

Article first published online 3 July 2015
Key Words: Colitis, disease susceptibility, IBD, interleukin-10-deficient mouse, microbiota, microflora
Abstract:
Complex mechanisms are pulling the strings to initiate the development of inflammatory bowel disease. Current evidence indicates that an interaction of genetic susceptibilities (polymorphisms), environmental factors, and the host microbiota leads to a dysregulation of the mucosal immune system. In the past decades, the interleukin-10–deficient mouse has served as an excellent model to mirror the multifactorial nature of this disease. Here, we want to review in detail the interplay of the genetic factors, immune aspects, and especially summarize and discuss the role of the microbiota contributing to colitis development in the interleukin-10–deficient mouse model of inflammatory bowel disease as a multihit model.
Homeostasis in the gut relies on a fine-tuned interplay of the host's protective immunity and the microbiota that generate the intestinal environment.1 Disruption of this homeostasis can lead to intestinal disorders such as inflammatory bowel disease (IBD). IBD is a chronic inflammatory disease of the gastrointestinal tract that encompasses Crohn's disease (CD) and ulcerative colitis. Epidemiological studies indicate that IBD affects more than 3.6 million people and that the number of cases has increased over recent years.2–4 Symptoms of IBD include primarily diarrhea, abdominal pain, and rectal bleeding. However, there are frequent extraintestinal manifestations that may affect the skin, joints, eyes, bones, and the hepatobiliary tract.5 The etiology of IBD still remains unknown although the current perception is that a genetically determined abnormal response of the mucosal immune system against the microorganisms of the normal intestinal flora leads to inflammation.6 However, although a genetic susceptibility seems to be the root cause for IBD development, genetic factors seem to explain only a certain percentage of the disease outcome. A total of 163 susceptibility loci have been identified so far, but even this huge quantity of loci has been postulated to explain only a minority of the disease risk variance suggesting that other factors contribute substantially to IBD pathogenesis.7 These factors may include innate, adaptive, and environmental mechanisms that add on a large, yet to be defined, scale to the development and course of IBD. Recent efforts to understand the complex nature of IBD development focused especially on the contribution of the presence and composition of the host microbiota.8 In this context, animal models of IBD have served as valuable tools to systematically determine and analyze the components pivotal for the development of this complex multifactorial disease.
INTERLEUKIN-10–DEFICIENT MOUSE MODEL OF IBD
A genetically engineered model that has been extensively used to dissect IBD etiology is the interleukin-10 (IL-10)–deficient mouse, which was generated in 1993 by Kühn et al.9 The generation was performed by use of targeted mutation disrupting the IL-10 gene by replacing a 500 base pair fragment of exon 1 with a termination codon and a neo expression cassette and by introducing a termination codon into exon 3 (Il10tm1Cgn, IL10−/−). The spontaneous onset of gut inflammation after weaning in mice with this mutation may be reversed by treatment with recombinant IL-10.10 Colitis in IL-10–deficient mice is characterized by histological findings that are similar to those of human IBD.9,11 In detail, the discontinuous and transmural inflammatory lesions are characterized by inflammatory cell infiltrates into the lamina propria and submucosa, epithelial hyperplasia, mucin depletion, crypt abscesses, ulcers, and thickening of the intestinal wall.11,12 Colitis development in mutated mice develops shortly after weaning and originates from the fact that IL-10 is a key mediator of gut homeostasis.
MECHANISM OF IL-10 DEFICIENCY
IL-10 is an immunoregulatory cytokine that is essential for the maintenance of intestinal homeostasis and that can be secreted by a wide range of immune cells such as macrophages, dendritic cells, and T cells (Fig. 1). It suppresses effector functions of Th1/Th17 cells as well as NK cells and macrophages, thereby modulating the cellular immune response.13–15 Subsequently, colitis in IL-10–deficient mice stems from an aberrant response of CD4+ Th1-like T cells and an excessive secretion of the proinflammatory cytokines IL-12, IL-17, and IFNγ.12,16 Although originally believed to be a purely Th1-mediated inflammation, Th17 cells were later on shown to participate in the inflammatory immune response.17 IL-12 was believed to be crucial for colitis induction as mice deficient for both IL-10 and IL-12p40 did not develop intestinal pathology and treatment of colitic IL-10–deficient mice with a monoclonal antibody directed against the IL-12p40 subunit's reduced intestinal inflammation.18 However, the finding that the IL-12p40 subunit can also assemble to form the cytokine IL-23 has led to a reassessment regarding the contribution of IL-12 and IL-23 to inflammation development.19 Several studies have now indicated that the T-cell–mediated colitis of IL-10–deficient mice also depends on IL-23 promoting inflammation through IL-17 and IL-6, underlining the fact that several mechanisms may contribute to colitis development.17,20,21 In this context, it has recently been demonstrated that colitis and intestinal inflammation in IL-10–deficient mice also depend on an IL-13Rα2–mediated attenuation of IL-13 activity, highlighting a role for IL-13 and its decoy receptor in the regulation of the Th1/Th17-mediated inflammation in IL-10–deficient mice.22 Interestingly, a recent study found evidence that the critical balance of the IL-10– and IL-12/23–seesaw is governed by the PI3K subunit p110δ, which controls the homeostatic interaction of antigen-presenting cells and T cells, and p110δ-deficient mice have also been shown to be prone to spontaneously occurring Th1- and Th17-mediated colitis.23,24 Therefore, it has been speculated that one mechanism responsible for the excessive inflammatory responses in IL-10 deficiency may be due to loss of induction of p110δ.23
FIGURE 1.
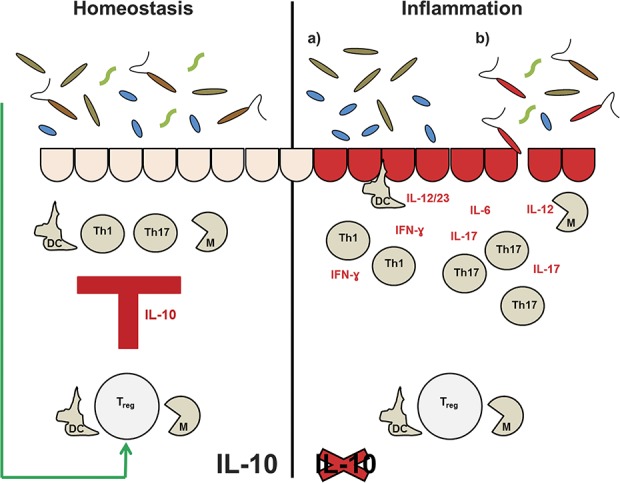
IL-10 maintains intestinal homeostasis (left side). It is produced by dendritic cells (DC) and macrophages (M), but the main source of IL-10 are Treg cells. Effector functions of the innate immune response (DC and M) and of the adaptive immune response (Th1/Th17 cells) are sufficiently suppressed by these cells through IL-10. Interestingly, the intestinal microbiota has been show to induce IL-10–producing Treg cells in an effort to establish a mutually beneficial commensalism. In IL-10 deficiency (right side), the response of Th1/Th17 cells is not counterbalanced anymore and colitis stems from an excessive secretion of proinflammatory cytokines. However, several hits have to find their target before colitis occurs. Either a misbalanced microbiota ([a] dysbiosis) contributes to colitis development or the presence of a specific pathogen initiates inflammation (b).
REGULATORY INFLUENCE OF IL-10 IN THE INTESTINAL INNATE AND ADAPTIVE IMMUNE RESPONSE
Immunologic tolerance to bacterial antigens in the intestine is primarily mediated by IL-10 and TGFβ produced by regulatory T cells.8,25 This is demonstrated by the finding that monoassociation of IL-10–deficient mice with Enterococcus faecalis led to chronic colitis and induced persistent NF-κB activation as well as TLR2 and subsequent proinflammatory gene expression in intestinal epithelial cells. Therefore, IL-10 seems to play a substantial role in regulating bacterial-induced epithelial NF-κB activation.25 Various studies have demonstrated that IL-10 operates on innate and on adaptive mechanisms to suppress inflammatory responses in the intestine, which we want to shortly highlight here. For a detailed overview on the control of intestinal inflammation by IL-10, see Kole and Maloy.26 Briefly, IL-10 effects on innate immune responses can be attributed mainly to macrophages and dendritic cells. In the gut, macrophages and dendritic cells are a major source of IL-10, and its production is induced by means of TLR-dependent and TLR-independent pathways. IL-10 has been shown to interfere with the maturation process of dendritic cells and to block the function of mature dendritic cells. Furthermore, IL-10 terminated macrophage activation and proliferation and inhibited macrophage autophagy, thereby maintaining the anergic phenotype of macrophages and dendritic cells that is characteristic for the state of intestinal homeostasis.13,26 In addition, IL-10 also seems to have protective effects on intestinal epithelial cells (IEC) as a proteome analysis of IEC derived from IL-10–deficient mice revealed defects in the response to endoplasmatic reticulum (ER) stress, energy metabolism, and apoptosis.27 Furthermore, IEC from IL-10–deficient mice and from patients with IBD displayed increased expression levels of the glucose-regulated ER stress protein 78, and IL-10 was shown to block inflammation-induced ER stress response mechanisms.28 However, the most substantial impacts of IL-10 may be attributed to effects on the adaptive immune response especially on the control of T-cell expansion and T-cell–mediated cytokine production. The maintenance of small intestinal and colonic tolerance by IL-10–producing regulatory T (Treg) cell subsets has been nicely reviewed recently.29 IL-10 may exert its effects on T cells directly by promoting the maintenance, expansion, and function of Treg cells or indirectly by depriving naive T cells of their appropriate stimuli.13,26 As mice with a Treg cell-specific deletion of IL-10 develop spontaneous colitis, Treg cells have been proposed as the critical source of IL-10 to maintain homeostasis at the environmental interface of the intestine.30 However, interestingly, recent studies indicate that macrophages play a central role in gut homeostasis, and the prevention of colitis as IL-10 receptor signaling was shown to drive macrophages to express homeostatic tolerogenic functions.31–33 Remarkably, protective effects of IL-10 in the gut seem to depend on microbial colonization.34 The differentiation of CD4+ Foxp3+ IL-10–producing cells can be induced in the intestine by the microbial product of a commensal bacterium, Bacteroides fragilis, thereby actively inducing mucosal tolerance.35 Recently, the induction of IL-10 production by indigenous Clostridium species and subsequent promotion of colonic Treg cells was described.36 Therefore, the induction of IL-10 by intestinal bacteria has been hypothesized as an important factor by which the intestinal microbiota establishes a mutually beneficial commensalism.26,37 In summary, IL-10 is one of the main factors to maintain intestinal homeostasis.
Therefore, the IL-10–deficient mouse model of IBD has been used widely as a valuable tool to dissect the complex mechanisms of this disease. However, analogous to the human situation, disease development in the IL-10–deficient mouse relies on genetic and on environmental factors, eminently the composition of the microflora.
GENETIC AND ENVIRONMENTAL FACTORS MODIFY DISEASE SUSCEPTIBILITY
A decade ago, Mähler and Leiter38 elucidated that genetic and environmental factors determined the course of colitis development in the IL-10–deficient mouse. Since then, a vast number of studies were conducted that focused on the differential contribution and interplay of these factors in the IL-10–deficient mouse model of IBD. Here, we want to review these studies and our own experiments, focusing especially on the role of the microbiota contributing to colitis development.
CONTRIBUTION OF THE GENETIC BACKGROUND TO COLITIS DEVELOPMENT IN THE IL-10–DEFICIENT MOUSE
Early on, it has been demonstrated that the genetic background of IL-10–deficient mice determines colitis susceptibility and severity. Several inbred mouse strains are highly susceptible to disease development (e.g., C3H/HeJBir and 129/SvEv), whereas others are relatively resistant (e.g., C57BL/6J and C57BL/10) indicating a significant contribution from genetic background modifiers.12,38–40 C3H/HeJBir (C3Bir-IL10−/−) susceptibility and C57BL/6J (B6-IL10−/−) resistance were analyzed by the use of segregating F2 and backcross populations derived from these 2 strains laying the foundation to further dissect IBD etiology.38,40–42 From an F2 analysis performed at the Jackson Laboratory and the reciprocal N2 crosses to the respective C3Bir-IL10−/− and B6-IL10−/− parental strains performed at the Hannover Medical School colitogenic quantitative trait loci (QTL) were identified and designated as Cdcs1-10 (Cdcs for Cytokine deficiency–induced colitis susceptibility).41,42 A major C3Bir-derived QTL, Cdcs1, was detected on chromosome 3 contributing to lesions in both cecum and colon and interacting epistatically or additively with multiple loci on other chromosomes.38 Furthermore, Cdcs1 was confirmed as a large effect QTL by using an F2 intercross of C3Bir-IL10−/− and B6-IL10−/− mice and lymphatic vascular changes as a trait.43 As peak linkage analyses indicated the presence of more than 1 genetic modifier in this region, reciprocal Cdcs1-congenic strains were generated to further characterize the Cdcs1 region.44 The transfer of Cdcs1 from the C3Bir to the B6 background rendered IL-10–deficient B6.C3Bir-Cdcs1-congenic mice susceptible and vice versa. Fine mapping of Cdcs1 using a total of 15 reciprocal subcongenic lines demonstrated that this QTL contains at least 3 subintervals (Cdcs1.1–Cdcs1.3) with independent genetic factors that confer innate hyporesponsiveness and adaptive hyperresponsiveness.44–46 Interestingly, the Cdcs1 region has independently been identified in several studies by the use of different colitis models and genetic backgrounds as a major locus regulating intestinal inflammation by both innate and adaptive immune mechanisms.47 To assess in detail the influence of this congenic element on the adaptive immune response, functional studies are currently performed by our group.48 In addition, the IL-10 model has demonstrated that even resistant backgrounds can contribute susceptibility genes once they are unmasked by segregation from major resistance loci. An example is Cdcs6 with the candidate gene Cd14, whose expression level determines protection from disease.49,50 Apart from the genetic background contributing to IBD development, it has early on been demonstrated that the presence and composition of the gut microbiota determines disease outcome as well. In this context, it is noteworthy that the overall histopathology scores for both the parental strain and their F1 hybrids were significantly lower in the Hannover Medical School facility than those determined for the Jackson Laboratory facility indicating that environmental factors, most likely differences in the composition of the microflora, modulated the genetic susceptibilities.38,51 Notably, these differences were observed without even sanitizing these mice. Therefore, to determine environmental factors interacting with the Cdcs1 locus, we performed embryotransfer-sanitation of C3Bir-IL10−/−, B6-IL10−/−, and the respective congenic mouse strains carrying the whole Cdcs1 region to maintain animals in noncolitogenic specific pathogen-free and germ-free environments. Then, to dissect Cdcs1 contribution to colitis susceptibility in interaction with environmental factors pivotal for colitis development, we used these sanitized mice for infection experiments. The outcome of these studies will be discussed in context with current literature and recent developments in the next section.
CONTRIBUTION OF THE MICROBIOTA TO COLITIS DEVELOPMENT IN THE IL-10–DEFICIENT MOUSE
Sellon et al52 demonstrated as early as 1998 that resident enteric bacteria are necessary for the development of spontaneous colitis in IL-10–deficient mice as colitis development was not observed under germ-free conditions. In addition, it was shown that the inflammatory response in both, adult and neonatal, germ-free IL-10–deficient mice is similar when colonized with a microbial flora indicating that the absence of IL-10 leads to a loss of tolerance towards bacterial antigens that is independent of the time at which microbial colonization occurs.53 Furthermore, it was early on shown that antibiotic treatment may prevent or attenuate colitis development in several mouse models of IBD.54–57 Treatment of neonatal IL-10–deficient mice with either neomycin/metronidazole or ciprofloxacin prevented the onset of colitis for up to 12 weeks indicating that an early alteration of the normal intestinal flora may affect colitis progression in later life.58,59 The use of antibiotics with a selective aerobic or anaerobic spectrum had different therapeutic effects in various sections of the colon of IL-10–deficient mice indicating that subsets of aerobic or anaerobic bacteria show regional differences in their capacity to mediate IL-10–deficient colitis.54 That bacterial sensing by intestinal immune cells, which is essential for inflammation development in IL-10–deficient mice has also been indicated by the finding that colitis development involves TLR-induced NF-κB activation derived mostly from mucosal immune cells.60 Furthermore, spontaneous colitis in IL-10–deficient mice requires MyD88 signaling, and IL-10−/− MyD88−/− double knockout mice do not develop colitis.61,62 Interestingly, it has been demonstrated that immunostimulatory DNA sequences derived from bacterial DNA ameliorated colonic inflammation in IL-10–deficient mice, suggesting that these potent activators of innate immune responses may have a physiologic anti-inflammatory role in the intestine.63 In addition, CpG motifs of bacterial DNA have been shown to exert a protective effect in mouse models of IBD by inducing an antigen-independent tolerance as the presence of a bacterial flora was not required.64 However, in IL-10–deficient mice raised under germ-free or specific pathogen-free conditions the introduction of specific bacteria either exerted a protective effect or promoted inflammation depending on the bacterial species.51 Briefly, members of the Lactobacillus and Bifidobacterium species attenuated colitis whereas E. faecalis and, in most studies, Helicobacter spp. exerted a proinflammatory effect.65–69 See Table 1 for an overview on microbiota effects in IL-10-deficient mice.
TABLE 1.
Microbiota Effects in the IL-10–deficient Mouse Model of IBD

However, it still remains controversial whether a specific individual pathogen induces inflammation, whether a disruption of the intestinal microbiota is the decisive factor leading to inflammation or whether these factors have to act in concert. Therefore, in general, the contribution of the microbiota towards colitis development in IL-10-deficient mice may be divided into effects due to specific (pathogenic) agents that may serve as colitogenic triggers and into effects that occur due to the general composition of the microbiota in terms of an imbalance of the commensal flora (dysbiosis).
CONTRIBUTION OF INFECTIOUS AGENTS (OR TRADITIONAL PATHOGENS) TO COLITIS DEVELOPMENT IN THE IL-10-DEFICIENT MOUSE
Various members of the microbiota including bacteria, viruses, yeasts and fungi have been speculated as potential IBD pathogens but their specific role and the respective contexts in which they may trigger inflammation have still to be defined. It has early on been demonstrated that the (mono-) association of a susceptible mouse strain like the IL-10-deficient mouse with a single commensal bacterial species, e.g., E. faecalis or Escherichia coli, can trigger inflammation (depending as well on a susceptible genetic background). Here, however, monoassociation selectively initiated immune-mediated intestinal inflammation with different colitis phenotypes and dual association was shown to have additive effects as this induced severe, early-onset pancolitis.68,70,71 Inoculation of IL-10-deficient mice with different Enterococcus species resulted in colonic inflammation and colonic gene expression changes similar to those detected in human IBD.72 The development of chronic colitis in IL-10-deficient mice mono-colonized with E. faecalis has in part been attributed to the interaction of this enteric bacterium with the IEC inducing protein changes that are involved in ER stress.27 In a recent study, germ-free IL-10-deficient mice were monoassociated with a probiotic E. faecalis and a colitogenic control strain. Proinflammatory responses were observed in both set-ups underlining the notion that even probiotic bacteria may trigger inflammation in an IBD-prone host like the IL-10-deficient mouse.73 On the other hand, probiotic bacteria have also been shown to enhance the epithelial barrier function in IL-10-deficient mice when administered for 4 weeks leading to a complete normalization of intestinal barrier integrity.81 Recently, another colitogenic bacterium, Bilophila wadsworthia, was identified in IL-10-deficient mice that were fed a diet high in saturated fat. Furthermore, the monoassociation of germ-free IL-10-deficient mice with B. wadsworthia also led to a proinflammatory Th1 cell-mediated colitis.78 In contrast, the monoassociation of germ-free IL-10-deficient mice with Helicobacter hepaticus, a classical colitogenic trigger to induce colitis, does not necessarily lead to inflammation.74 However, when maintained under specific pathogen-free conditions, H. hepaticus was shown to trigger colitis development in IL-10–deficient mice.66,67 On contrary, other studies could not report induction of colitis by H. hepaticus.74,75 Mähler and Leiter already proposed in 2002 that the divergent results of H. hepaticus ability to induce colitis in IL-10–deficient mice may depend on the interactions of this bacterium with other members of the microbiota.38 This is corroborated by the finding that, as mentioned above, germ-free IL-10–deficient mice did not develop colitis when infected with H. hepaticus. And, it was recently shown that after cocolonization of mice with Lactobacillus reuteri, H. hepaticus again served as a colitogenic trigger-inducing thyphlocolitis.75 Furthermore, this notion was recently underlined by a study demonstrating that the susceptibility of IL-10–deficient mice to H. hepaticus–induced intestinal pathology was correlated with significant differences in the composition of the intestinal microbiota.82 Interestingly, the microbiota composition of susceptible mice included the presence of B. wadsworthia and L. reuteri, both of which have been shown to have colitogenic potential in IL-10–deficient mice.75,78,82 Furthermore, it has been speculated that H. hepaticus–induced inflammation may rely on the presence of bacteria capable of inducing lymphoid structures as infection of IL-10–deficient mice with H. hepaticus leads to a strong adaptive immune response.18,26,67 Another recent study confirmed that an intestinal microbiota is needed for the development of colitis in IL-10–deficient mice infected with H. hepaticus and demonstrated that alteration of the microbiota by antibiotic treatment can modulate the disease phenotype. However, colitis initiation and progression was possible in the presence of several different microbial communities.83 It also has to be considered that H. hepaticus–induced inflammation in itself will affect and modulate the microbiota composition of the host, which in turn may depend both on the host immune response and the characteristics of the microbiota and which both might vary in a mouse strain-specific manner.76
As described earlier, a major colitogenic QTL, Cdcs1, was detected by using the IL-10–deficient mouse model of IBD, and this locus has been hypothesized to contribute to colitis development in this model not only by conferring innate hyporesponsiveness and adaptive hyperresponsiveness but also by interacting specifically with the intestinal microbiota. Next, we want to dissect Cdcs1 contribution to colitis susceptibility in interaction with environmental factors pivotal for colitis development.
GENETIC × MICROBIAL INTERACTIONS: ELUCIDATING Cdcs1 CONTRIBUTION TO COLITIS DEVELOPMENT IN INTERACTION WITH THE MICROBIOTA
As mentioned above, we used sanitized C3Bir-IL10−/−, B6-IL10−/−, and the respective Cdcs1-congenic IL-10–deficient mice for infection experiments with H. hepaticus and treatment with DSS and murine norovirus (MNV). After infection with H. hepaticus, the C3Bir haplotype, as expected, conferred a more severe inflammation in the cecum upon infection and BCR3 mice (originally resistant but now carrying the Cdcs1 haplotype of the C3Bir mice) showed severe inflammation in the cecum as well. However, unexpectedly, C3Bir mice demonstrated a milder form of inflammation in the colon than B6 mice, and H. hepaticus infection alone was not sufficient to restore the original more severe phenotypes. Therefore, Cdcs1 modified H. hepaticus–induced inflammation, but the original strain-specific response seemed to be driven by the composition of the microbiota. Subsequently, microbiota composition was determined by T-RFLP demonstrating that H. hepaticus–induced inflammation led to a strain-dependent decrease in bacterial richness and diversity (with C3Bir showing less richness and diversity than B6) and to strain-specific blooms of specific bacterial families correlating with colitis susceptibility.76 Therefore, the influence of the mouse strain-dependent, and especially Cdcs1-dependent, differences of the microbiota composition on colitis susceptibility should be the focus of future studies. To further dissect Cdcs1 contribution to colitis development, DSS treatment was used for colitis induction in sanitized IL-10–deficient and congenic mice. Again, the discrepancy to the original phenotype was detected with B6 mice displaying a more severe inflammation in the colon. However, BCR3 and BCR2 mice (both carrying the Cdcs1 haplotype of C3Bir mice but with differential interval lengths) were most susceptible and demonstrated the highest scores for inflammation (own observation). These findings again underline that the microflora not only modifies the expression and degree of inflammation in the IL-10–deficient mouse model of IBD but also modifies mouse strain-specific susceptibilities. This, however, is at least partly caused by genetic modifiers of the Cdcs1 region as they interact with environmental factors. Interestingly, this is corroborated by several QTL studies where the Cdcs1 region was independently replicated by use of different models, most of which were depending on microbial factors.47 Finally, we were recently able to demonstrate that an MNV infection can provide a colitogenic stimulus in the IL-10–deficient mouse that depends on the presence of the commensal microflora.77 Here, MNV infection of germ-free IL-10–deficient mice did not trigger inflammation, but colonization of germ-free mice with an altered Schaedler flora before MNV infection was sufficient to induce colitis. That nonbacterial members of the microbiota such as noroviruses may contribute to or initiate the development of IBD has been widely discussed, especially because it was recently shown that norovirus infection leads to the development of colitis in mice with a mutation in the CD susceptibility gene Atg16L1.84–86 Remarkably, norovirus infection has been demonstrated to aggravate IBD symptoms in human patients as well.87 However, so far, no evidence has been found for a viral infection to be the sole causative factor for the development of IBD but rather the combination of specific genetic susceptibilities and the presence of viral factors, probably in interaction with the endogenous microflora, contribute to IBD development. Our own observations regarding Cdcs1 contribution to colitis development in interaction with the microbiota underline this notion as the genetic susceptibility may be abolished or fundamentally reversed when the composition of the microbiota changes.
CONTRIBUTION OF AN INTESTINAL DYSBIOSIS TO COLITIS DEVELOPMENT IN THE IL-10–DEFICIENT MOUSE
It has early on been postulated that the development of IBD does not stem from a single causative factor but rather that an interaction of genetic susceptibilities, environmental factors, and the host microbiota leads to a dysregulation of the mucosal immune system. However, only recently, the contribution of a misbalanced microbiota has become the focus of attention in this complex disease. It can even be speculated that the effect of microbiota composition and diversity may have similar effects or may even be a more important contributor to colitis development than the effect of pathogen colonization.88 However, it is currently accepted that an imbalance between putative harmful versus protective bacterial species may promote intestinal inflammation.89,90 In this context, the reduction of anaerobic bacteria such as Bacteroides species, Eubacterium species, and Lactobacillus species has been reported in patients with IBD.91 Furthermore, fewer Firmicutes bacteria were found in IBD samples with a simultaneous increase in Bacteroidetes and, in CD samples only, Enterobacteriaceae.92 Other metagenomic studies have shown a decrease especially in the Firmicutes and the Bacteroidetes phyla in patients with CD.93–95 Interestingly, B. fragilis has been shown to exhibit a protective effect on colitis development in mice infected with H. hepaticus. As studies with IL-10–deficient mice indicated, the protective effect was demonstrated to be mediated through a functional requirement for IL-10–producing T cells.96 Therefore, the decrease in the richness of Bacteroidetes species may contribute to inflammation. However, commensal Bacteroides species have also been demonstrated to induce colitis in mice deficient for both, IL-10 and TGFβ.79 In addition, several other bacteria have been shown to be relevant to the dysbiosis in IBD as they independently induced intestinal inflammation in IL-10–deficient mice, including certain E. coli and E. faecalis strains.70,71 It has early on been shown that IL-10–deficient mice, similar to patients with CD, exhibit alterations in the species and levels of bacteria colonizing the colon compared with controls. Furthermore, reduced levels of probiotic bacteria were detected and, when administered early on, demonstrated to exert a protective effect.59,97 Also, the diversity of cecal bacteria was found to be altered before and after the onset of colitis in IL-10–deficient mice.98 Another study also reported changes in the diversity and composition of the intestinal microbiota with a progressive decrease in diversity and richness and an increase of Proteobacteria and E. coli during the onset of inflammation in IL-10–deficient mice.99 A recent study focusing on the changes in the intestinal steroid profile of IL-10–deficient mice during the onset of colitis detected a reduced microbial diversity compared with wild-type controls, which was accompanied by a proliferation of E. coli and Enterococcus gallinarum.100 Therefore, the substantial loss of bacterial diversity is believed to be not only a contributing factor for colitis development in the IL-10–deficient mouse but also to be a dominant factor in IBD pathogenesis.101 Microbiome studies with the largest pediatric cohort to date revealed an axis defined by an increased abundance of Enterobacteriaceae, Pasteurellaceae, Veillonellaceae, and Fusobacteriacea and a decreased abundance of Erysipelotrichales, Bacteroidales, and Clostridiales correlating strongly with the disease status.102 In this context, studies elucidating the impact of introducing a limited flora into germ-free and/or IBD susceptible mice such as the IL-10–deficient mouse are mandatory to systematically analyze microbiota contribution to IBD development. An elegant study determining microbiota–host interactions at a complex yet defined level used germ-free IL-10–deficient and wild-type mice to demonstrate that a simplified human microbiota consortium induced colitis in these mice.80 Furthermore, immunodominant bacterial species were identified in these “humanized” mice that induced Th1 and Th17 immune responses independent of luminal and mucosal bacterial concentrations. Also, a recent study described the influence of an altered diet on the microbiota and subsequent colitis development in IL-10–deficient but not wild-type mice. Here, it was shown that dietary fat–induced taurocholic acid promotes the expansion of the sulfate-reducing pathobiont B. wadsworthia driving colitis in IL-10–deficient mice.78 In summary, although indications for a correlation between the composition of the microbiota and the development of IBD are piling up, final evidence for a causative role of the host microbiota in IBD pathogenesis has yet to be provided.85 In this context, using the IL-10–deficient mouse model of IBD will help to complete our understanding of the dynamic role of the microbiota in this complex disease.
IL-10–DEFICIENT MOUSE SERVES AS A MULTIHIT MODEL
We have elaborated in detail on the interplay of genetic factors, immune aspects, and the role of the microbiota contributing to colitis development in the IL-10–deficient mouse, working out the facets of this excellent mirror of the multifactorial disease of IBD. The IL-10–deficient mouse serves as a multihit model where a colitogenic trigger initiates the inflammatory process, strain-specific genetic factors determine the dysregulation of the mucosal immune response, and where the microflora modifies these susceptibilities and responses (Fig. 2). The IL-10–deficient mouse is an excellent model to further define the hypothesis elaborated by Maloy and Powrie37 that the induction and perpetuation of intestinal inflammation may require the convergence of several abnormalities affecting intestinal homeostasis and that only when a specific threshold is reached inflammation manifests. As the entirety of recent studies indicate that IL-10 is a crucial regulatory cytokine suppressing inflammation because of the dysregulation of the microbial homeostasis,26 further utilization of this model will help to provide much-needed insights not only into IBD-related microbiota–host interactions but also into the primary versus secondary role of dysbiosis in this disease.
FIGURE 2.
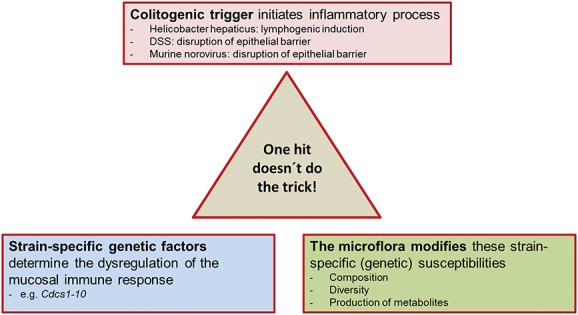
A multi-hit model: the IL-10–deficient mouse.
Footnotes
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Izcue A, Coombes JL, Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu Rev Immunol. 2009;27:313–338. [DOI] [PubMed] [Google Scholar]
- 2.Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. [DOI] [PubMed] [Google Scholar]
- 3.Ng SC, Bernstein CN, Vatn MH, et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut. 2013;62:630–649. [DOI] [PubMed] [Google Scholar]
- 4.Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54.e42; quiz e30. [DOI] [PubMed] [Google Scholar]
- 5.Larsen S, Bendtzen K, Nielsen OH. Extraintestinal manifestations of inflammatory bowel disease: epidemiology, diagnosis, and management. Ann Med. 2010;42:97–114. [DOI] [PubMed] [Google Scholar]
- 6.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. [DOI] [PubMed] [Google Scholar]
- 7.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. [DOI] [PubMed] [Google Scholar]
- 9.Kühn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. [DOI] [PubMed] [Google Scholar]
- 10.Steidler L, Hans W, Schotte L, et al. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science. 2000;289:1352–1355. [DOI] [PubMed] [Google Scholar]
- 11.Bleich A, Mahler M, Most C, et al. Refined histopathologic scoring system improves power to detect colitis QTL in mice. Mamm Genome. 2004;15:865–871. [DOI] [PubMed] [Google Scholar]
- 12.Berg DJ, Davidson N, Kuhn R, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paul G, Khare V, Gasche C. Inflamed gut mucosa: downstream of interleukin-10. Eur J Clin Invest. 2012;42:95–109. [DOI] [PubMed] [Google Scholar]
- 14.Moore KW, O'Garra A, de Waal Malefyt R, et al. Interleukin-10. Annu Rev Immunol. 1993;11:165–190. [DOI] [PubMed] [Google Scholar]
- 15.Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol. 2006;6:379–386. [DOI] [PubMed] [Google Scholar]
- 16.Davidson NJ, Leach MW, Fort MM, et al. T helper cell 1-type CD4+ T cells, but not B cells, mediate colitis in interleukin 10-deficient mice. J Exp Med. 1996;184:241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kullberg MC, Rothfuchs AG, Jankovic D, et al. Helicobacter hepaticus-induced colitis in interleukin-10-deficient mice: cytokine requirements for the induction and maintenance of intestinal inflammation. Infect Immun. 2001;69:4232–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. [DOI] [PubMed] [Google Scholar]
- 20.Kullberg MC, Jankovic D, Feng CG, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hue S, Ahern P, Buonocore S, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson MS, Ramalingam TR, Rivollier A, et al. Colitis and intestinal inflammation in IL10-/- mice results from IL-13Ralpha2-mediated attenuation of IL-13 activity. Gastroenterology. 2011;140:254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uno JK, Rao KN, Matsuoka K, et al. Altered macrophage function contributes to colitis in mice defective in the phosphoinositide-3 kinase subunit p110delta. Gastroenterology. 2010;139:1642–1653, 1653.e1641–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinbach EC, Kobayashi T, Russo SM, et al. Innate PI3K p110delta regulates Th1/Th17 development and microbiota-dependent colitis. J Immunol. 2014;192:3958–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruiz PA, Shkoda A, Kim SC, et al. IL-10 gene-deficient mice lack TGF-beta/Smad-mediated TLR2 degradation and fail to inhibit proinflammatory gene expression in intestinal epithelial cells under conditions of chronic inflammation. Ann N Y Acad Sci. 2006;1072:389–394. [DOI] [PubMed] [Google Scholar]
- 26.Kole A, Maloy KJ. Control of intestinal inflammation by interleukin-10. Curr Top Microbiol Immunol. 2014;380:19–38. [DOI] [PubMed] [Google Scholar]
- 27.Werner T, Shkoda A, Haller D. Intestinal epithelial cell proteome in IL-10 deficient mice and IL-10 receptor reconstituted epithelial cells: impact on chronic inflammation. J Proteome Res. 2007;6:3691–3704. [DOI] [PubMed] [Google Scholar]
- 28.Shkoda A, Ruiz PA, Daniel H, et al. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. 2007;132:190–207. [DOI] [PubMed] [Google Scholar]
- 29.Veenbergen S, Samsom JN. Maintenance of small intestinal and colonic tolerance by IL-10-producing regulatory T cell subsets. Curr Opin Immunol. 2012;24:269–276. [DOI] [PubMed] [Google Scholar]
- 30.Rubtsov YP, Rasmussen JP, Chi EY, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. [DOI] [PubMed] [Google Scholar]
- 31.Mantovani A, Marchesi F. IL-10 and macrophages orchestrate gut homeostasis. Immunity. 2014;40:637–639. [DOI] [PubMed] [Google Scholar]
- 32.Shouval DS, Biswas A, Goettel JA, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zigmond E, Bernshtein B, Friedlander G, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40:720–733. [DOI] [PubMed] [Google Scholar]
- 34.Pils MC, Bleich A, Prinz I, et al. Commensal gut flora reduces susceptibility to experimentally induced colitis via T-cell-derived interleukin-10. Inflamm Bowel Dis. 2011;17:2038–2046. [DOI] [PubMed] [Google Scholar]
- 35.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. [DOI] [PubMed] [Google Scholar]
- 38.Mähler M, Leiter EH. Genetic and environmental context determines the course of colitis developing in IL-10-deficient mice. Inflamm Bowel Dis. 2002;8:347–355. [DOI] [PubMed] [Google Scholar]
- 39.Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. [DOI] [PubMed] [Google Scholar]
- 40.Bristol IJ, Farmer MA, Cong Y, et al. Heritable susceptibility for colitis in mice induced by IL-10 deficiency. Inflamm Bowel Dis. 2000;6:290–302. [DOI] [PubMed] [Google Scholar]
- 41.Farmer MA, Sundberg JP, Bristol IJ, et al. A major quantitative trait locus on chromosome 3 controls colitis severity in IL-10-deficient mice. Proc Natl Acad Sci U S A. 2001;98:13820–13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahler M, Most C, Schmidtke S, et al. Genetics of colitis susceptibility in IL-10-deficient mice: backcross versus F2 results contrasted by principal component analysis. Genomics. 2002;80:274–282. [DOI] [PubMed] [Google Scholar]
- 43.Jurisic G, Sundberg JP, Bleich A, et al. Quantitative lymphatic vessel trait analysis suggests Vcam1 as candidate modifier gene of inflammatory bowel disease. Genes Immun. 2010;11:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beckwith J, Cong Y, Sundberg JP, et al. Cdcs1, a major colitogenic locus in mice, regulates innate and adaptive immune response to enteric bacterial antigens. Gastroenterology. 2005;129:1473–1484. [DOI] [PubMed] [Google Scholar]
- 45.Bleich A, Buchler G, Beckwith J, et al. Cdcs1 a major colitis susceptibility locus in mice; subcongenic analysis reveals genetic complexity. Inflamm Bowel Dis. 2010;16:765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ermann J, Garrett WS, Kuchroo J, et al. Severity of innate immune-mediated colitis is controlled by the cytokine deficiency-induced colitis susceptibility-1 (Cdcs1) locus. Proc Natl Acad Sci U S A. 2011;108:7137–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buettner M, Bleich A. Mapping colitis susceptibility in mouse models: distal chromosome 3 contains major loci related to Cdcs1. Physiol Genomics. 2013;45:925–930. [DOI] [PubMed] [Google Scholar]
- 48.Bruesch I, Keubler LM, Messner B, et al. Relation of Cdcs1 and the development of a colitogenic T cell population. Paper presented at: 7th Seeon Conference: Microbiota, Probiota and Host. Monastery Seeon, Germany; 2014:29.
- 49.de Buhr MF, Hedrich HJ, Westendorf AM, et al. Analysis of Cd14 as a genetic modifier of experimental inflammatory bowel disease (IBD) in mice. Inflamm Bowel Dis. 2009;15:1824–1836. [DOI] [PubMed] [Google Scholar]
- 50.de Buhr MF, Mahler M, Geffers R, et al. Cd14, Gbp1, and Pla2g2a: three major candidate genes for experimental IBD identified by combining QTL and microarray analyses. Physiol Genomics. 2006;25:426–434. [DOI] [PubMed] [Google Scholar]
- 51.Bleich A, Mahler M. Environment as a critical factor for the pathogenesis and outcome of gastrointestinal disease: experimental and human inflammatory bowel disease and helicobacter-induced gastritis. Pathobiology. 2005;72:293–307. [DOI] [PubMed] [Google Scholar]
- 52.Sellon RK, Tonkonogy S, Schultz M, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sydora BC, Tavernini MM, Wessler A, et al. Lack of interleukin-10 leads to intestinal inflammation, independent of the time at which luminal microbial colonization occurs. Inflamm Bowel Dis. 2003;9:87–97. [DOI] [PubMed] [Google Scholar]
- 54.Hoentjen F, Harmsen HJ, Braat H, et al. Antibiotics with a selective aerobic or anaerobic spectrum have different therapeutic activities in various regions of the colon in interleukin 10 gene deficient mice. Gut. 2003;52:1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–5744. [PubMed] [Google Scholar]
- 56.Tamagawa H, Hiroi T, Mizushima T, et al. Therapeutic effects of roxithromycin in interleukin-10-deficient colitis. Inflamm Bowel Dis. 2007;13:547–556. [DOI] [PubMed] [Google Scholar]
- 57.Hans W, Scholmerich J, Gross V, et al. The role of the resident intestinal flora in acute and chronic dextran sulfate sodium-induced colitis in mice. Eur J Gastroenterol Hepatol. 2000;12:267–273. [DOI] [PubMed] [Google Scholar]
- 58.Madsen KL, Doyle JS, Tavernini MM, et al. Antibiotic therapy attenuates colitis in interleukin 10 gene-deficient mice. Gastroenterology. 2000;118:1094–1105. [DOI] [PubMed] [Google Scholar]
- 59.Madsen KL. Inflammatory bowel disease: lessons from the IL-10 gene-deficient mouse. Clin Invest Med. 2001;24:250–257. [PubMed] [Google Scholar]
- 60.Karrasch T, Kim JS, Muhlbauer M, et al. Gnotobiotic IL-10-/-;NF-kappa B(EGFP) mice reveal the critical role of TLR/NF-kappa B signaling in commensal bacteria-induced colitis. J Immunol. 2007;178:6522–6532. [DOI] [PubMed] [Google Scholar]
- 61.Uronis JM, Muhlbauer M, Herfarth HH, et al. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4:e6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity. 2006;25:319–329. [DOI] [PubMed] [Google Scholar]
- 63.Rachmilewitz D, Karmeli F, Takabayashi K, et al. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology. 2002;122:1428–1441. [DOI] [PubMed] [Google Scholar]
- 64.Bleich A, Janus LM, Smoczek A, et al. CpG motifs of bacterial DNA exert protective effects in mouse models of IBD by antigen-independent tolerance induction. Gastroenterology. 2009;136:278–287. [DOI] [PubMed] [Google Scholar]
- 65.Schultz M, Veltkamp C, Dieleman LA, et al. Lactobacillus plantarum 299V in the treatment and prevention of spontaneous colitis in interleukin-10-deficient mice. Inflamm Bowel Dis. 2002;8:71–80. [DOI] [PubMed] [Google Scholar]
- 66.Burich A, Hershberg R, Waggie K, et al. Helicobacter-induced inflammatory bowel disease in IL-10- and T cell-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2001;281:G764–G778. [DOI] [PubMed] [Google Scholar]
- 67.Kullberg MC, Ward JM, Gorelick PL, et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect Immun. 1998;66:5157–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balish E, Warner T. Enterococcus faecalis induces inflammatory bowel disease in interleukin-10 knockout mice. Am J Pathol. 2002;160:2253–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McCarthy J, O'Mahony L, O'Callaghan L, et al. Double blind, placebo controlled trial of two probiotic strains in interleukin 10 knockout mice and mechanistic link with cytokine balance. Gut. 2003;52:975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim SC, Tonkonogy SL, Albright CA, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. [DOI] [PubMed] [Google Scholar]
- 71.Kim SC, Tonkonogy SL, Karrasch T, et al. Dual-association of gnotobiotic IL-10-/- mice with 2 nonpathogenic commensal bacteria induces aggressive pancolitis. Inflamm Bowel Dis. 2007;13:1457–1466. [DOI] [PubMed] [Google Scholar]
- 72.Barnett MP, McNabb WC, Cookson AL, et al. Changes in colon gene expression associated with increased colon inflammation in interleukin-10 gene-deficient mice inoculated with Enterococcus species. BMC Immunol. 2010;11:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoffmann M, Messlik A, Kim SC, et al. Impact of a probiotic Enterococcus faecalis in a gnotobiotic mouse model of experimental colitis. Mol Nutr Food Res. 2011;55:703–713. [DOI] [PubMed] [Google Scholar]
- 74.Dieleman LA, Arends A, Tonkonogy SL, et al. Helicobacter hepaticus does not induce or potentiate colitis in interleukin-10-deficient mice. Infect Immun. 2000;68:5107–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Whary MT, Taylor NS, Feng Y, et al. Lactobacillus reuteri promotes Helicobacter hepaticus-associated typhlocolitis in gnotobiotic B6.129P2-IL-10(tm1Cgn) (IL-10(-/-)) mice. Immunology. 2011;133:165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Buchler G, Wos-Oxley ML, Smoczek A, et al. Strain-specific colitis susceptibility in IL10-deficient mice depends on complex gut microbiota-host interactions. Inflamm Bowel Dis. 2012;18:943–954. [DOI] [PubMed] [Google Scholar]
- 77.Basic M, Keubler LM, Buettner M, et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm Bowel Dis. 2014;20:431–443. [DOI] [PubMed] [Google Scholar]
- 78.Devkota S, Wang Y, Musch MW, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bloom SM, Bijanki VN, Nava GM, et al. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe. 2011;9:390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eun CS, Mishima Y, Wohlgemuth S, et al. Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10-/- mice. Infect Immun. 2014;82:2239–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Madsen K, Cornish A, Soper P, et al. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology. 2001;121:580–591. [DOI] [PubMed] [Google Scholar]
- 82.Yang I, Eibach D, Kops F, et al. Intestinal microbiota composition of interleukin-10 deficient C57BL/6J mice and susceptibility to Helicobacter hepaticus-induced colitis. PloS One. 2013;8:e70783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nagalingam NA, Robinson CJ, Bergin IL, et al. The effects of intestinal microbial community structure on disease manifestation in IL-10-/- mice infected with Helicobacter hepaticus. Microbiome. 2013;1:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cadwell K, Patel KK, Maloney NS, et al. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hunter P. The secret garden's gardeners. Research increasingly appreciates the crucial role of gut viruses for human health and disease. EMBO Rep. 2013;14:683–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Khan RR, Lawson AD, Minnich LL, et al. Gastrointestinal norovirus infection associated with exacerbation of inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2009;48:328–333. [DOI] [PubMed] [Google Scholar]
- 88.Bleich A, Hansen AK. Time to include the gut microbiota in the hygienic standardisation of laboratory rodents. Comp Immunol Microbiol Infect Dis. 2012;35:81–92. [DOI] [PubMed] [Google Scholar]
- 89.Tamboli CP, Neut C, Desreumaux P, et al. Dysbiosis as a prerequisite for IBD. Gut. 2004;53:1057. [PMC free article] [PubMed] [Google Scholar]
- 90.Sartor RB. Induction of mucosal immune responses by bacteria and bacterial components. Curr Opin Gastroenterol. 2001;17:555–561. [DOI] [PubMed] [Google Scholar]
- 91.Ott SJ, Musfeldt M, Wenderoth DF, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Walker AW, Sanderson JD, Churcher C, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carriere J, Darfeuille-Michaud A, Nguyen HT. Infectious etiopathogenesis of Crohn's disease. World J Gastroenterol. 2014;20:12102–12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Martinez-Medina M, Aldeguer X, Gonzalez-Huix F, et al. Abnormal microbiota composition in the ileocolonic mucosa of Crohn's disease patients as revealed by polymerase chain reaction-denaturing gradient gel electrophoresis. Inflamm Bowel Dis. 2006;12:1136–1145. [DOI] [PubMed] [Google Scholar]
- 95.Mondot S, Kang S, Furet JP, et al. Highlighting new phylogenetic specificities of Crohn's disease microbiota. Inflamm Bowel Dis. 2011;17:185–192. [DOI] [PubMed] [Google Scholar]
- 96.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. [DOI] [PubMed] [Google Scholar]
- 97.Madsen KL, Doyle JS, Jewell LD, et al. Lactobacillus species prevents colitis in interleukin 10 gene-deficient mice. Gastroenterology. 1999;116:1107–1114. [DOI] [PubMed] [Google Scholar]
- 98.Knoch B, Nones K, Barnett MP, et al. Diversity of caecal bacteria is altered in interleukin-10 gene-deficient mice before and after colitis onset and when fed polyunsaturated fatty acids. Microbiology. 2010;156:3306–3316. [DOI] [PubMed] [Google Scholar]
- 99.Maharshak N, Packey CD, Ellermann M, et al. Altered enteric microbiota ecology in interleukin 10-deficient mice during development and progression of intestinal inflammation. Gut Microbes. 2013;4:316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wohlgemuth S, Keller S, Kertscher R, et al. Intestinal steroid profiles and microbiota composition in colitic mice. Gut Microbes. 2011;2:159–166. [DOI] [PubMed] [Google Scholar]
- 101.Basso PJ, Fonseca MT, Bonfa G, et al. Association among genetic predisposition, gut microbiota, and host immune response in the etiopathogenesis of inflammatory bowel disease. Braz J Med Biol Res. 2014;47:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]