

Abstract
S-nitrosoglutathione (GSNO) is an endogenous nitric oxide (NO) carrier that plays a critical role in redox based NO signaling. Recent studies have reported that GSNO regulates the activities of STAT3 and NF-κB via S-nitrosylation dependent mechanisms. Since STAT3 and NF-κB are key transcription factors involved in tumor progression, chemoresistance, and metastasis of head and neck cancer, we investigated the effect of GSNO in cell culture and mouse xenograft models of head and neck squamous cell carcinoma (HNSCC). For the cell culture studies, three HNSCC cell lines were tested (SCC1, SCC14a and SCC22a). All three cell lines had constitutively activated (phosphorylated) STAT3 (Tyr705). GSNO treatment of these cell lines reversibly decreased the STAT3 phosphorylation in a concentration dependent manner. GSNO treatment also decreased the basal and cytokine-stimulated activation of NF-κB in SCC14a cells and reduced the basal low degree of nitrotyrosine by inhibition of inducible NO synthase (iNOS) expression. The reduced STAT3/NF-κB activity by GSNO treatment was correlated with the decreased cell proliferation and increased apoptosis of HNSCC cells. In HNSCC mouse xenograft model, the tumor growth was reduced by systemic treatment with GSNO and was further reduced when the treatment was combined with radiation and cisplatin. Accordingly, GSNO treatment also resulted in decreased levels of phosphorylated STAT3. In summary, these studies demonstrate that GSNO treatment blocks the NF-κB and STAT3 pathways which are responsible for cell survival, proliferation and that GSNO mediated mechanisms complement cispaltin and radiation therapy, and thus could potentiate the therapeutic effect in HNSCC.
Keywords: S-Nitrosoglutathione, STAT3, NF-κB, Head and neck cancer, Radiotherapy, Cisplatin
Graphical abstract
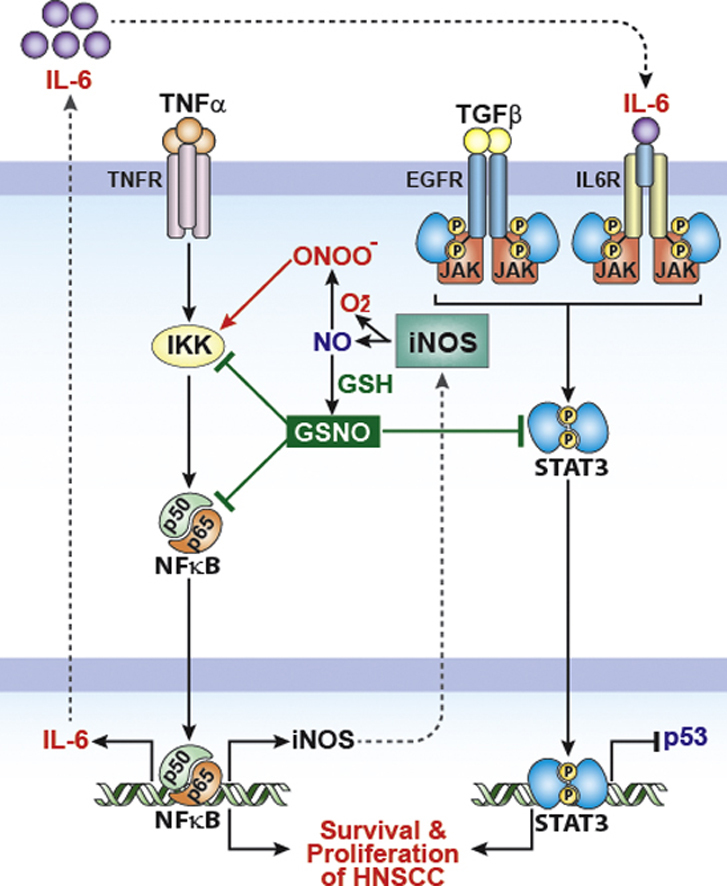
Highlights
-
•
S-nitrosoglutathione (GSNO) inhibits activations of STAT3 and NF-κB in HNSCC cells.
-
•
GSNO induces cell cycle arrest and apoptosis of HNSCC cells.
-
•
GSNO decreases iNOS and VEGF production in HNSCC cells.
-
•
GSNO enhances efficacy of chemoradiation therapy in HNSCC mouse xenograft model.
1. Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer and one of the leading causes of cancer deaths worldwide. Recent advances in the field of HNSCC include development of free flaps for reconstruction after surgery or intensity modulated radiation therapy for targeted radiotherapy [1,2]. However, the prognosis for these patients has not improved significantly. Therefore, better agents are needed to improve therapeutic outcomes.
Aberrant or constitutive activation of STAT3 and NF-κB has been detected in many human malignancies including HNSCC [3,4,6]. STAT3 and NF-κB play key roles in the regulation of immune/inflammatory responses, but growing evidence also supports a major role in oncogenesis [3,7]. Molecular targeting of STAT3 by various methods, such as interfering in dimerization and siRNA approaches, has been shown to inhibit tumor growth in preclinical models of human cancer [8,9]. However, these are not considered drug candidates because of uncertain feasibility of these approaches in clinical practice. NF-κB has been implicated in cancer progression by activation genes that stimulate cancer cell proliferation and survival, angiogenesis, and metastases, and thus is considered an interesting therapeutic target for treatment of cancer [4]. However, presently there is no effective treatment strategy for managing the aberrant NF-κB activity in cancers.
While studying the inflammatory pathways in cell culture and animal models of multiple sclerosis (experimental autoimmune encephalomyelitis) and stroke (middle cerebral artery occlusion), our laboratory reported that NF-κB activation under inflammatory conditions is inhibited by S-nitrosylated glutathione (GSNO) treatment [10,11]. Since NF-κB has been also reported to be regulated by glutathionylation [12,13], these studies document the roles of the cellular GSNO as well as redox potential in regulation of NF-κB pathways. In addition, we have also recently reported that STAT3 activation induced by IL-6 is inhibited by endogenously generated nitric oxide (NO) from inducible NO synthase (iNOS) or exogenously supplemented GSNO via S-nitrosylation of Cysteine 259 residue of STAT3 molecule, documenting that STAT3 S-nitrosylation is a physiological regulation [14]. We have also reported that inhibition of STAT3 by GSNO is implicated in TH17 immunomodulation under EAE conditions [15] and anti-proliferation of microglia and cancer cells [16,17]. GSNO is an endogenous NO carrier which plays a crucial role in redox based nitrosylation of protein thiols (S-nitrosylation) in health and disease [18] and is now recognized as an important cell signaling mechanism [19]. Although NF-κB and STAT3 have been implicated in tumorigenesis, metastasis, and chemoresistance [7,20], the biological function of NO and GSNO in regulation of NF-κB and STAT3 in cancer cells and their associated cell signaling pathways leading to cancer progression are not understood well at present.
NO, synthesized by nitric oxide synthases (NOS), is shown to have both pro- and anti-tumor activity [21–28]. These multifactorial effects may be manifested via a range of chemical modifications caused by different properties of the source of NO levels and its metabolites (NO2, NO+, and NO•) in nitration, nitrosation or nitrosylation of target molecules as well as the redox environment of the tumor. However, the observed chemosensitization of tumor by NO in doxorubicin resistance by nitroglycerine [27,28], efficacies of NO-nonsteroidal anti-inflammatory drugs [29] as well as direct efficacy of NO compounds in epithelial to messenchymal transition in cancer cells [25], and inhibition of cell proliferation or tumor growth by GSNO treatment in multiple myeloma cell lines [16] and ovarian cancer xenografted mouse models [17] suggests that NO compounds may be considered as potential therapeutic agents for blocking the metastasis cascade and for treatment of patients with refractory cancer. Understanding the different actions of NO and its metabolites in cancer at the molecular level can provide insights in devising potential strategies for cancer treatment.
Based on the fact that STAT3 and NF-κB are involved in tumor growth, angiogenesis and invasion and that GSNO, an endogenous nitrosylating agent, can inhibit activation of STAT3 [15] and NF-κB [10,30], we investigated the efficacy of GSNO in head and neck cancer using HNSCC cells in vitro and in vivo xenograft models. The studies described in this manuscript document that GSNO treatment inhibits cell survival and proliferation in in vitro cultures of HNSCC cells and also inhibits tumor growth in the xenograft animal model. Moreover, GSNO mediated mechanisms against tumor growth complement ionizing radiation and cisplatin treatments to provide greater efficacy. This study for the first time, documents that GSNO mediated inactivation of STAT3 signaling pathway may prove to be of therapeutic potential in the treatment of head and neck cancer.
2. Materials and methods
2.1. Cell cultures
HNSCC cell lines UM-SCC-1 (retromolar trigone/floor of the mouth; SCC1), UM-SCC-14A (SCC of anterior floor of the mouth; SCC14a), and UM-SCC-22A (SCC of hypopharynx; SCC22a) were obtained from Dr. Thomas Carey (Department of Otolaryngology/Head and Neck Surgery, University of Michigan) [31]. These cells lines were cultured in Dulbecco's Modified Eagle's Medium/Ham's F-12 (50/50) (Mediatech Inc., Manassas, VA) with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/mL streptomycin. The cells were grown at 37 °C under 5% CO2/95% air.
2.2. Cell survival, cell proliferation, and cell cycle assay
The cells in 96-well culture plates in serum free medium were treated with different doses of drugs/radiation. The fractions of the surviving cells were measured by colorimetric assay using WST-8 tetrazolium salt (Cayman Chemical, Ann Arbor, MI). The amount of formazan dye generated from WST-8 by the activity of dehydrogenases in cells was measured by SpectraMax 190 ELISA reader (Molecular Devices, Sunnyvale, CA) at 450 nm. For cell proliferation assay, the cells were treated with 5-bromo-2′-deoxyuridine (BrdU; 10 μM) for 4 h and BrdU incorporated into DNA was measured by colormetric BrdU assay kit (Cell Signaling Technology, Danvers, MA) as per manufacturer's instruction. Cell cycle analysis was performed by flow cytometry following the staining of cells with propidium iodide. Briefly, the ethanol fixed cells were treated with RNase solution and then stained with propidium iodide (50 μg/ml). The samples were analyzed by FACSCalibur™ flow cytometer with Cell QuestTM software (BD Biosciences, San Jose, CA). Expression of cell survival and cell cycle regulators was measured by Western blot analysis using antibodies specific to Bcl-xL, cIAP, and c-Myc (Cell Signaling Technology).
2.3. Assay for STAT3 activation
The activities of STAT3 were analyzed as Western analysis for phosphorylated (Tyr705) STAT3 (pSTAT3) and total STAT3 with specific antibodies (Cell Signaling Technologies, Danvers, MA). For immunofluorescent staining of pSTAT3/STAT3, SCC14a cells cultured on chamber slides (LabTek, Nunc, Inc., Naperville, IL) were fixed in cold methanol and incubated with primary antibodies against phospho- or pan-STAT3 and then secondary antibody (goat anti-Rabbit IgG conjugated with DyLight 488 or 594) (Jackson Immunoresearch Laboratories Inc. West Grove, PA). 4′,6-diamidino-2-phenylindole (DAPI) was used for nuclei counter-stain. Immunofluorescence images were captured by fluorescent microscope equipped with Olympus digital camera (Olympus BX-60, Goleta, CA) and quantified by using Image-Pro 6.2 (Media Cybernetics, Silver Springs, MD). Identical illumination, microscope, and camera settings were used to obtain images for quantification.
2.4. Assay for NF-κB activation and HIF-1α accumulation
Nuclear and cytoplasmic extracts from SCC14a cells treated with GSNO and/or cytokines were prepared using a previously published method [32]. The cytoplasmic and nuclear levels of p65 NF-κB and HIF-1α were analyzed by Western analysis using specific antibodies (Cell Signaling Technologies). H3 histone and β-actin were used for internal loading control for nuclear and cytoplasmic proteins. The nuclear protein extracts were also used for the gel-shift assay for detection of NF-κB DNA binding activities. The gel-shift assay for NF-κB DNA binding activity was performed as described previously [32].
2.5. Generation of HSCC xenograft model using athymic nude mouse and drug treatment
All animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care & Use Committee (IACUC) of the Medical University of South Carolina (Permit number: AR2830). The male athymic nude mouse (Hsd:Athymic Nude-Foxn1nu) of age 5-6 weeks (Harlan Laboratories, Wilmington, MA) were randomly allocated in eight groups (control, radiation, cisplatin, radiation+cisplatin, GSNO, GSNO+radiation, GSNO+cisplatin, GSNO+radiation+cisplatin; n=6). For bioluminescent imaging, SCC14a were stably transfected with firefly luciferase in pcDNA3 plasmid. Tumors were induced by injecting 0.1 ml luciferase expressing SCC14a cell (1×106) suspension into the flank region, sub-cutaneously. The cells were allowed to form solid tumor for two weeks before starting the measurement of the tumor size. The tumor size was then measured in all animals starting from the third week and every week after that. Treatments were started at fourth week of the tumor cell inoculation. For radiotherapy, 8 Gy single dose was treated to the flank region bearing tumor with Clinac 21EX Linear Accelerator once a week for three weeks. For cisplatin chemotherapy, the mice were treated with 2 mg/kg cisplatin in PBS intraperitoneally once a week at about 4–6 h before radiotherapy. For GSNO treatment, the mice were treated with 10 mg/kg GSNO in PBS for five days a week intraperitoneally. The GSNO treatments continued for 6 weeks. In one set of animals, GSNO treatment was withdrawn at 3 weeks of treatment initiation.
2.6. Bioluminescent imaging
Non-invasive measurement of tumor size was performed by bioluminescent imaging of firefly luciferase expressing SCC14a cells every week. Mice were anesthetized by inhalation of 1–2% isofluorane (Aerrane; Baxter Healthcare Corp, Deerfield, IL) and intraperitoneally injected with 200 µL d-luciferin (150 mg/kg; Biosynth AG, Staad, Switzerland). Images were acquired every 2 min for 30 min (10 s exposure/image). Xenogen IVIS 200 bioluminescent/fluorescent imaging system (PerkinElmer, Waltham, MA) was used and the images obtained were analyzed by Living Image® software (PerkinElmer), and the photons emitted was measured as p/s/cm2/sr and calculated as a ratio to the control.
2.7. Statistical analysis
Statistical analysis was performed with Graphpad Prism5. Values are expressed as mean±standard error mean. Comparisons among means of groups were made with a two-tailed Student's t-test for unpaired variables. Multiple comparisons were performed using one-way ANOVA followed by Bonferroni test.
3. Results
3.1. GSNO treatment decreases STAT3 activity (phosphorylation) in HNSCC cells
Based on the fact that activated STAT3 (phosphorylated STAT3) is involved in tumor growth, angiogenesis and invasion and that GSNO, an endogenous nitrosylating agent, can inhibit activation of STAT3 via S-nitrosylation of Cysteine 259 residue [15–17], we examined the effect of GSNO treatment on activation of STAT3 in three human oral cancer cell lines; SCC1, SCC14a and SCC22a. All three cell lines were treated with different concentrations of GSNO twice (12 h interval) and changing the media and harvested at 24 h. Fig. 1A shows that all these HNSCC cell lines had constitutively phosphorylated STAT3 (pSTAT3). GSNO treatment for 24 h decreased the basal pSTAT3 levels in a dose dependent manner. Further, single treatment of SCC14a cells with GSNO resulted in the reversal of the STAT3 phosphorylation over 24 h, indicating that GSNO is continuously required for its inhibitory activity on STAT3 activation and that this effect is reversible (Fig. 1B). The representative images of the STAT3 and pSTAT3 immuno staining of cells with or without GSNO treatment also reveal the inhibition of STAT3 phosphorylation by GSNO (Fig. 1C). These observations suggest that GSNO mediated inhibition of STAT3 activation may inhibit the pro-oncogenic pathways (such as proliferation and survival) in cancer cells.
Fig. 1.

Effect of GSNO treatment on STAT3 activation in HNSCC cells. (A) The HNSCC cells were treated with different concentrations of GSNO for 24 h and then levels of total or phosphorylated (Tyr705) STAT3 (pSTAT3) (i) and their ratios (ii) were analyzed by Western analysis. (B) GSNO mediated reversible inhibition of STAT3 phosphorylation was analyzed in SCC14a cell lines after single treatment with 250 µM GSNO. (C) SCC14a cells were treated with 250 μM GSNO for 3 h and then fixed and labeled with STAT3 or pSTAT3 antibodies (i) and the intensities of fluorescence were quantified (ii). The results in bar and line graphs were given as mean of more than three independent experiments. The vertical lines indicate the standard error of mean; *P<0.05; ***P<0.001; n.s. (not significant) compared with vehicle treated control groups.
3.2. GSNO treatment decreases cell survival and proliferation and inhibits cell cycle progression in HNSCC cells
Next, we investigated the effect of GSNO on proliferation/survival of HNSCC cells. Fig. 2A shows that GSNO treatment decreased the cell number in a concentration dependent manner. We also examined the effect of GSNO treatment on HNSCC cell proliferation by BrdU incorporation assay. As shown in Fig. 2B, GSNO treatment decreased the proliferation of all three HNSCC cell lines tested. The effect of GSNO treatment on cell cycle of SCC14a cells was carried out by flow cytometry of propidium iodide stained cells (Fig. 2C-i). Fig. 2C-ii shows that GSNO treatment significantly increased the percentage of cells in the S and G2 phase while decreasing the proportion of cells in G1 Phase as compared to untreated controls, indicating that the GSNO mediated mechanisms induce cell cycle arrest in S and G2 phases. In addition to cell cycle arrest, GSNO treatment also increased the number of apoptotic cells (Fig. 2C-iii). Consistent with the observed increases in apoptosis and cell cycle arrest, GSNO treatment decreased the expression levels of cell survival and cycle regulators, such as Bcl-xL, cIAP, and c-Myc, which are known to be regulated by STAT3 and NF-κB [33–36]. These studies indicate that GSNO is a potential regulator of cell cycle and cell survival pathways in HNSCC cells.
Fig. 2.

Effect of GSNO treatment on HNSCC cell survival, proliferation, and cell cycle progression. Following the treatment of SCC1, SCC14a and SCC22a HNSCC cell lines with different concentrations of GSNO, the cell survival (A) and proliferation (B) assay were performed as mentioned in the methods. SCC14a cells were treated with 250 μM GSNO for 24 h and then the cell cycle analysis by propidium iodide staining (C-i) was performed. The percentage of cells in each cell cycle phase (C-ii) and apoptosis (C-iii) and effect of GSNO on cell survival and cell cycle regulators (e.g. Bcl-xL, cIAP, and c-Myc) (C-iv) were analyzed by flow cytometry and Western analysis. The results in bar and line graphs were given as mean of at least three independent experiments. The vertical lines indicate the standard deviation; ***P<0.001 compared with vehicle treated control groups.
3.3. GSNO treatment enhances radiation induced HNSCC cell death
Next, we studied whether GSNO mediated mechanisms will complement effect of radiation therapy. To determine whether the GSNO treatment would complement radiotherapy, HNSCC cells were treated with 10 Gy radiation and then immediately treated with GSNO (250 µM; 12 h interval with change of medium) for 24 h, and analyzed for status of STAT3 activity (Fig. 3A). In addition, time course of HNSCC cell survival was also evaluated following the treatment with GSNO and radiation (Fig. 3B). In all three test cell lines, GSNO as well as radiation treatments inhibited STAT3 activities (phosphorylation) (Fig. 3A). Fig. 3B for cell survival assay shows greater efficacy with combination of GSNO and radiation than individual treatments. These results indicate that GSNO treatment of HNSCC cells enhances radiation-induced growth inhibition by downregulating STAT3 activity.
Fig. 3.

Effect of GSNO treatment and radiation on STAT3 activities and HNSCC cell survival. (A) Following the treatment of SCC1, SCC14a and SCC22a HNSCC cell lines with 10 Gy radiation and 250 μM GSNO for 24 h, the levels of STAT3 and their phosphorylated forms were analyzed by Western analysis using specific antibodies. The densitometry values as compared to that of control calculated by using NIH-Image-J. (B) HNSCC cell lines were treated with 10 Gy radiation and 250 μM GSNO for indicated time and the cell survival was analyzed as described under Section 2. The results in bar and line graphs were given as mean of at least three independent experiments. The vertical lines indicate the standard deviation; *P<0.05; **P<0.01; compared with vehicle (VHC; dimethylsulfoxide) treated control groups.
3.4. GSNO inhibits NF-κB activity and iNOS expression but increases HIF1-α levels without inducing VEGF secretion
NF-κB is a crucial transcription factor for cancer cell survival and chemoresistance [7]. To assess the role of GSNO in NF-κB activation in HNSCC cells, SCC14a cells were treated with GSNO for 3 h and then treated with proinflammatory cytokines (25 ng/ml TNF-α, 25ng/IL-1β, and 25 ng/ml IFN-γ) for 15 min. Fig. 4A shows that GSNO treatment inhibited the NF-κB activation measured as decreased nuclear translocalization of p65 (NF-κB) (Fig. 4A-i) as well as its DNA binding activity (Fig. 4A-ii) under both control and cytokine stimulated conditions.
Fig. 4.

Effects of GSNO treatment on NF-κB activities and expressions of iNOS, HIF-1α, and VEGF. (A) Following the treatment with GSNO (250 μM) for 3 h, the SCC14a cells were treated with cytokine mix (TNFα, IL-1β, and IFN-γ; 25 ng/ml each) for 20 min. The nuclear and cytoplasmic levels of p65 NF-κB (i) and nuclear NF-κB DNA binding activities (ii) were analyzed by Western and gel-shift assay. H3 histone (H3) and β-actin were used as an internal loading control. (B) Following the treatment of SCC14a cells with GSNO, cytokines, and/or L-NAME (250 μM) for 12hrs, the expression of iNOS (i) and cellular nitro-tyrosine (N-Tyr) (ii) levels were investigated by Western analysis using specific antibodies. (C) Time course effect of GSNO (250 μM) on cellular HIF-1α levels was analyzed by Western blot. (D) GSNO induced inhibition of VEGF secretion in SCC14a cell cultures were analyzed. SCC14a cells were pretreated with GSNO (250 μM for 3 h) and then treated with/without cytokine mix (TNFα, IL-1β, and IFNγ; 25 ng/ml each/14 h incubation). Following the incubation, human VEGF levels were analyzed by ELISA assay. The results in bar and line graphs were given as mean of at least three independent experiments. The vertical lines indicate the standard deviation; *P<0.05; **P<0.01; ***P<0.001; compared with vehicle treated control groups. (D) Two way ANOVA was used for statistical analysis for differences between control vs. cytokine treated groups (###P<0.001).
The inducible form of NO synthase (iNOS) has been implicated both in anti-cancer and pro-cancer cell signaling processes [37,38]. iNOS produces NO as well as superoxide anion radical (O2•−) depending on the cellular redox status and substrate and cofactor availabilities. High levels of NO and O2•− are described to trigger apoptosis of cancer cells [39–41]; however their sustained low levels are reported to contribute to cancer cell survival by promoting cell death resistance signals [41,42], thus low and sustained expression of iNOS could participate in promotion of cancer cell survival. Fig. 4B shows that SCC14a cells under normal conditions express low and sustained levels of iNOS. GSNO treatment decreased the basal expression of iNOS in a dose dependent manner. In addition, the basal levels of protein nitrotyrosine, observed in cancer cells, was also decreased by treatment with GSNO to the levels observed in the cells treated with NOS inhibitor (l-Nω-nitroarginine methyl ester), thereby suggesting that GSNO may be effective on the inhibition of basal expression of iNOS as well as ONOO− generation from NO and O2•− in HNSCC cells. Interestingly, the treatment of SCC14a cells with proinflammatory cytokines did not result in increased expression of iNOS in spite of NF-κB activation (Fig. 4A), indicating a mechanism for limiting the overexpression of iNOS gene under proinflammatory conditions in HNSCC cell line. The basis of these derangements in signaling for inflammatory regulation of iNOS gene is not understood at present, but these observations document that GSNO mediated mechanisms inhibit the induction of iNOS as well as NO associated toxicity as nitrotyrosine formation.
Hypoxic inducible factor-1α (HIF-1α) activation and tumor-angiogenesis by vascular endothelial growth factor (VEGF) release were described as potential mechanisms for NO-mediated tumorigenesis and metastasis [22,43,44]. As NO is known to stabilize HIF-1α by S-nitrosylation dependent mechanisms [44], we observed that GSNO treatment also increased HIF-1α levels in both nucleus and cytoplasm (Fig. 4C). Although HIF-1α is one of prerequisites for induction of VEGF gene expression [22,43], we did not observe increase in VEGF secretion by GSNO treatment (250 μM for 17 h) of SCC14a cells (Fig. 4D). Instead, GSNO treatment slightly, but significantly, decreased VEGF secretion by SCC14a cells under normal conditions. Proinflammatory cytokines are known to induce VEGF gene expression in various cell types [45,46]. GSNO pretreatment of SCC14a cells, 3 h prior to cytokine treatment (TNF-α, IL-1β, and IFN-γ; 25 ng/ml each/14 h incubation), also inhibited cytokine-induced VEGF secretion (Fig. 4D). These data indicate that regardless of activation of HIF-1α, exogenous GSNO does not induce VEGF-dependent angiogenesis.
3.5. GSNO enhances efficacy of radiation and cisplatin therapy in HNSCC cancer xenograft mouse model
An animal xenograft model was generated by injection of nude mice with luciferase labeled SCC14a into the flank. Tumors were allowed to establish for 4 weeks prior to initiation of treatments. The mice were then treated with radiotherapy (8 Gy; one a week for three times) or cisplatin (2 mg/kg body weight; one a week for three times) or GSNO (10 mg/kg body weight; one a day till 10th week following the transplantation) individually or in combination as described in methods section (n=11 per group). The tumor size was measured by bioluminescent imaging (Fig. 5A and B). GSNO treatment significantly decreased the tumor size in the HNSCC xenografted animals as compared to radiation or cisplatin treatments alone. GSNO treatment in combination with radiotherapy and/or cisplatin further decreased the growth of tumor. Because GSNO inhibited STAT3 phosphorylation reversibly, we examined whether GSNO treatment inhibits tumor growth reversibly. Fig. 5B shows that discontinuation of GSNO treatment at 7th week in the mice treated with cisplatin and radiation reversibly increased the tumor growth, indicating that GSNO treatment increased the therapeutic potential of cisplatin and radiotherapy in a reversible manner.
Fig. 5.

Effects of radiation, cisplatin and GSNO and their combinations on tumor growth in HNSCC xenografted mice. (A) Nude mice were injected with luciferase labeled SCC14a cells (106) and the tumor growth was evaluated by fluorescence intensity after 6 weeks of treatments with the respective protocol. (B) The animals were treated with radiation, cisplatin, and/or GSNO 4 weeks after inoculation of cancer cells, and the time-course changes in tumor size (fluorescence intensity) were measured. To investigate the reversibility in GSNO-inhibited tumor growth, GSNO treatment was discontinued for a group mice treated with cisplatin and radiation after 7 weeks (solid red diamond). The data are mean±standard error from six animals in each group.
3.6. GSNO treatment with radiation and chemotherapy inhibits STAT3 activation and enhances tumor cell death in HNSCC xenografted mice
STAT3 activation increases cell cycle progression, proliferation, and survival of tumor cells by upregulating the expression and activities of its down-stream genes, such as VEGF, and Bcl-2 [5,47]. To examine the effect of GSNO on STAT3 activation and STAT3 regulated gene expression, tumor tissues harvested from xenografted mice treated with vehicle, cisplatin, radiation, and/or GSNO (10th week following the transplantation) were used for Western analysis for phospho/total-STAT3, VEGF, and Bcl-2. All experiments were performed at least three times (total n=6 mice per group). Consistent with our cell culture studies (Figs. 1 and 2), GSNO treatment decreased pSTAT3 levels in tumor tissues harvested from HNSCC xenograft mouse model (Fig. 6A). Accordingly, GSNO treatment also reduced the expression of tumor growth regulators, such as VEGF and Bcl-2, thereby suggesting a potential inhibitory effect of GSNO on STAT3-mediated tumor progression in HNSCC. Next we investigated the effect of GSNO treatment on tumor cell survival in combination with radiation and/or cisplatin treated xenograft mice. Fig. 6B shows that GSNO treatment alone and in combination with other treatments significantly increased the apoptosis reflected as increased TUNEL positive cells. The above observations indicate that GSNO controls the growth of cancer and also complements radiotherapy and chemotherapy for a greater degree of efficacy.
Fig. 6.

Effects of radiation, cisplatin and GSNO and their combinations on STAT3 activity and its downstream protein expression in HNSCC xenografted mice. (A) Western analysis of the tumor tissues harvested from control and experimental animals for STAT3, pSTAT3, VEGF, Bcl-2, or β-actin was performed. (B) The tumor tissues were subjected to TUNEL assay for detection of the cells undergoing apoptosis. The sections were counterstained with DAPI for detection of nuclei. (i) The number of TUNEL positive cells was counted in 10 different regions of the slides (two regions from each animal) (ii). The results in bar graphs were given as mean of at least three independent experiments. The vertical lines indicate the standard deviation; *P<0.05; **P<0.01; ***P<0.001; compared with vehicle treated control groups.
4. Discussion
Studies from our laboratory and others have documented that activities of STAT3 and NF-κB are regulated by redox based post-translational modification mediated by NO (S-nitrosylation) [10,11,15,16,48,49]. STAT3 plays key roles in the regulation of gene expression for cell survival (i.e. Bcl-xL, cIAP, survivin, and Mcl-1), cell cycle (i.e. c-myc, CDK2, Cyclin-E, CDK1, and Cyclin-B), and tumor angiogenesis (i.e. vascular endothelial growth factor; VEGF) [33,34,50–57]. Similarly, NF-κB also participates in regulation of gene expression for cell survival (i.e. cIAP, Bcl-2, Bcl-xL, and XIAP), cell cycle (i.e. c-Myc and Cyclin D), and multidrug resistance (e.g. MDR1) [35,36,58–60]. Studies described here document that treatment with GSNO, an endogenous S-nitrosothiol compound, may hold potential to target aberrant STAT3 and NF-κB activation in HNSCC and may complement conventional cancer treatments, such as radiation and cisplatin, to achieve greater efficacy against tumor growth. These conclusions are based on the following observations: (1) GSNO treatment of HNSCC cells in vitro reversibly inhibited the activation of STAT3. (2) The inhibition of STAT3 activation by GSNO treatment correlated with the inhibition of HNSCC cell proliferation, cell cycle progression; and cell survival (3) GSNO treatment with radiation in in vitro cultures inhibited the STAT3 activation to a higher degree than GSNO or radiation treatment alone. (4) GSNO treatment alone or in combination with cisplatin and radiation reversibly reduced the tumor mass in HNSCC xenograft mouse model. These observations clearly document that GSNO treatment reversibly inhibit the activation of STAT3 and NF-κB and tumor growth in in vitro cell culture model and in vivo xenograft model of HNSCC. Since STAT3 modulates various physiological functions, its persistent and irreversible inhibition may cause side effects on other physiological functions. In this aspect, the reversible inhibition of STAT3 by GSNO is particularly beneficial for clinical cancer treatment.
STAT3 activation in HNSCC cells was found to be mediated by the gp130 cytokine receptor [61,62]. Activation of gp130 is primarily driven by the inflammatory cytokine IL-6 which is produced by HNSCC cells themselves [63]. Interestingly, production of IL-6 by HNSCC cells requires NF-κB activity as the inhibition of NF-κB led to down-regulation of IL-6 expression [61,64], thus documenting the importance of inhibition of cross-talk between the NF-κB and the STAT3 signaling system in HNSCC therapeutics. In this study, we observed that GSNO treatment inhibited not only STAT3 (Fig. 1) but also NF-κB (Fig. 4), thus indicating the efficacy of GSNO-mediated S-nitrosylation mechanism on inhibition of cross-talk between STAT3 and NF-κB pathways in HNSCC.
NOS expression has been observed in many human cancers where NO produced by NOS have been reported to be both pro- and anti-tumor [21–27]. In xenograft mouse model of hepatoma, implantation of eNOS-overexpressing hepatoma cells as well as injection of NO donor into wild-type hepatoma-derived tumors resulted in increased hepatoma cell death and reduced tumor cell growth [65]. In addition, NO compounds, such as diethylamine dinitric oxide [25] and nitroglycerin [28], NO-nonsteroidal anti-inflammatory drugs [29] have been reported to enhance chemosensitization of tumor. GSNO treatment was reported to inhibit cell growth and promote apoptosis in colon cancer cell lines [66] and nitrosylcobalamin promoted cell death via nitrosylation of Apo2L/TRAIL receptor DR4 in several human cancer cell lines [26]. In this study, we demonstrated that treatment of HNSCC cells with GSNO, an endogenous NO carrier molecule, inhibited proliferation and survival of HNSCC cells via inhibiting activities of STAT3 and NF-κB, suggesting the role of NOS and thus NO/GSNO in inhibition of cancer growth and survival.
NOS produces not only NO but also O2•− depending on cellular redox conditions and cofactor (tetrahydrobiopterine/BH4) availability [67]. NOS coupled with BH4 generates NO and l-citrulline from l-arginine. However, under oxidative stress conditions and limiting availability of cofactor and/or l-arginine, uncoupled NOS generates O2•− more than NO and also leads to production of peroxynitrite (ONOO−). Formation of high amounts of O2•− and ONOO− is known to trigger cancer cells into apoptosis [39–41]. On the other hand, their sustained low levels could contribute to cancer cell survival by promoting cell death resistance signals via activation of NF-κB [41,42,68,69]. In turn, this low and sustained expression of iNOS could participate in promotion of cancer cell survival via O2•−/ONOO− induced NF-κB activation. GSNO is known to inhibit activation of NF-κB, a prerequisite for iNOS expression, via S-nitrosylation dependent mechanisms [10,11,48,70,71]. Accordingly, we observed that GSNO treatment attenuated sustained low-level activation of NF-κB (Fig. 4A) and iNOS expression (Fig. 4B-i). In addition, GSNO treatment attenuated sustained low levels of protein nitrotyrosine content in SCC14a cells (Fig. 4B-ii). The observed inhibition of protein nitrotyrosine content in SCC14a cells by iNOS inhibitor (L-NAME) suggests the possible role of GSNO mediated mechanisms in attenuation of iNOS-dependent sustained low-level of nitrosative stress.
NO has been reported to stabilize HIF-1α, key transcription factors for VEGF expression [22,43] under normoxic conditions by S-nitrosylation dependent mechanisms [44]. Therefore, HIF-1α activation and induction of tumor-angiogenesis via VEGF release were described as potential mechanisms for NO-mediated tumorigenesis and metastasis [22,43,44]. Indeed, GSNO was reported to induce VEGF expression in cultured keratinocytes during the cutaneous wound repair [72]. Consistent with the previous studies, we also observed that GSNO treatment induced cellular and nuclear accumulation of HIF-1α (Fig. 4C). However, GSNO treatment decreased the VEGF expression both in in vitro cell culture study (Fig. 4D) and in tumor tissues from HNSCC xenografted mice (Fig. 6A). VEGF expression is regulated by several transcription factors including AP-1, STAT-3, LXR, Sp1, AP-2 and Egr-1 in addition to HIF-1α [47]. Among them AP-1, Sp1, Egr-1, and STAT3 have been reported to be inhibited by S-nitrosylation dependent mechanisms [16,73] (Figs. 1, 3, and 6), suggesting that GSNO-mediated regulation of VEGF expression is more complex than the simple activation of HIF-1α by S-nitrosylation. Constitutive STAT3 activity is reported to up-regulate VEGF expression and tumor angiogenesis [74]. Since GSNO inhibits STAT3 activity (phosphorylation of Tyr705) via S-nitrosylation of its Cys259 residue (Fig. 1) [16], we speculate that GSNO inhibits VEGF expression via inhibiting STAT3 activity. The detailed molecular mechanisms underlying the role of GSNO in regulation of VEGF expression would be further elucidated in future studies.
A combination of systemic cisplatin and loco-regional radiotherapy are widely used in head and neck cancer. However, patients treated with chemoradiation can eventually relapse from minimal residual cancer in a similar way to patients who are primarily treated with surgery. In addition, the majority of relapsed cancers develop cisplatin-resistance. Cisplatin resistance is complex and involves several mechanisms. Increased activities of Akt/mTOR pathways were reported to participate in cisplatin resistance in ovarian cancer [75]. In addition, the increased expressions and activities of STAT3 and NF-κB have been also associated with cisplatin resistance via upregulating the cancer cell survival factors (i.e. Bcl-2 and Bcl-xL) and multidrug resistance molecules [76,77]. In summary, this study reports that GSNO efficiently regulates STAT3 and NF-κB activities in HNSCC cell lines (SCC1, SCC14a and SCC22a) in cell culture as well as in tumor tissues from radiation, cisplatin, and GSNO treated xenograft mice. The efficacy of GSNO treatment in attenuation of tumor growth by itself or in combination with cisplatin and/or radiation suggests a potential utility of GSNO-mediated mechanisms not only as stand alone cancer therapy but also in management of cancer relapse as a maintenance drug following chemoradiation therapy and cisplatin resistance.
Acknowledgments
In part, this research was supported by grants from VA, United States (BX-002829 and BX-001062) and NIH, United States (NS-37766 and NS-72511).
Footnotes
This article belongs to a special issue on Nitric Oxide and Cancer, edited by Jordi Muntané and Benjamin Bonavida.
Contributor Information
Je-Seong Won, Email: wonj@musc.edu.
Inderjit Singh, Email: singhi@musc.edu.
References
- 1.Koukourakis M.I., Tsoutsou P.G., Karpouzis A., Tsiarkatsi M., Karapantzos I. Radiochemotherapy with cetuximab, cisplatin, and amifostine for locally advanced head and neck cancer: a feasibility study. Int. J. Radiat. Oncol. Biol. Phys. 2010;77:9–15. doi: 10.1016/j.ijrobp.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 2.Amornphimoltham P., Patel V., Leelahavanichkul K., Abraham R.T., Gutkind J.S. A retroinhibition approach reveals a tumor cell-autonomous response to rapamycin in head and neck cancer. Cancer Res. 2008;68:1144–1153. doi: 10.1158/0008-5472.CAN-07-1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aggarwal B.B., Sethi G., Ahn K.S., Sandur S.K., Pandey M.K. Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: modern target but ancient solution. Ann. N.Y. Acad. Sci. 2006;1091:151–169. doi: 10.1196/annals.1378.063. [DOI] [PubMed] [Google Scholar]
- 4.Dolcet X., Llobet D., Pallares J., Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446:475–482. doi: 10.1007/s00428-005-1264-9. [DOI] [PubMed] [Google Scholar]
- 5.Yu H., Jove R. The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 6.Lun M., Zhang P.L., Pellitteri P.K., Law A., Kennedy T.L. Nuclear factor-kappaB pathway as a therapeutic target in head and neck squamous cell carcinoma: pharmaceutical and molecular validation in human cell lines using Velcade and siRNA/NF-kappaB. Ann. Clin. Lab. Sci. 2005;35:251–258. [PubMed] [Google Scholar]
- 7.Wilczynski J., Duechler M., Czyz M. Targeting NF-kappaB and HIF-1 pathways for the treatment of cancer: Part I. Arch. Immunol. Ther. Exp. (Warsz) 2011;59:289–299. doi: 10.1007/s00005-011-0131-4. [DOI] [PubMed] [Google Scholar]
- 8.Lai S.Y., Johnson F.M. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resist. Updates. 2010;13:67–78. doi: 10.1016/j.drup.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Yu H., Pardoll D., Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prasad R., Giri S., Nath N., Singh I., Singh A.K. GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. Glia. 2007;55:65–77. doi: 10.1002/glia.20436. [DOI] [PubMed] [Google Scholar]
- 11.Khan M., Sekhon B., Giri S., Jatana M., Gilg A.G. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J. Cereb. Blood Flow Metab. 2005;25:177–192. doi: 10.1038/sj.jcbfm.9600012. [DOI] [PubMed] [Google Scholar]
- 12.Pineda-Molina E., Klatt P., Vazquez J., Marina A., Garcia de Lacoba M. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 13.Reynaert N.L., van der Vliet A., Guala A.S., McGovern T., Hristova M. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Won J.S., Kim J., Annamalai B., Shunmugavel A., Singh I. Protective role of S-nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J. Alzheimers Dis. 2013;34:621–635. doi: 10.3233/JAD-121786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nath N., Morinaga O., Singh I. S-nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 2010;5:240–251. doi: 10.1007/s11481-009-9187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J., Won J.S., Singh A.K., Sharma A.K., Singh I. STAT3 regulation by S-nitrosylation: implication for inflammatory disease. Antioxid. Redox Signal. 2014;20:2514–2527. doi: 10.1089/ars.2013.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giri S., Rattan R., Deshpande M., Maguire J.L., Johnson Z. Preclinical therapeutic potential of a nitrosylating agent in the treatment of ovarian cancer. PLoS One. 2014;9:e97897. doi: 10.1371/journal.pone.0097897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaston B.M., Carver J., Doctor A., Palmer L.A. S-nitrosylation signaling in cell biology. Mol. Interv. 2003;3:253–263. doi: 10.1124/mi.3.5.253. [DOI] [PubMed] [Google Scholar]
- 19.Martinez-Ruiz A., Lamas S. S-nitrosylation: a potential new paradigm in signal transduction. Cardiovasc. Res. 2004;62:43–52. doi: 10.1016/j.cardiores.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 20.Egloff A.M., Grandis J.R. Response to combined molecular targeting: defining the role of P-STAT3. Clin. Cancer Res. 2011;17:393–395. doi: 10.1158/1078-0432.CCR-10-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korde Choudhari S., Sridharan G., Gadbail A., Poornima V. Nitric oxide and oral cancer: a review. Oral Oncol. 2012;48:475–483. doi: 10.1016/j.oraloncology.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Muntane J., la Mata M.D. Nitric oxide and cancer. World J. Hepatol. 2010;2:337–344. doi: 10.4254/wjh.v2.i9.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z. Protein S-nitrosylation and cancer. Cancer Lett. 2012;320:123–129. doi: 10.1016/j.canlet.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 24.Switzer C.H., Glynn S.A., Cheng R.Y., Ridnour L.A., Green J.E. S-nitrosylation of egfr and src activates an oncogenic signaling network in human basal-like breast cancer. Mol. Cancer Res. 2012;10:1203–1215. doi: 10.1158/1541-7786.MCR-12-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baritaki S., Huerta-Yepez S., Sahakyan A., Karagiannides I., Bakirtzi K. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle. 2010;9:4931–4940. doi: 10.4161/cc.9.24.14229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang Z., Bauer J.A., Morrison B., Lindner D.J. Nitrosylcobalamin promotes cell death via S nitrosylation of Apo2L/TRAIL receptor DR4. Mol. Cell. Biol. 2006;26:5588–5594. doi: 10.1128/MCB.00199-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Luca A., Moroni N., Serafino A., Primavera A., Pastore A. Treatment of doxorubicin-resistant MCF7/Dx cells with nitric oxide causes histone glutathionylation and reversal of drug resistance. Biochem. J. 2011;440:175–183. doi: 10.1042/BJ20111333. [DOI] [PubMed] [Google Scholar]
- 28.Yasuda H., Nakayama K., Watanabe M., Suzuki S., Fuji H. Nitroglycerin treatment may enhance chemosensitivity to docetaxel and carboplatin in patients with lung adenocarcinoma. Clin. Cancer Res. 2006;12:6748–6757. doi: 10.1158/1078-0432.CCR-06-1124. [DOI] [PubMed] [Google Scholar]
- 29.Rigas B. Novel agents for cancer prevention based on nitric oxide. Biochem. Soc. Trans. 2007;35:1364–1368. doi: 10.1042/BST0351364. [DOI] [PubMed] [Google Scholar]
- 30.Fortenberry J.D., Owens M.L., Chen N.X., Brown L.A. S-nitrosoglutathione inhibits TNF-alpha-induced NFkappaB activation in neutrophils. Inflamm. Res. 2001;50:89–95. doi: 10.1007/s000110050729. [DOI] [PubMed] [Google Scholar]
- 31.Brenner J.C., Graham M.P., Kumar B., Saunders L.M., Kupfer R. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 2010;32:417–426. doi: 10.1002/hed.21198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Won J.S., Im Y.B., Khan M., Singh A.K., Singh I. The role of neutral sphingomyelinase produced ceramide in lipopolysaccharide-mediated expression of inducible nitric oxide synthase. J. Neurochem. 2004;88:583–593. doi: 10.1046/j.1471-4159.2003.02165.x. [DOI] [PubMed] [Google Scholar]
- 33.Bhattacharya S., Ray R.M., Johnson L.R. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem. J. 2005;392:335–344. doi: 10.1042/BJ20050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barre B., Vigneron A., Coqueret O. The STAT3 transcription factor is a target for the Myc and riboblastoma proteins on the Cdc25A promoter. J. Biol. Chem. 2005;280:15673–15681. doi: 10.1074/jbc.M413203200. [DOI] [PubMed] [Google Scholar]
- 35.Aggarwal B.B. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 36.La Rosa F.A., Pierce J.W., Sonenshein G.E. Differential regulation of the c-myc oncogene promoter by the NF-kappa B rel family of transcription factors. Mol. Cell. Biol. 1994;14:1039–1044. doi: 10.1128/mcb.14.2.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y.K., Hsuen S.S., Lin L.M. Expression of inducible nitric oxide synthase in human oral premalignant epithelial lesions. Arch. Oral Biol. 2002;47:387–392. doi: 10.1016/s0003-9969(02)00011-0. [DOI] [PubMed] [Google Scholar]
- 38.Harada K., Supriatno, Kawaguchi S., Tomitaro O., Yoshida H. Overexpression of iNOS gene suppresses the tumorigenicity and metastasis of oral cancer cells. In Vivo. 2004;18:449–455. [PubMed] [Google Scholar]
- 39.Brune B. The intimate relation between nitric oxide and superoxide in apoptosis and cell survival. Antioxid. Redox Signal. 2005;7:497–507. doi: 10.1089/ars.2005.7.497. [DOI] [PubMed] [Google Scholar]
- 40.Choi B.M., Pae H.O., Jang S.I., Kim Y.M., Chung H.T. Nitric oxide as a pro-apoptotic as well as anti-apoptotic modulator. J. Biochem. Mol. Biol. 2002;35:116–126. doi: 10.5483/bmbrep.2002.35.1.116. [DOI] [PubMed] [Google Scholar]
- 41.Fukumura D., Kashiwagi S., Jain R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 42.Pervaiz S., Clement M.V. Superoxide anion: oncogenic reactive oxygen species? Int. J. Biochem. Cell Biol. 2007;39:1297–1304. doi: 10.1016/j.biocel.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 43.Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;9(Suppl. 5):S10–S17. doi: 10.1634/theoncologist.9-90005-10. [DOI] [PubMed] [Google Scholar]
- 44.Li F., Sonveaux P., Rabbani Z.N., Liu S., Yan B. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell. 2007;26:63–74. doi: 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ristimaki A., Narko K., Enholm B., Joukov V., Alitalo K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J. Biol. Chem. 1998;273:8413–8418. doi: 10.1074/jbc.273.14.8413. [DOI] [PubMed] [Google Scholar]
- 46.Alagappan V.K., McKay S., Widyastuti A., Garrelds I.M., Bogers A.J. Proinflammatory cytokines upregulate mRNA expression and secretion of vascular endothelial growth factor in cultured human airway smooth muscle cells. Cell Biochem. Biophys. 2005;43:119–129. doi: 10.1385/CBB:43:1:119. [DOI] [PubMed] [Google Scholar]
- 47.Pages G., Pouyssegur J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene—a concert of activating factors. Cardiovasc. Res. 2005;65:564–573. doi: 10.1016/j.cardiores.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 48.Reynaert N.L., Ckless K., Korn S.H., Vos N., Guala A.S. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. USA. 2004;101:8945–8950. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marshall H.E., Hess D.T., Stamler J.S. S-nitrosylation: physiological regulation of NF-kappaB. Proc. Natl. Acad. Sci. USA. 2004;101:8841–8842. doi: 10.1073/pnas.0403034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Platt D.H., Bartoli M., El-Remessy A.B., Al-Shabrawey M., Lemtalsi T. Peroxynitrite increases VEGF expression in vascular endothelial cells via STAT3. Free Radic. Biol. Med. 2005;39:1353–1361. doi: 10.1016/j.freeradbiomed.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 51.Lanuti P., Bertagnolo V., Pierdomenico L., Bascelli A., Santavenere E. Enhancement of TRAIL cytotoxicity by AG-490 in human ALL cells is characterized by downregulation of cIAP-1 and cIAP-2 through inhibition of Jak2/Stat3. Cell Res. 2009;19:1079–1089. doi: 10.1038/cr.2009.80. [DOI] [PubMed] [Google Scholar]
- 52.Buettner R., Mora L.B., Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002;8:945–954. [PubMed] [Google Scholar]
- 53.Darnell J.E., Jr. STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 54.Taga T., Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- 55.Bharadwaj U., Li M., Chen C., Yao Q. Mesothelin-induced pancreatic cancer cell proliferation involves alteration of cyclin E via activation of signal transducer and activator of transcription protein 3. Mol. Cancer Res. 2008;6:1755–1765. doi: 10.1158/1541-7786.MCR-08-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jarnicki A., Putoczki T., Ernst M. Stat3: linking inflammation to epithelial cancer-more than a “gut” feeling? Cell Div. 2010;5:14. doi: 10.1186/1747-1028-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niu G., Wright K.L., Ma Y., Wright G.M., Huang M. Role of Stat3 in regulating p53 expression and function. Mol. Cell. Biol. 2005;25:7432–7440. doi: 10.1128/MCB.25.17.7432-7440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bentires-Alj M., Barbu V., Fillet M., Chariot A., Relic B. NF-kappaB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene. 2003;22:90–97. doi: 10.1038/sj.onc.1206056. [DOI] [PubMed] [Google Scholar]
- 59.Shishodia S., Aggarwal B.B. Nuclear factor-kappaB activation: a question of life or death. J. Biochem. Mol. Biol. 2002;35:28–40. doi: 10.5483/bmbrep.2002.35.1.028. [DOI] [PubMed] [Google Scholar]
- 60.Baichwal V.R., Baeuerle P.A. Activate NF-kappa B or die? Curr. Biol. 1997;7:R94–R96. doi: 10.1016/s0960-9822(06)00046-7. [DOI] [PubMed] [Google Scholar]
- 61.Squarize C.H., Castilho R.M., Sriuranpong V., Pinto D.S., Jr, Gutkind J.S. Molecular cross-talk between the NFkappaB and STAT3 signaling pathways in head and neck squamous cell carcinoma. Neoplasia. 2006;8:733–746. doi: 10.1593/neo.06274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee T.L., Yeh J., Van Waes C., Chen Z. Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to interleukin-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carcinomas. Mol. Cancer Ther. 2006;5:8–19. doi: 10.1158/1535-7163.MCT-05-0069. [DOI] [PubMed] [Google Scholar]
- 63.Sriuranpong V., Park J.I., Amornphimoltham P., Patel V., Nelkin B.D. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003;63:2948–2956. [PubMed] [Google Scholar]
- 64.Duffey D.C., Chen Z., Dong G., Ondrey F.G., Wolf J.S. Expression of a dominant-negative mutant inhibitor-kappaBalpha of nuclear factor-kappaB in human head and neck squamous cell carcinoma inhibits survival, proinflammatory cytokine expression, and tumor growth in vivo. Cancer Res. 1999;59:3468–3474. [PubMed] [Google Scholar]
- 65.Gonzalez R., Ferrin G., Aguilar-Melero P., Ranchal I., Linares C.I. Targeting hepatoma using nitric oxide donor strategies. Antioxid. Redox Signal. 2013;18:491–506. doi: 10.1089/ars.2011.4476. [DOI] [PubMed] [Google Scholar]
- 66.Liu Q., Chan S.T., Mahendran R. Nitric oxide induces cyclooxygenase expression and inhibits cell growth in colon cancer cell lines. Carcinogenesis. 2003;24:637–642. doi: 10.1093/carcin/bgg014. [DOI] [PubMed] [Google Scholar]
- 67.Okazaki T., Otani H., Shimazu T., Yoshioka K., Fujita M. Reversal of inducible nitric oxide synthase uncoupling unmasks tolerance to ischemia/reperfusion injury in the diabetic rat heart. J. Mol. Cell. Cardiol. 2011;50:534–544. doi: 10.1016/j.yjmcc.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 68.Morgan M.J., Liu Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hattori Y., Kasai K., Gross S.S. NO suppresses while peroxynitrite sustains NF-kappaB: a paradigm to rationalize cytoprotective and cytotoxic actions attributed to NO. Cardiovasc. Res. 2004;63:31–40. doi: 10.1016/j.cardiores.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 70.Marshall H.E., Stamler J.S. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 71.DelaTorre A., Schroeder R.A., Kuo P.C. Alteration of NF-kappa B p50 DNA binding kinetics by S-nitrosylation. Biochem. Biophys. Res. Commun. 1997;238:703–706. doi: 10.1006/bbrc.1997.7279. [DOI] [PubMed] [Google Scholar]
- 72.Frank S., Stallmeyer B., Kampfer H., Kolb N., Pfeilschifter J. Nitric oxide triggers enhanced induction of vascular endothelial growth factor expression in cultured keratinocytes (HaCaT) and during cutaneous wound repair. FASEB J. 1999;13:2002–2014. [PubMed] [Google Scholar]
- 73.Sha Y., Marshall H.E. S-nitrosylation in the regulation of gene transcription. Biochim. Biophys. Acta. 2012;1820:701–711. doi: 10.1016/j.bbagen.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Niu G., Wright K.L., Huang M., Song L., Haura E. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 75.Peng D.J., Wang J., Zhou J.Y., Wu G.S. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2010;394:600–605. doi: 10.1016/j.bbrc.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gu F., Ma Y., Zhang Z., Zhao J., Kobayashi H. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol. Rep. 2010;23:671–676. doi: 10.3892/or_00000683. [DOI] [PubMed] [Google Scholar]
- 77.Morais C., Gobe G., Johnson D.W., Healy H. Inhibition of nuclear factor kappa B transcription activity drives a synergistic effect of pyrrolidine dithiocarbamate and cisplatin for treatment of renal cell carcinoma. Apoptosis. 2010;15:412–425. doi: 10.1007/s10495-009-0414-y. [DOI] [PubMed] [Google Scholar]