaling in human hepatoma cells: analysis of phosphoinositide 3'-kinase/Akt signaling. Biometals. 2009;22:307–316. doi: 10.1007/s10534-008-9167-2. [DOI] [PubMed] [Google Scholar]
- 84.Plum L.M., Brieger A., Engelhardt G., Hebel S., Nessel A., Arlt M., Kaltenberg J., Schwaneberg U., Huber M., Rink L., Haase H. PTEN-inhibition by zinc ions augments interleukin-2-mediated Akt phosphorylation. Metallomics. 2014 doi: 10.1039/c3mt00197k. [DOI] [PubMed] [Google Scholar]
- 85.Jomova K., Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 86.Schwerdtle T., Walter I., Mackiw I., Hartwig A. Induction of oxidative DNA damage by arsenite and its trivalent and pentavalent methylated metabolites in cultured human cells and isolated DNA. Carcinogenesis. 2003;24:967–974. doi: 10.1093/carcin/bgg018. [DOI] [PubMed] [Google Scholar]
- 87.Cai B., Xia Z. p38 MAP kinase mediates arsenite-induced apoptosis through FOXO3a activation and induction of Bim transcription. Apoptosis. 2008;13:803–810. doi: 10.1007/s10495-008-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hamann I., Klotz L.O. Arsenite-induced stress signaling: Modulation of the phosphoinositide 3'-kinase/Akt/FoxO signaling cascade. Redox Biol. 2013;1:104–109. doi: 10.1016/j.redox.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hamann I., Petroll K., Hou X., Anwar-Mohamed A., El-Kadi A.O., Klotz L.O. Acute and long-term effects of arsenite in HepG2 cells: modulation of insulin signaling. Biometals. 2014;27:317–332. doi: 10.1007/s10534-014-9714-y. [DOI] [PubMed] [Google Scholar]
- 90.Long L.H., Lan A.N., Hsuan F.T., Halliwell B. Generation of hydrogen peroxide by “antioxidant” beverages and the effect of milk addition. Is cocoa the best beverage? Free Radic. Res. 1999;31:67–71. doi: 10.1080/10715769900300611. [DOI] [PubMed] [Google Scholar]
- 91.Long L.H., Clement M.V., Halliwell B. Artifacts in cell culture: rapid generation of hydrogen peroxide on addition of (−)-epigallocatechin, (−)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly used cell culture media. Biochem. Biophys. Res. Commun. 2000;273:50–53. doi: 10.1006/bbrc.2000.2895. [DOI] [PubMed] [Google Scholar]
- 92.Bartholome A., Kampkötter A., Tanner S., Sies H., Klotz L.O. Epigallocatechin gallate-induced modulation of FoxO signaling in mammalian cells and C. elegans: FoxO stimulation is masked via PI3K/Akt activation by hydrogen peroxide formed in cell culture. Arch. Biochem. Biophys. 2010;501:58–64. doi: 10.1016/j.abb.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 93.Essers M.A., Weijzen S., de Vries-Smits A.M., Saarloos I., de Ruiter N.D., Bos J.L., Burgering B.M. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang H., Regan K.M., Lou Z., Chen J., Tindall D.J. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314:294–297. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- 95.Huo X., Liu S., Shao T., Hua H., Kong Q., Wang J., Luo T., Jiang Y. GSK3 protein positively regulates type I insulin-like growth factor receptor through forkhead transcription factors FOXO1/3/4. J. Biol. Chem. 2014;289:24759–24770. doi: 10.1074/jbc.M114.580738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Himpel S., Tegge W., Frank R., Leder S., Joost H.G., Becker W. Specificity determinants of substrate recognition by the protein kinase DYRK1A. J. Biol. Chem. 2000;275:2431–2438. doi: 10.1074/jbc.275.4.2431. [DOI] [PubMed] [Google Scholar]
- 97.Aranda S., Laguna A., de la Luna S. DYRK family of protein kinases: evolutionary relationships, biochemical properties, and functional roles. FASEB J. 2011;25:449–462. doi: 10.1096/fj.10-165837. [DOI] [PubMed] [Google Scholar]
- 98.Woods Y.L., Rena G., Morrice N., Barthel A., Becker W., Guo S., Unterman T.G., Cohen P. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J. 2001;355:597–607. doi: 10.1042/bj3550597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bain J., McLauchlan H., Elliott M., Cohen P. The specificities of protein kinase inhibitors: an update. Biochem. J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Obsil T., Obsilova V. Structure/function relationships underlying regulation of FOXO transcription factors. Oncogene. 2008;27:2263–2275. doi: 10.1038/onc.2008.20. [DOI] [PubMed] [Google Scholar]
- 101.Brunet A., Sweeney L.B., Sturgill J.F., Chua K.F., Greer P.L., Lin Y., Tran H., Ross S.E., Mostoslavsky R., Cohen H.Y., Hu L.S., Cheng H.L., Jedrychowski M.P., Gygi S.P., Sinclair D.A., Alt F.W., Greenberg M.E. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 102.Daitoku H., Hatta M., Matsuzaki H., Aratani S., Ohshima T., Miyagishi M., Nakajima T., Fukamizu A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Motta M.C., Divecha N., Lemieux M., Kamel C., Chen D., Gu W., Bultsma Y., McBurney M., Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 104.Frescas D., Valenti L., Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- 105.Perrot V., Rechler M.M. The coactivator p300 directly acetylates the forkhead transcription factor Foxo1 and stimulates Foxo1-induced transcription. Mol. Endocrinol. 2005;19:2283–2298. doi: 10.1210/me.2004-0292. [DOI] [PubMed] [Google Scholar]
- 106.Jing E., Gesta S., Kahn C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007;6:105–114. doi: 10.1016/j.cmet.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nakae J., Oki M., Cao Y. The FoxO transcription factors and metabolic regulation. FEBS Lett. 2008;582:54–67. doi: 10.1016/j.febslet.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 108.Matsuzaki H., Daitoku H., Hatta M., Aoyama H., Yoshimochi K., Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 2005;102:11278–11283. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dansen T.B., Smits L.M., van Triest M.H., de Keizer P.L., van Leenen D., Koerkamp M.G., Szypowska A., Meppelink A., Brenkman A.B., Yodoi J., Holstege F.C., Burgering B.M. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat. Chem. Biol. 2009;5:664–672. doi: 10.1038/nchembio.194. [DOI] [PubMed] [Google Scholar]
- 110.Rodgers J.T., Lerin C., Haas W., Gygi S.P., Spiegelman B.M., Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 111.Nasrin N., Ogg S., Cahill C.M., Biggs W., Nui S., Dore J., Calvo D., Shi Y., Ruvkun G., Alexander-Bridges M.C. DAF-16 recruits the CREB-binding protein coactivator complex to the insulin-like growth factor binding protein 1 promoter in HepG2 cells. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10412–10417. doi: 10.1073/pnas.190326997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mahmud D.L., Deb M.G.A., Platanias D.K., Uddin L.C., Wickrema S. Phosphorylation of forkhead transcription factors by erythropoietin and stem cell factor prevents acetylation and their interaction with coactivator p300 in erythroid progenitor cells. Oncogene. 2002;21:1556–1562. doi: 10.1038/sj.onc.1205230. [DOI] [PubMed] [Google Scholar]
- 113.Spiegelman B.M., Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–167. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 114.Fukuoka M., Daitoku H., Hatta M., Matsuzaki H., Umemura S., Fukamizu A. Negative regulation of forkhead transcription factor AFX (Foxo4) by CBP-induced acetylation. Int. J. Mol. Med. 2003;12:503–508. [PubMed] [Google Scholar]
- 115.van der Horst A., de Vries-Smits A.M., Brenkman A.B., van Triest M.H., van den Broek N., Colland F., Maurice M.M., Burgering B.M. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat. Cell Biol. 2006;8:1064–1073. doi: 10.1038/ncb1469. [DOI] [PubMed] [Google Scholar]
- 116.Wang F., Chan C.H., Chen K., Guan X., Lin H.K., Tong Q. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene. 2012;31:1546–1557. doi: 10.1038/onc.2011.347. [DOI] [PubMed] [Google Scholar]
- 117.Huang H., Regan K.M., Wang F., Wang D., Smith D.I., van Deursen J.M., Tindall D.J. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc. Natl. Acad. Sci. U.S.A. 2005;102:1649–1654. doi: 10.1073/pnas.0406789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Storz P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid. Redox Signal. 2011;14:593–605. doi: 10.1089/ars.2010.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stone J.R., Yang S. Hydrogen peroxide: a signaling messenger. Antioxid. Redox Signal. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 120.Zschauer T.C., Matsushima S., Altschmied J., Shao D., Sadoshima J., Haendeler J. Interacting with thioredoxin-1 – disease or no disease? Antioxid. Redox Signal. 2013;18:1053–1062. doi: 10.1089/ars.2012.4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Putker M., Vos H.R., van Dorenmalen K., de Ruiter H., Duran A.G., Snel B., Burgering B.M., Vermeulen M., Dansen T.B. Evolutionary acquisition of cysteines determines FOXO paralog-specific redox signaling. Antioxid. Redox Signal. 2015;22:15–28. doi: 10.1089/ars.2014.6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Imai S., Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell. Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.van der Horst A., Tertoolen L.G., de Vries-Smits L.M., Frye R.A., Medema R.H., Burgering B.M. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J. Biol. Chem. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 124.Tanno M., Kuno A., Yano T., Miura T., Hisahara S., Ishikawa S., Shimamoto K., Horio Y. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J. Biol. Chem. 2010;285:8375–8382. doi: 10.1074/jbc.M109.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Alcendor R.R., Gao S., Zhai P., Zablocki D., Holle E., Yu X., Tian B., Wagner T., Vatner S.F., Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 126.Hasegawa K., Wakino S., Yoshioka K., Tatematsu S., Hara Y., Minakuchi H., Washida N., Tokuyama H., Hayashi K., Itoh H. Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem. Biophys. Res. Commun. 2008;372:51–56. doi: 10.1016/j.bbrc.2008.04.176. [DOI] [PubMed] [Google Scholar]
- 127.Hori Y.S., Kuno A., Hosoda R., Horio Y. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. PLoS One. 2013;8:e73875. doi: 10.1371/journal.pone.0073875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kitamura Y.I., Kitamura T., Kruse J.P., Raum J.C., Stein R., Gu W., Accili D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 129.Brent M.M., Anand R., Marmorstein R. Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure. 2008;16:1407–1416. doi: 10.1016/j.str.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang Y., Hou H., Haller E.M. Nicosia SV, Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–1032. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hatta M., Cirillo L.A. Chromatin opening and stable perturbation of core histone:DNA contacts by FoxO1. J. Biol. Chem. 2007;282:35583–35593. doi: 10.1074/jbc.M704735200. [DOI] [PubMed] [Google Scholar]
- 132.Daitoku H., Sakamaki J., Fukamizu A. Regulation of FoxO transcription factors by acetylation and protein–protein interactions. Biochim. Biophys. Acta. 2011;1813:1954–1960. doi: 10.1016/j.bbamcr.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 133.Gabrielsen J.S., Gao Y., Simcox J.A., Huang J., Thorup D., Jones D., Cooksey R.C., Gabrielsen D., Adams T.D., Hunt S.C., Hopkins P.N., Cefalu W.T., McClain D.A. Adipocyte iron regulates adiponectin and insulin sensitivity. J. Clin. Investig. 2012;122:3529–3540. doi: 10.1172/JCI44421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang F., Nguyen M., Qin F.X., Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–514. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 135.Wang F., Tong Q. SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1's repressive interaction with PPARgamma. Mol. Biol. Cell. 2009;20:801–808. doi: 10.1091/mbc.E08-06-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rose G., Dato S., Altomare K., Bellizzi D., Garasto S., Greco V., Passarino G., Feraco E., Mari V., Barbi C., BonaFe M., Franceschi C., Tan Q., Boiko S., Yashin A.I., De Benedictis G. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp. Gerontol. 2003;38:1065–1070. doi: 10.1016/s0531-5565(03)00209-2. [DOI] [PubMed] [Google Scholar]
- 137.Bellizzi D., Rose G., Cavalcante P., Covello G., Dato S., De Rango F., Greco V., Maggiolini M., Feraco E., Mari V., Franceschi C., Passarino G., De Benedictis G. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. 2005;85:258–263. doi: 10.1016/j.ygeno.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 138.Schwer B., North B.J., Frye R.A., Ott M., Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Scher M.B., Vaquero A., Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007;21:920–928. doi: 10.1101/gad.1527307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jacobs K.M., Pennington J.D., Bisht K.S., Aykin-Burns N., Kim H.S., Mishra M., Sun L., Nguyen P., Ahn B.H., Leclerc J., Deng C.X., Spitz D.R., Gius D. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int. J. Biol. Sci. 2008;4:291–299. doi: 10.7150/ijbs.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sundaresan N.R., Gupta M., Kim G., Rajamohan S.B., Isbatan A., Gupta M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ni Y.G., Berenji K., Wang N., Oh M., Sachan N., Dey A., Cheng J., Lu G., Morris D.J., Castrillon D.H., Gerard R.D., Rothermel B.A., Hill J.A. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation. 2006;114:1159–1168. doi: 10.1161/CIRCULATIONAHA.106.637124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chiang W.C., Tishkoff D.X., Yang B., Wilson-Grady J., Yu X., Mazer T., Eckersdorff M., Gygi S.P., Lombard D.B., Hsu A.L. C. elegans SIRT6/7 homolog SIR-2.4 promotes DAF-16 relocalization and function during stress. PLoS Genet. 2012;8:e1002948. doi: 10.1371/journal.pgen.1002948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tao R., Xiong X., DePinho R.A., Deng C.X., Dong X.C. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J. Biol. Chem. 2013;288:29252–29259. doi: 10.1074/jbc.M113.481473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tao R., Xiong X., DePinho R.A., Deng C.X., Dong X.C. Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J. Lipid Res. 2013;54:2745–2753. doi: 10.1194/jlr.M039339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nowak K., Killmer K., Gessner C., Lutz W. E2F-1 regulates expression of FOXO1 and FOXO3a. Biochim. Biophys. Acta. 2007;1769:244–252. doi: 10.1016/j.bbaexp.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 147.Trimarchi J.M., Lees J.A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell. Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 148.Shan B., Lee W.H. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol. Cell. Biol. 1994;14:8166–8173. doi: 10.1128/mcb.14.12.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.DeGregori J., Leone G., Miron A., Jakoi L., Nevins J.R. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Qin X.Q., Livingston D.M., Kaelin W.G., Jr., Adams P.D. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc. Natl. Acad. Sci. U.S.A. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Angelis E., Zhao P., Zhang R., Goldhaber J.I., Maclellan W.R. The role of E2F-1 and downstream target genes in mediating ischemia/reperfusion injury in vivo. J. Mol. Cell. Cardiol. 2011;51:919–926. doi: 10.1016/j.yjmcc.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sengupta A., Molkentin J.D., Paik J.H., DePinho R.A., Yutzey K.E. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J. Biol. Chem. 2011;286:7468–7478. doi: 10.1074/jbc.M110.179242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shats I., Gatza M.L., Liu B., Angus S.P., You L., Nevins J.R. FOXO transcription factors control E2F1 transcriptional specificity and apoptotic function. Cancer Res. 2013;73:6056–6067. doi: 10.1158/0008-5472.CAN-13-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dimri G.P., Itahana K., Acosta M., Campisi J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol. Cell. Biol. 2000;20:273–285. doi: 10.1128/mcb.20.1.273-285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nogueira V., Park Y., Chen C.C., Xu P.Z., Chen M.L., Tonic I., Unterman T., Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–470. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Xie Q., Peng S., Tao L., Ruan H., Yang Y., Li T.M., Adams U., Meng S., Bi X., Dong M.Q., Yuan Z. E2F transcription factor 1 regulates cellular and organismal senescence by inhibiting Forkhead box O transcription factors. J. Biol. Chem. 2014;289:34205–34213. doi: 10.1074/jbc.M114.587170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zheng S., Moehlenbrink J., Lu Y.C., Zalmas L.P., Sagum C.A., Carr S., McGouran J.F., Alexander L., Fedorov O., Munro S., Kessler B., Bedford M.T., Yu Q., La Thanque N.B. Arginine methylation-dependent reader–writer interplay governs growth control by E2F-1. Mol. Cell. 2013;52:37–51. doi: 10.1016/j.molcel.2013.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kurinna S., Stratton S.A., Tsai W.W., Akdemir K.C., Gu W., Singh P., Goode T., Darlington G.J., Barton M.C. Direct activation of forkhead box O3 by tumor suppressors p53 and p73 is disrupted during liver regeneration in mice. Hepatology. 2010;52:1023–1032. doi: 10.1002/hep.23746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Renault V.M., Thekkat P.U., Hoang K.L., White J.L., Brady C.A., Kenzelmann Broz D., Venturelli O.S., Johnson T.M., Oskoui P.R., Xuan Z., Santo E.E., Zhang M.Q., Vogel H., Attardi L.D., Brunet A. The pro-longevity gene FoxO3 is a direct target of the p53 tumor suppressor. Oncogene. 2011;30:3207–3221. doi: 10.1038/onc.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vogelstein B., Lane D., Levine A.J. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 161.Trotman L.C., Alimonti A., Scaglioni P.P., Koutcher J.A., Cordon-Cardo C., Pandolfi P.P. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–527. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Essaghir A., Dif N., Marbehant C.Y., Coffer P.J., Demoulin J.B. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J. Biol. Chem. 2009;284:10334–10342. doi: 10.1074/jbc.M808848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhu W.L., Tong H., Teh J.T., Wang M. Forkhead box protein O3 transcription factor negatively regulates autophagy in human cancer cells by inhibiting forkhead box protein O1 expression and cytosolic accumulation. PLoS One. 2014;9:e115087. doi: 10.1371/journal.pone.0115087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bakker W.J., Harris I.S., Mak T.W. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol. Cell. 2007;28:941–953. doi: 10.1016/j.molcel.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 165.Samarin J., Wessel J., Cicha I., Kroening S., Warnecke C., Goppelt-Struebe M. FoxO proteins mediate hypoxic induction of connective tissue growth factor in endothelial cells. J. Biol. Chem. 2010;285:4328–4336. doi: 10.1074/jbc.M109.049650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wondisford A.R., Xiong L., Chang E., Meng S., Meyers D.J., Li M., Cole P.A., He L. Control of Foxo1 gene expression by co-activator P300. J. Biol. Chem. 2014;289:4326–4333. doi: 10.1074/jbc.M113.540500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lützner N., Kalbacher H., Krones-Herzig A., Rösl F. FOXO3 is a glucocorticoid receptor target and regulates LKB1 and its own expression based on cellular AMP levels via a positive autoregulatory loop. PLoS One. 2012;7:e42166. doi: 10.1371/journal.pone.0042166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Qin W., Pan J., Qin Y., Lee D.N., Bauman W.A., Cardozo C. Identification of functional glucocorticoid response elements in the mouse FoxO1 promoter. Biochem. Biophys. Res. Commun. 2014;450:979–983. doi: 10.1016/j.bbrc.2014.06.080. [DOI] [PubMed] [Google Scholar]
- 169.Berry F.B., Skarie J.M., Mirzayans F., Fortin Y., Hudson T.J., Raymond V., Link B.A., Walter M.A. FOXC1 is required for cell viability and resistance to oxidative stress in the eye through the transcriptional regulation of FOXO1A. Hum. Mol. Genet. 2008;17:490–505. doi: 10.1093/hmg/ddm326. [DOI] [PubMed] [Google Scholar]
- 170.Shen M., Lin F., Zhang J., Tang Y., Chen W.K., Liu H. Involvement of the up-regulated FoxO1 expression in follicular granulosa cell apoptosis induced by oxidative stress. J. Biol. Chem. 2012;287:25727–25740. doi: 10.1074/jbc.M112.349902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nakae J., Kitamura T., Kitamura Y., Biggs W.H., 3rd, Arden K.C., Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell. 2003;4:119–129. doi: 10.1016/s1534-5807(02)00401-x. [DOI] [PubMed] [Google Scholar]
- 172.Liu G.S., Chan E.C., Higuchi M., Dusting G.J., Jiang F. Redox mechanisms in regulation of adipocyte differentiation: beyond a general stress response. Cells. 2012;1:976–993. doi: 10.3390/cells1040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Higuchi M., Dusting G.J., Peshavariya H., Jiang F., Hsiao S.T., Chan E.C., Liu G.S. Differentiation of human adipose-derived stem cells into fat involves reactive oxygen species and Forkhead box O1 mediated upregulation of antioxidant enzymes. Stem Cells Dev. 2013;22:878–888. doi: 10.1089/scd.2012.0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Christian M., Zhang X., Schneider-Merck T., Unterman T.G., Gellersen B., White J.O., Brosens J.J. Cyclic AMP-induced forkhead transcription factor, FKHR, cooperates with CCAAT/enhancer-binding protein beta in differentiating human endometrial stromal cells. J. Biol. Chem. 2002;277:20825–20832. doi: 10.1074/jbc.M201018200. [DOI] [PubMed] [Google Scholar]
- 175.Labied S., Kajihara T., Madureira P.A., Fusi L., Jones M.C., Higham J.M., Varshochi R., Francis J.M., Zoumpoulidou G., Essafi A., Fernandez de Mattos S., Lam E.W., Brosens J.J. Progestins regulate the expression and activity of the forkhead transcription factor FOXO1 in differentiating human endometrium. Mol. Endocrinol. 2006;20:35–44. doi: 10.1210/me.2005-0275. [DOI] [PubMed] [Google Scholar]
- 176.Kajihara T., Jones M., Fusi L., Takano M., Feroze-Zaidi F., Pirianov G., Mehmet H., Ishihara O., Higham J.M., Lam E.W., Brosens J.J. Differential expression of FOXO1 and FOXO3a confers resistance to oxidative cell death upon endometrial decidualization. Mol. Endocrinol. 2006;20:2444–2455. doi: 10.1210/me.2006-0118. [DOI] [PubMed] [Google Scholar]
- 177.Takano M., Lu Z., Goto T., Fusi L., Higham J., Francis J., Withey A., Hardt J., Cloke B., Stavropoulou A.V., Ishihara O., Lam E.W., Unterman T.G., Brosens J.J., Kim J.J. Transcriptional cross talk between the forkhead transcription factor forkhead box O1A and the progesterone receptor coordinates cell cycle regulation and differentiation in human endometrial stromal cells. Mol. Endocrinol. 2007;21:2334–2349. doi: 10.1210/me.2007-0058. [DOI] [PubMed] [Google Scholar]
- 178.Imae M., Fu Z., Yoshida A., Noguchi T., Kato H. Nutritional and hormonal factors control the gene expression of FoxOs, the mammalian homologues of DAF-16. J. Mol. Endocrinol. 2003;30:253–262. doi: 10.1677/jme.0.0300253. [DOI] [PubMed] [Google Scholar]
- 179.Furuyama T., Yamashita H., Kitayama K., Higami Y., Shimokawa I., Mori N. Effects of aging and caloric restriction on the gene expression of Foxo1, 3, and 4 (FKHR, FKHRL1, and AFX) in the rat skeletal muscles. Microsc. Res. Tech. 2002;59:331–334. doi: 10.1002/jemt.10213. [DOI] [PubMed] [Google Scholar]
- 180.Zhu X., Hart R., Chang M.S., Kim J.W., Lee S.Y., Cao Y.A., Mock D., Ke E., Saunders B., Alexander A., Grossoehme J., Lin K.M., Yan Z., Hsueh R., Lee J., Scheuermann R.H., Fruman D.A., Seaman W., Subramaniam S., Sternweis P., Simon M.I., Choi S. Analysis of the major patterns of B cell gene expression changes in response to short-term stimulation with 33 single ligands. J. Immunol. 2004;173:7141–7149. doi: 10.4049/jimmunol.173.12.7141. [DOI] [PubMed] [Google Scholar]
- 181.Hinman R.M., Bushanam J.N., Nichols W.A., Satterthwaite A.B. B cell receptor signaling down-regulates forkhead box transcription factor class O 1 mRNA expression via phosphatidylinositol 3-kinase and Bruton's tyrosine kinase. J. Immunol. 2007;178:740–747. doi: 10.4049/jimmunol.178.2.740. [DOI] [PubMed] [Google Scholar]
- 182.Wang W., Furneaux H., Cheng H., Caldwell M.C., Hutter D., Liu Y., Holbrook N., Gorospe M. HuR regulates p21 mRNA stabilization by UV light. Mol. Cell. Biol. 2000;20:760–769. doi: 10.1128/mcb.20.3.760-769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tran H., Maurer F., Nagamine Y. Stabilization of urokinase and urokinase receptor mRNAs by HuR is linked to its cytoplasmic accumulation induced by activated mitogen-activated protein kinase-activated protein kinase 2. Mol. Cell. Biol. 2003;23:7177–7188. doi: 10.1128/MCB.23.20.7177-7188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fernau N.S., Fugmann D., Leyendecker M., Reimann K., Grether-Beck S., Galban S., Ale-Agha N., Krutmann J., Klotz L.O. Role of HuR and p38MAPK in ultraviolet B-induced post-transcriptional regulation of COX-2 expression in the human keratinocyte cell line HaCaT. J. Biol. Chem. 2010;285:3896–3904. doi: 10.1074/jbc.M109.081430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bhattacharyya S.N., Habermacher R., Martine U., Closs E.I., Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 186.Song I.S., Tatebe S., Dai W., Kuo M.T. Delayed mechanism for induction of gamma-glutamylcysteine synthetase heavy subunit mRNA stability by oxidative stress involving p38 mitogen-activated protein kinase signaling. J. Biol. Chem. 2005;280:28230–28240. doi: 10.1074/jbc.M413103200. [DOI] [PubMed] [Google Scholar]
- 187.Abdelmohsen K., Kuwano Y., Kim H.H., Gorospe M. Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: implications for cellular senescence. Biol. Chem. 2008;389:243–255. doi: 10.1515/BC.2008.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Subbaramaiah K., Marmo T.P., Dixon D.A., Dannenberg A.J. Regulation of cyclooxgenase-2 mRNA stability by taxanes: evidence for involvement of p38, MAPKAPK-2, and HuR. J. Biol. Chem. 2003;278:37637–37647. doi: 10.1074/jbc.M301481200. [DOI] [PubMed] [Google Scholar]
- 189.Lafarga V., Cuadrado A., Lopez de Silanes I., Bengoechea R., Fernandez-Capetillo O., Nebreda A.R. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol. Cell. Biol. 2009;29:4341–4351. doi: 10.1128/MCB.00210-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Benoit R.M., Meisner N.C., Kallen J., Graff P., Hemmig R., Cebe R., Ostermeier C., Widmer H., Auer M. The x-ray crystal structure of the first RNA recognition motif and site-directed mutagenesis suggest a possible HuR redox sensing mechanism. J. Mol. Biol. 2010;397:1231–1244. doi: 10.1016/j.jmb.2010.02.043. [DOI] [PubMed] [Google Scholar]
- 191.Li Y., Yu J., Du D., Fu S., Chen Y., Yu F., Gao P. Involvement of post-transcriptional regulation of FOXO1 by HuR in 5-FU-induced apoptosis in breast cancer cells. Oncol. Lett. 2013;6:156–160. doi: 10.3892/ol.2013.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yu F., Jin L., Yang G., Ji L., Wang F., Lu Z. Post-transcriptional repression of FOXO1 by QKI results in low levels of FOXO1 expression in breast cancer cells. Oncol. Rep. 2014;31:1459–1465. doi: 10.3892/or.2013.2957. [DOI] [PubMed] [Google Scholar]
- 193.Yang J., Li T., Gao C., Lv X., Liu K., Song H., Xing Y., Xi T. FOXO1 3′UTR functions as a ceRNA in repressing the metastases of breast cancer cells via regulating miRNA activity. FEBS Lett. 2014;588:3218–3224. doi: 10.1016/j.febslet.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 194.Lin H., Dai T., Xiong H., Zhao X., Chen X., Yu C., Li J., Wang X., Song L. Unregulated miR-96 induces cell proliferation in human breast cancer by downregulating transcriptional factor FOXO3a. PLoS One. 2010;5:e15797. doi: 10.1371/journal.pone.0015797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wu Z., Sun H., Zeng W., He J., Mao X. Upregulation of MircoRNA-370 induces proliferation in human prostate cancer cells by downregulating the transcription factor FOXO1. PLoS One. 2012;7:e45825. doi: 10.1371/journal.pone.0045825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wu L., Li H., Jia C.Y., Cheng W., Yu M., Peng M., Zhu Y., Zhao Q., Dong Y.W., Shao K., Wu A., Wu X.Z. MicroRNA-223 regulates FOXO1 expression and cell proliferation. FEBS Lett. 2012;586:1038–1043. doi: 10.1016/j.febslet.2012.02.050. [DOI] [PubMed] [Google Scholar]
- 197.Fan C., Liu S., Zhao Y., Han Y., Yang L., Tao G., Li Q., Zhang L. Upregulation of miR-370 contributes to the progression of gastric carcinoma via suppression of FOXO1. Biomed. Pharmacother. 2013;67:521–526. doi: 10.1016/j.biopha.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 198.Ling N., Gu J., Lei Z., Li M., Zhao J., Zhang H.T., Li X. microRNA-155 regulates cell proliferation and invasion by targeting FOXO3a in glioma. Oncol. Rep. 2013;30:2111–2118. doi: 10.3892/or.2013.2685. [DOI] [PubMed] [Google Scholar]
- 199.Tan C., Liu S., Tan S., Zeng X., Yu H., Li A., Bei C., Qiu X. Polymorphisms in microRNA target sites of forkhead box O genes are associated with hepatocellular carcinoma. PLoS One. 2015;10:e0119210. doi: 10.1371/journal.pone.0119210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Puigserver P., Rhee J., Donovan J., Walkey C.J., Yoon J.C., Oriente F., Kitamura Y., Altomonte J., Dong H., Accili D., Spiegelman B.M. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 201.Sandri M., Lin J., Handschin C., Yang W., Arany Z.P., Lecker S.H., Goldberg A.L., Spiegelman B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Geng T., Li P., Yin X., Yan Z. PGC-1alpha promotes nitric oxide antioxidant defenses and inhibits FOXO signaling against cardiac cachexia in mice. Am. J. Pathol. 2011;178:1738–1748. doi: 10.1016/j.ajpath.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wilcox C.S., Pearlman A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol. Rev. 2008;60:418–469. doi: 10.1124/pr.108.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Yoon H.E., Kim S.J., Kim S.J., Chung S., Shin S.J. Tempol attenuates renal fibrosis in mice with unilateral ureteral obstruction: the role of PI3K-Akt-FoxO3a signaling. J. Korean Med. Sci. 2014;29:230–237. doi: 10.3346/jkms.2014.29.2.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chung H.W., Lim J.H., Kim M.Y., Shin S.J., Chung S., Choi B.S., Kim H.W., Kim Y.S., Park C.W., Chang Y.S. High-fat diet-induced renal cell apoptosis and oxidative stress in spontaneously hypertensive rat are ameliorated by fenofibrate through the PPARalpha-FoxO3a-PGC-1alpha pathway. Nephrol. Dial. Transplant. 2012;27:2213–2225. doi: 10.1093/ndt/gfr613. [DOI] [PubMed] [Google Scholar]
- 206.Exil V., Ping L., Yu Y., Chakraborty S., Caito S.W., Wells K.S., Karki P., Lee E., Aschner M. Activation of MAPK and FoxO by manganese (Mn) in rat neonatal primary astrocyte cultures. PLoS One. 2014;9:e94753. doi: 10.1371/journal.pone.0094753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Olmos Y., Valle I., Borniquel S., Tierrez A., Soria E., Lamas S., Monsalve M. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. J. Biol. Chem. 2009;284:14476–14484. doi: 10.1074/jbc.M807397200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Liu X., Greer C., Secombe J. KDM5 interacts with Foxo to modulate cellular levels of oxidative stress. PLoS Genet. 2014;10:e1004676. doi: 10.1371/journal.pgen.1004676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Araujo J., Breuer P., Dieringer S., Krauss S., Dorn S., Zimmermann K., Pfeifer A., Klockgether T., Wuellner U., Evert B.O. FOXO4-dependent upregulation of superoxide dismutase-2 in response to oxidative stress is impaired in spinocerebellar ataxia type 3. Hum. Mol. Genet. 2011;20:2928–2941. doi: 10.1093/hmg/ddr197. [DOI] [PubMed] [Google Scholar]
- 210.Scherz-Shouval R., Shvets E., Fass E., Shorer H., Gil L., Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 212.Hariharan N., Maejima Y., Nakae J., Paik J., Depinho R.A., Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 2010;107:1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S.J., Di Lisi R., Sandri C., Zhao J., Goldberg A.L., Schiaffino S., Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 214.Lee D., Goldberg A.L. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J. Biol. Chem. 2013;288:30515–30526. doi: 10.1074/jbc.M113.489716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zhao Y., Yang J., Liao W., Liu X., Zhang H., Wang S., Wang D., Feng J., Yu L., Zhu W.G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010;12:665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 216.Peserico A., Chiacchiera F., Grossi V., Matrone A., Latorre D., Simonatto M., Fusella A., Ryall J.G., Finley L.W., Haigis M.C., Villani G., Puri P.L., Sartorelli V., Simone C. A novel AMPK-dependent FoxO3A-SIRT3 intramitochondrial complex sensing glucose levels. Cell. Mol. Life Sci. 2013;70:2015–2029. doi: 10.1007/s00018-012-1244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Chen I.C., Chiang W.F., Chen P.F., Chiang H.C. STRESS-responsive deacetylase SIRT3 is up-regulated by areca nut extract-induced oxidative stress in human oral keratinocytes. J. Cell. Biochem. 2014;115:328–339. doi: 10.1002/jcb.24667. [DOI] [PubMed] [Google Scholar]
- 218.Tseng A.H., Shieh S.S., Wang D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013;63:222–234. doi: 10.1016/j.freeradbiomed.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 219.Tseng A.H., Wu L.H., Shieh S.S., Wang D.L. SIRT3 interactions with FOXO3 acetylation, phosphorylation and ubiquitinylation mediate endothelial cell responses to hypoxia. Biochem. J. 2014;464:157–168. doi: 10.1042/BJ20140213. [DOI] [PubMed] [Google Scholar]
- 220.Mercken E.M., Crosby S.D., Lamming D.W., JeBailey L., Krzysik-Walker S., Villareal D.T., Capri M., Franceschi C., Zhang Y., Becker K., Sabatini D.M., de Cabo R., Fontana L. Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell. 2013;12:645–651. doi: 10.1111/acel.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Cristancho A.G., Lazar M.A. Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell. Biol. 2011;12:722–734. doi: 10.1038/nrm3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ducluzeau P.H., Priou M., Weitheimer M., Flamment M., Duluc L., Iacobazi F., Soleti R., Simard G., Durand A., Rieusset J., Andriantsitohaina R., Malthiery Y. Dynamic regulation of mitochondrial network and oxidative functions during 3T3-L1 fat cell differentiation. J. Physiol. Biochem. 2011;67:285–296. doi: 10.1007/s13105-011-0074-6. [DOI] [PubMed] [Google Scholar]
- 223.Lee H., Lee Y.J., Choi H., Ko E.H., Kim J.W. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J. Biol. Chem. 2009;284:10601–10609. doi: 10.1074/jbc.M808742200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rajalin A.M., Micoogullari M., Sies H., Steinbrenner H. Upregulation of the thioredoxin-dependent redox system during differentiation of 3T3-L1 cells to adipocytes. Biol. Chem. 2014;395:667–677. doi: 10.1515/hsz-2014-0102. [DOI] [PubMed] [Google Scholar]
- 225.Kelly T., Yang W., Chen C.S., Reynolds K., He J. Global burden of obesity in 2005 and projections to 2030. Int. J. Obes. (Lond.) 2008;32:1431–1437. doi: 10.1038/ijo.2008.102. [DOI] [PubMed] [Google Scholar]
- 226.Wild S., Roglic G., Green A., Sicree R., King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 227.Rochette L., Zeller M., Cottin Y., Vergely C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta. 2014;1840:2709–2729. doi: 10.1016/j.bbagen.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 228.Tiganis T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol. Sci. 2011;32:82–89. doi: 10.1016/j.tips.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 229.Tiedge M., Lortz S., Drinkgern J., Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46:1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- 230.Sena C.M., Pereira A.M., Seica R. Endothelial dysfunction – a major mediator of diabetic vascular disease. Biochim. Biophys. Acta. 2013;1832:2216–2231. doi: 10.1016/j.bbadis.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 231.Nakae J., Biggs W.H., 3rd, Kitamura T., Cavenee W.K., Wright C.V., Arden K.C., Accili D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat. Genet. 2002;32:245–253. doi: 10.1038/ng890. [DOI] [PubMed] [Google Scholar]
- 232.Altomonte J., Cong L., Harbaran S., Richter A., Xu J., Meseck M., Dong H.H. Foxo1 mediates insulin action on apoC-III and triglyceride metabolism. J. Clin. Investig. 2004;114:1493–1503. doi: 10.1172/JCI19992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Gross D.N., van den Heuvel A.P., Birnbaum M.J. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- 234.Wang Y., Zhou Y., Graves D.T. FOXO transcription factors: their clinical significance and regulation. Biomed. Res. Int. 2014:925350. doi: 10.1155/2014/925350. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zhang K., Li L., Qi Y., Zhu X., Gan B., DePinho R.A., Averitt T., Guo S. Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology. 2012;153:631–646. doi: 10.1210/en.2011-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Behl Y., Krothapalli P., Desta T., Roy S., Graves D.T. FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes. 2009;58:917–925. doi: 10.2337/db08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Alblowi J., Kayal R.A., Siqueira M., McKenzie E., Krothapalli N., McLean J., Conn J., Nikolajczyk B., Einhorn T.A., Gerstenfeld L., Graves D.T. High levels of tumor necrosis factor-alpha contribute to accelerated loss of cartilage in diabetic fracture healing. Am. J. Pathol. 2009;175:1574–1585. doi: 10.2353/ajpath.2009.090148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kim-Muller J.Y., Zhao S., Srivastava S., Mugabo Y., Noh H.L., Kim Y.R., Madiraju S.R., Ferrante A.W., Skolnik E.Y., Prentki M., Accili D. Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab. 2014;20:593–602. doi: 10.1016/j.cmet.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Massillon D., Barzilai N., Chen W., Hu M., Rossetti L. Glucose regulates in vivo glucose-6-phosphatase gene expression in the liver of diabetic rats. J Biol Chem. 1996;271:9871–9874. doi: 10.1074/jbc.271.17.9871. [DOI] [PubMed] [Google Scholar]
- 240.Yoon J.C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C.R., Granner D.K., Newgard C.B., Spiegelman B.M. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 241.Housley M.P., Udeshi N.D., Rodgers J.T., Shabanowitz J., Puigserver P., Hunt D.F., Hart G.W. A PGC-1alpha-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J. Biol. Chem. 2009;284:5148–5157. doi: 10.1074/jbc.M808890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Speckmann B., Sies H., Steinbrenner H. Attenuation of hepatic expression and secretion of selenoprotein P by metformin. Biochem. Biophys. Res. Commun. 2009;387:158–163. doi: 10.1016/j.bbrc.2009.06.143. [DOI] [PubMed] [Google Scholar]
- 243.Misu H., Takamura T., Takayama H., Hayashi H., Matsuzawa-Nagata N., Kurita S., Ishikura K., Ando H., Takeshita Y., Ota T., Sakurai M., Yamashita T., Mizukoshi E., Yamashita T., Honda M., Miyamoto K., Kubota T., Kubota N., Kadowaki T., Kim H.J., Lee I.K., Minokoshi Y., Saito Y., Takahashi K., Yamada Y., Takakura N., Kaneko S. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010;12:483–495. doi: 10.1016/j.cmet.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 244.Zhang W., Hietakangas V., Wee S., Lim S.C., Gunaratne J., Cohen S.M. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 2013;27:441–449. doi: 10.1101/gad.201731.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Haeusler R.A., Hartil K., Vaitheesvaran B., Arrieta-Cruz I., Knight C.M., Cook J.R., Kammoun H.L., Febbraio M.A., Gutierrez-Juarez R., Kurland I.J., Accili D. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat. Commun. 2014;5:5190. doi: 10.1038/ncomms6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Sugita S., Arai Y., Tonooka A., Hama N., Totoki Y., Fujii T., Aoyama T., Asanuma H., Tsukahara T., Kaya M., Shibata T., Hasegawa T. A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am. J. Surg. Pathol. 2014;38:1571–1576. doi: 10.1097/PAS.0000000000000286. [DOI] [PubMed] [Google Scholar]
- 247.Xie L., Ushmorov A., Leithauser F., Guan H., Steidl C., Farbinger J., Pelzer C., Vogel M.J., Maier H.J., Gascoyne R.D., Moller P., Wirth T. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood. 2012;119:3503–3511. doi: 10.1182/blood-2011-09-381905. [DOI] [PubMed] [Google Scholar]
- 248.Korani M., Fallah S., Tehranian A., Nourbakhsh M., Samadikuchaksaraei A., Pour M.S., Maleki J. The evaluation of the FOXO1, KLF9 and YT521 genes expression in human endometrial cancer. Clin. Lab. 2013;59:483–489. doi: 10.7754/clin.lab.2012.120626. [DOI] [PubMed] [Google Scholar]
- 249.Goto T., Takano M., Hirata J., Tsuda H. The involvement of FOXO1 in cytotoxic stress and drug-resistance induced by paclitaxel in ovarian cancers. Br. J. Cancer. 2008;98:1068–1075. doi: 10.1038/sj.bjc.6604279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Paik J.H., Kollipara R., Chu G., Ji H., Xiao Y., Ding Z., Miao L., Tothova Z., Horner J.W., Carrasco D.R., Jiang S., Gilliland D.G., Chin L., Wong W.H., Castrillon D.H., DePinho R.A. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Qinyu L., Long C., Zhen-dong D., Min-min S., Wei-ze W., Wei-ping Y., Cheng-hong P. FOXO6 promotes gastric cancer cell tumorigenicity via upregulation of C-myc. FEBS Lett. 2013;587:2105–2111. doi: 10.1016/j.febslet.2013.05.027. [DOI] [PubMed] [Google Scholar]
- 252.Aykin-Burns N., Ahmad I.M., Zhu Y., Oberley L.W., Spitz D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009;418:29–37. doi: 10.1042/BJ20081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sun W., Kalen A.L., Smith B.J., Cullen J.J., Oberley L.W. Enhancing the antitumor activity of adriamycin and ionizing radiation. Cancer Res. 2009;69:4294–4300. doi: 10.1158/0008-5472.CAN-09-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Harris I.S., Blaser H., Moreno J., Treloar A.E., Gorrini C., Sasaki M., Mason J.M., Knobbe C.B., Rufini A., Halle M., Elia A.J., Wakeham A., Tremblay M.L., Melino G., Done S., Mak T.W. PTPN12 promotes resistance to oxidative stress and supports tumorigenesis by regulating FOXO signaling. Oncogene. 2014;33:1047–1054. doi: 10.1038/onc.2013.24. [DOI] [PubMed] [Google Scholar]
- 255.Wang G.J., Liu G.H., Ye Y.W., Fu Y., Zhang X.F. The role of microRNA-1274a in the tumorigenesis of gastric cancer: accelerating cancer cell proliferation and migration via directly targeting FOXO4. Biochem. Biophys. Res. Commun. 2015;459:629–635. doi: 10.1016/j.bbrc.2015.02.160. [DOI] [PubMed] [Google Scholar]
- 256.Mao X.P., Zhang L.S., Huang B., Zhou S.Y., Liao J., Chen L.W., Qiu S.P., Chen J.X. Mir-135a enhances cellular proliferation through post-transcriptionally regulating PHLPP2 and FOXO1 in human bladder cancer. J. Transl. Med. 2015;13:86. doi: 10.1186/s12967-015-0438-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kim S.Y., Yoon J., Ko Y.S., Chang M.S., Park J.W., Lee H.E., Kim M.A., Kim J.H., Kim W.H., Lee B.L. Constitutive phosphorylation of the FOXO1 transcription factor in gastric cancer cells correlates with microvessel area and the expressions of angiogenesis-related molecules. BMC Cancer. 2011;11:264. doi: 10.1186/1471-2407-11-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Pramanik K.C., Fofaria N.M., Gupta P., Srivastava S.K. CBP-mediated FOXO-1 acetylation inhibits pancreatic tumor growth by targeting SirT. Mol. Cancer Ther. 2014;13:687–698. doi: 10.1158/1535-7163.MCT-13-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Peck B., Ferber E.C., Schulze A. Antagonism between FOXO and MYC regulates cellular powerhouse. Front. Oncol. 2013;3:96. doi: 10.3389/fonc.2013.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ferber E.C., Peck B., Delpuech O., Bell G.P., East P., Schulze A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012;19:968–979. doi: 10.1038/cdd.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Jensen K.S., Binderup T., Jensen K.T., Therkelsen I., Borup R., Nilsson E., Multhaupt H., Bouchard C., Quistorff B., Kjaer A., Landberg G., Staller P. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 2011;30:4554–4570. doi: 10.1038/emboj.2011.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Loguercio C., Federico A. Oxidative stress in viral and alcoholic hepatitis. Free Radic. Biol. Med. 2003;34:1–10. doi: 10.1016/s0891-5849(02)01167-x. [DOI] [PubMed] [Google Scholar]
- 263.Diesen D.L., Kuo P.C. Nitric oxide and redox regulation in the liver: Part I. General considerations and redox biology in hepatitis. J. Surg. Res. 2010;162:95–109. doi: 10.1016/j.jss.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Dey A., Cederbaum A.I. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–S74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 265.Muriel P. Role of free radicals in liver diseases. Hepatol. Int. 2009;3:526–536. doi: 10.1007/s12072-009-9158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Tao G.Z., Lehwald N., Jang K.Y., Baek J., Xu B., Omary M.B., Sylvester K.G. Wnt/beta-catenin signaling protects mouse liver against oxidative stress-induced apoptosis through the inhibition of forkhead transcription factor FoxO3. J. Biol. Chem. 2013;288:17214–17224. doi: 10.1074/jbc.M112.445965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Kim P.J., Plescia J., Clevers H., Fearon E.R., Altieri D.C. Survivin and molecular pathogenesis of colorectal cancer. Lancet. 2003;362:205–209. doi: 10.1016/S0140-6736(03)13910-4. [DOI] [PubMed] [Google Scholar]
- 268.Tapia J.C., Torres V.A., Rodriguez D.A., Leyton L., Quest A.F. Casein kinase 2 (CK2) increases survivin expression via enhanced beta-catenin-T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc. Natl. Acad. Sci. U.S.A. 2006;103:15079–15084. doi: 10.1073/pnas.0606845103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Dehner M., Hadjihannas M., Weiske J., Huber O., Behrens J. Wnt signaling inhibits Forkhead box O3a-induced transcription and apoptosis through up-regulation of serum- and glucocorticoid-inducible kinase 1. J. Biol. Chem. 2008;283:19201–19210. doi: 10.1074/jbc.M710366200. [DOI] [PubMed] [Google Scholar]
- 270.Tenbaum S.P., Ordonez-Moran P., Puig I., Chicote I., Arques O., Landolfi S., Fernandez Y., Herance J.R., Gispert J.D., Mendizabal L., Aguilar S., Ramon y Cajal S., Schwartz S., Jr., Vivancos A., Espin E., Rojas S., Baselga J., Tabernero J., Munoz A., Palmer H.G. beta-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat. Med. 2012;18:892–901. doi: 10.1038/nm.2772. [DOI] [PubMed] [Google Scholar]
- 271.Yan Y., Lackner M.R. FOXO3a and beta-catenin co-localization: double trouble in colon cancer? Nat. Med. 2012;18:854–856. doi: 10.1038/nm.2799. [DOI] [PubMed] [Google Scholar]
- 272.Yamaza H., Komatsu T., Wakita S., Kijogi C., Park S., Hayashi H., Chiba T., Mori R., Furuyama T., Mori N., Shimokawa I. FoxO1 is involved in the antineoplastic effect of calorie restriction. Aging Cell. 2010;9:372–382. doi: 10.1111/j.1474-9726.2010.00563.x. [DOI] [PubMed] [Google Scholar]
- 273.Masui K., Tanaka K., Akhavan D., Babic I., Gini B., Matsutani T., Iwanami A., Liu F., Villa G.R., Gu Y., Campos C., Zhu S., Yang H., Yong W.H., Cloughesy T.F., Mellinghoff I.K., Cavenee W.K., Shaw R.J., Mischel P.S. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18:726–739. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Mihaylova M.M., Vasquez D.S., Ravnskjaer K., Denechaud P.D., Yu R.T., Alvarez J.G., Downes M., Evans R.M., Montminy M., Shaw R.J. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–621. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Senf S.M., Sandesara P.B., Reed S.A., Judge A.R. p300 Acetyltransferase activity differentially regulates the localization and activity of the FOXO homologues in skeletal muscle. Am. J. Physiol. Cell Physiol. 2011;300:C1490–C1501. doi: 10.1152/ajpcell.00255.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Bertaggia E., Coletto L., Sandri M. Posttranslational modifications control FoxO3 activity during denervation. Am. J. Physiol. Cell Physiol. 2012;302:C587–C596. doi: 10.1152/ajpcell.00142.2011. [DOI] [PubMed] [Google Scholar]
- 277.Beharry A.W., Sandesara P.B., Roberts B.M., Ferreira L.F., Senf S.M., Judge A.R. HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J. Cell Sci. 2014;127:1441–1453. doi: 10.1242/jcs.136390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Sandri M., Barberi L., Bijlsma A.Y., Blaauw B., Dyar K.A., Milan G., Mammucari C., Meskers C.G., Pallafacchina G., Paoli A., Pion D., Roceri M., Romanello V., Serrano A.L., Toniolo L., Larsson L., Maier A.B., Munoz-Canoves P., Musaro A., Pende M., Reggiani C., Rizzuto R., Schiaffino S. Signalling pathways regulating muscle mass in ageing skeletal muscle: the role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology. 2013;14:303–323. doi: 10.1007/s10522-013-9432-9. [DOI] [PubMed] [Google Scholar]
- 279.Owusu-Ansah E., Song W., Perrimon N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 2013;155:699–712. doi: 10.1016/j.cell.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Ambrogini E., Almeida M., Martin-Millan M., Paik J.H., Depinho R.A., Han L., Goellner J., Weinstein R.S., Jilka R.L., O'Brien C.A., Manolagas S.C. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 2010;11:136–146. doi: 10.1016/j.cmet.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Rached M.T., Kode A., Xu L., Yoshikawa Y., Paik J.H., Depinho R.A., Kousteni S. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 2010;11:147–160. doi: 10.1016/j.cmet.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Garrett I.R., Boyce B.F., Oreffo R.O., Bonewald L., Poser J., Mundy G.R. Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J. Clin. Investig. 1990;85:632–639. doi: 10.1172/JCI114485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Lee N.K., Choi Y.G., Baik J.Y., Han S.Y., Jeong D.W., Bae Y.S., Kim N., Lee S.Y. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106:852–859. doi: 10.1182/blood-2004-09-3662. [DOI] [PubMed] [Google Scholar]
- 284.Bhatt N.Y., Kelley T.W., Khramtsov V.V., Wang Y., Lam G.K., Clanton T.L., Marsh C.B. Macrophage-colony-stimulating factor-induced activation of extracellular-regulated kinase involves phosphatidylinositol 3-kinase and reactive oxygen species in human monocytes. J. Immunol. 2002;169:6427–6434. doi: 10.4049/jimmunol.169.11.6427. [DOI] [PubMed] [Google Scholar]
- 285.Manolagas S.C. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010;31:266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Bartell S.M., Kim H.N., Ambrogini E., Han L., Iyer S., Serra Ucer S., Rabinovitch P., Jilka R.L., Weinstein R.S., Zhao H., O'Brien C.A., Manolagas S.C., Almeida M. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat. Commun. 2014;5:3773. doi: 10.1038/ncomms4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Akasaki Y., Hasegawa A., Saito M., Asahara H., Iwamoto Y., Lotz M.K. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthr. Cartil. 2014;22:162–170. doi: 10.1016/j.joca.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Akasaki Y., Alvarez-Garcia O., Saito M., Carames B., Iwamoto Y., Lotz M.K. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol. 2014;66:3349–3358. doi: 10.1002/art.38868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Jallali N., Ridha H., Thrasivoulou C., Underwood C., Butler P.E., Cowen T. Vulnerability to ROS-induced cell death in ageing articular cartilage: the role of antioxidant enzyme activity. Osteoarthrit. Cartil. 2005;13:614–622. doi: 10.1016/j.joca.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 290.Renault V.M., Rafalski V.A., Morgan A.A., Salih D.A., Brett J.O., Webb A.E., Villeda S.A., Thekkat P.U., Guillerey C., Denko N.C., Palmer T.D., Butte A.J., Brunet A. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell. 2009;5:527–539. doi: 10.1016/j.stem.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Paik J.H., Ding Z., Narurkar R., Ramkissoon S., Muller F., Kamoun W.S., Chae S.S., Zheng H., Ying H., Mahoney J., Hiller D., Jiang S., Protopopov A., Wong W.H., Chin L., Ligon K.L., DePinho R.A. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell. 2009;5:540–553. doi: 10.1016/j.stem.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Neri C. Role and therapeutic potential of the pro-longevity factor FOXO and its regulators in neurodegenerative disease. Front. Pharmacol. 2012;3:15. doi: 10.3389/fphar.2012.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Tothova Z., Kollipara R., Huntly B.J., Lee B.H., Castrillon D.H., Cullen D.E., McDowell E.P., Lazo-Kallanian S., Williams I.R., Sears C., Armstrong S.A., Passegue E., DePinho R.A., Gilliland D.G. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 294.Wang X., Wang Z., Chen Y., Huang X., Hu Y., Zhang R., Ho M.S., Xue L. FoxO mediates APP-induced AICD-dependent cell death. Cell Death Dis. 2014;5:e1233. doi: 10.1038/cddis.2014.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Belanger M., Magistretti P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009;11:281–295. doi: 10.31887/DCNS.2009.11.3/mbelanger. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Owusu-Ansah E., Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Tsuchiya K., Westerterp M., Murphy A.J., Subramanian V., Ferrante A.W., Jr., Tall A.R., Accili D. Expanded granulocyte/monocyte compartment in myeloid-specific triple FoxO knockout increases oxidative stress and accelerates atherosclerosis in mice. Circ. Res. 2013;112:992–1003. doi: 10.1161/CIRCRESAHA.112.300749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Mortuza R., Chen S., Feng B., Sen S., Chakrabarti S. High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS One. 2013;8:e54514. doi: 10.1371/journal.pone.0054514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Zhang S., Zhao Y., Xu M., Yu L., Zhao Y., Chen J., Yuan Y., Zheng Q., Niu X. FoxO3a modulates hypoxia stress induced oxidative stress and apoptosis in cardiac microvascular endothelial cells. PLoS One. 2013;8:e80342. doi: 10.1371/journal.pone.0080342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Yiu W.H., Mead P.A., Jun H.S., Mansfield B.C., Chou J.Y. Oxidative stress mediates nephropathy in type Ia glycogen storage disease. Lab. Investig. 2010;90:620–629. doi: 10.1038/labinvest.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Lu Q., Zhai Y., Cheng Q., Liu Y., Gao X., Zhang T., Wei Y., Zhang F., Yin X. The Akt-FoxO3a-manganese superoxide dismutase pathway is involved in the regulation of oxidative stress in diabetic nephropathy. Exp. Physiol. 2013;98:934–945. doi: 10.1113/expphysiol.2012.068361. [DOI] [PubMed] [Google Scholar]
- 302.Ponugoti B., Xu F., Zhang C., Tian C., Pacios S., Graves D.T. FOXO1 promotes wound healing through the up-regulation of TGF-beta1 and prevention of oxidative stress. J. Cell Biol. 2013;203:327–343. doi: 10.1083/jcb.201305074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Ponugoti B., Dong G., Graves D.T. Role of forkhead transcription factors in diabetes-induced oxidative stress. Exp. Diabetes Res. 2012:939751. doi: 10.1155/2012/939751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Hanks S.K., Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 305.Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 306.Pearce L.R., Komander D., Alessi D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell. Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 307.Rena G., Guo S., Cichy S.C., Unterman T.G., Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 1999;274:17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 308.Guo S., Rena G., Cichy S., He X., Cohen P., Unterman T. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem. 1999;274:17184–17192. doi: 10.1074/jbc.274.24.17184. [DOI] [PubMed] [Google Scholar]
- 309.Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S., Anderson M.J., Arden K.C., Blenis J., Greenberg M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 310.Kops G.J., de Ruiter N.D., De Vries-Smits A.M., Powell D.R., Bos J.L., Burgering B.M. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- 311.Takaishi H., Konishi H., Matsuzaki H., Ono Y., Shirai Y., Saito N., Kitamura T., Ogawa W., Kasuga M., Kikkawa U., Nishizuka Y. Regulation of nuclear translocation of forkhead transcription factor AFX by protein kinase B. Proc. Natl. Acad. Sci. U.S.A. 1999;96:11836–11841. doi: 10.1073/pnas.96.21.11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Jacobs F.M., van der Heide L.P., Wijchers P.J., Burbach J.P., Hoekman M.F., Smidt M.P. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J. Biol. Chem. 2003;278:35959–35967. doi: 10.1074/jbc.M302804200. [DOI] [PubMed] [Google Scholar]
- 313.van der Heide L.P., Jacobs F.M., Burbach J.P., Hoekman M.F., Smidt M.P. FoxO6 transcriptional activity is regulated by Thr26 and Ser184, independent of nucleo-cytoplasmic shuttling. Biochem. J. 2005;391:623–629. doi: 10.1042/BJ20050525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Brunet A., Park J., Tran H., Hu L.S., Hemmings B.A., Greenberg M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol. Cell. Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Lee J.W., Chen H., Pullikotil P., Quon M.J. Protein kinase A-alpha directly phosphorylates FoxO1 in vascular endothelial cells to regulate expression of vascular cellular adhesion molecule-1 mRNA. J. Biol. Chem. 2011;286:6423–6432. doi: 10.1074/jbc.M110.180661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Kannan N., Neuwald A.F. Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2alpha. Protein Sci. 2004;13:2059–2077. doi: 10.1110/ps.04637904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Clavel S., Siffroi-Fernandez S., Coldefy A.S., Boulukos K., Pisani D.F., Derijard B. Regulation of the intracellular localization of Foxo3a by stress-activated protein kinase signaling pathways in skeletal muscle cells. Mol. Cell. Biol. 2010;30:470–480. doi: 10.1128/MCB.00666-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Yuan Z., Becker E.B., Merlo P., Yamada T., DiBacco S., Konishi Y., Schaefer E.M., Bonni A. Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science. 2008;319:1665–1668. doi: 10.1126/science.1152337. [DOI] [PubMed] [Google Scholar]
- 319.Rena G., Woods Y.L., Prescott A.R., Peggie M., Unterman T.G., Williams M.R., Cohen P. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J. 2002;21:2263–2271. doi: 10.1093/emboj/21.9.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Kim S., Kim Y., Lee J., Chung J. Regulation of FOXO1 by TAK1-Nemo-like kinase pathway. J. Biol. Chem. 2010;285:8122–8129. doi: 10.1074/jbc.M110.101824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Vielhaber E., Virshup D.M. Casein kinase I: from obscurity to center stage. IUBMB Life. 2001;51:73–78. doi: 10.1080/15216540117461. [DOI] [PubMed] [Google Scholar]
- 322.Chow K.T., Timblin G.A., McWhirter S.M., Schlissel M.S. MK5 activates Rag transcription via Foxo1 in developing B cells. J. Exp. Med. 2013;210:1621–1634. doi: 10.1084/jem.20130498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Kress T.R., Cannell I.G., Brenkman A.B., Samans B., Gaestel M., Roepman P., Burgering B.M., Bushell M., Rosenwald A., Eilers M. The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol. Cell. 2011;41:445–457. doi: 10.1016/j.molcel.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 324.Lehtinen M.K., Yuan Z., Boag P.R., Yang Y., Villen J., Becker E.B., DiBacco S., de la Iglesia N., Gygi S., Blackwell T.K., Bonni A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 325.Hu M.C., Lee D.F., Xia W., Golfman L.S., Ou-Yang F., Yang J.Y., Zou Y., Bao S., Hanada N., Saso H., Kobayashi R., Hung M.C. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 326.So C.W., Cleary M.L. MLL-AFX requires the transcriptional effector domains of AFX to transform myeloid progenitors and transdominantly interfere with forkhead protein function. Mol. Cell. Biol. 2002;22:6542–6552. doi: 10.1128/MCB.22.18.6542-6552.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wang F., Marshall C.B., Yamamoto K., Li G.Y., Plevin M.J., You H., Mak T.W., Ikura M. Biochemical and structural characterization of an intramolecular interaction in FOXO3a and its binding with p53. J. Mol. Biol. 2008;384:590–603. doi: 10.1016/j.jmb.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 328.Boura E., Silhan J., Herman P., Vecer J., Sulc M., Teisinger J., Obsilova V., Obsil T. Both the N-terminal loop and wing W2 of the forkhead domain of transcription factor Foxo4 are important for DNA binding. J. Biol. Chem. 2007;282:8265–8275. doi: 10.1074/jbc.M605682200. [DOI] [PubMed] [Google Scholar]
- 329.Obsilova V., Vecer J., Herman P., Pabianova A., Sulc M., Teisinger J., Boura E., Obsil T. 14-3-3 Protein interacts with nuclear localization sequence of forkhead transcription factor FoxO4. Biochemistry. 2005;44:11608–11617. doi: 10.1021/bi050618r. [DOI] [PubMed] [Google Scholar]
- 330.Biggs W.H., 3rd, Meisenhelder J., Hunter T., Cavenee W.K., Arden K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Brunet A., Kanai F., Stehn J., Xu J., Sarbassova D., Frangioni J.V., Dalal S.N., DeCaprio J.A., Greenberg M.E., Yaffe M.B. 14-3-3 Transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J. Cell Biol. 2002;156:817–828. doi: 10.1083/jcb.200112059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Brownawell A.M., Kops G.J., Macara I.G., Burgering B.M. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol. Cell. Biol. 2001;21:3534–3546. doi: 10.1128/MCB.21.10.3534-3546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M., Murphy M.R., O'Leary N.A., Pujar S., Rajput B., Rangwala S.H., Riddick L.D., Shkeda A., Sun H., Tamez P., Tully R.E., Wallin C., Webb D., Weber J., Wu W., DiCuccio M., Kitts P., Maglott D.R., Murphy T.D., Ostell J.M. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]