

Abstract
Meganema perideroedes Gr1T is a filamentous bacterium isolated from an activated sludge wastewater treatment plant where it is implicated in poor sludge settleability (bulking). M. perideroedes is the sole described species of the genus Meganema and of the proposed novel family “Meganemaceae”. Here we describe the features of the type strain Gr1T along with its annotated genome sequence. The 3,409,949 bp long draft genome consists of 22 scaffolds with 3,033 protein-coding and 59 RNA genes and is a part of Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes KMG project. Notably, genome annotation indicated the potential for facultative methylotrophy. However, the ability to utilize methanol as a carbon source could not be empirically demonstrated for the type strain or for in situ Meganema spp. strains.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-015-0013-1) contains supplementary material, which is available to authorized users.
Keywords: Activated sludge, Bulking, Facultative methylotroph, Filamentous, Meganema, Meganemaceae, Wastewater
Introduction
Strain Gr1T (= DSM 15528 = ATCC BAA-740) is the type strain of Meganema perideroedes in the monospecific genus Meganema [1]. M. perideroedes is a filamentous bacterium isolated from an activated sludge WWTP in Denmark. All current isolates, along with 16S rRNA gene clone sequences in public databases, were isolated from activated sludge related sources (see Figure 1). High abundance of the filamentous form in these systems, though rarely reported, is associated with the sludge settleability problems known as bulking, and is therefore undesired [1,2]. Meganema spp. are often detected in lab-scale SBR systems optimized for PHA production for valuable bioplastics manufacture [3-8], and have a relatively high capacity for intracellular storage of such compounds [9], making them of potential biotechnological interest. Here we describe the features of the type strain Gr1T along with its annotated genome sequence. The 3,409,949 bp long draft genome consists of 22 scaffolds with the 3,033 protein-coding and 59 RNA genes and is a part of Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes (KMG) project.
Figure 1.
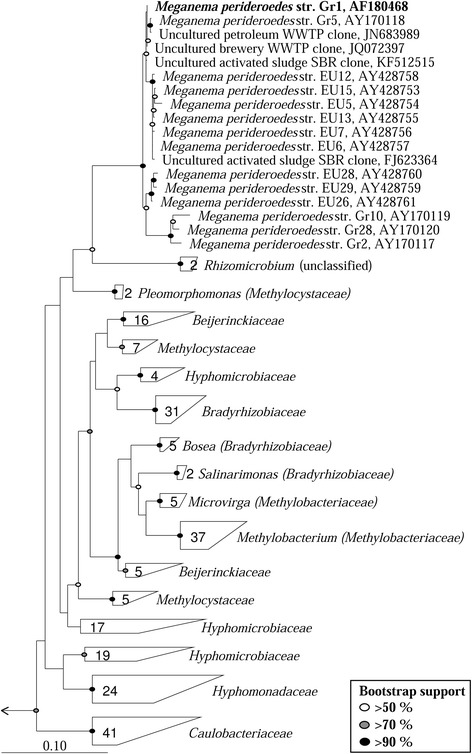
Maximum-likelihood phylogenetic tree of 16S rRNA genes for all Meganema isolates and closely related species in the LTP database (LTPs111) [ 10 ] constructed using the ARB software [ 13 ]. The 1342 bp long sequence fragment of the unique 16S rRNA gene copy in the genome is identical with the previously published 16S rRNA gene sequence for the Gr1T strain (AF18048). A 20% maximum frequency filter was applied in order to remove hypervariable positions. Included are all uncultured clone sequences from the NCBI database which share ≥94% sequence similarity with the Gr1T strain (all were ≥ 98%), with sequences from the same study clustered at ≥ 99% similarity and a representative included. Bootstrap values, calculated from 100 re-samplings, are indicated for branches with > 50% support. Scale bar represents substitutions per nucleotide. The family Fusobacteriaceae was used as the out-group.
Classification and features
M. perideroedes Gr1T was initially reported to be affiliated with the Methylobacterium/Xanthobacter group based on the common major fatty acid C18:1ω7c [1], which presumably led to its later classification to the family Methylobacteriaceae in ‘The All-Species Living Tree Project Database’ (release LTPs111) [10]. However, in The Prokaryotes Manual 4thEdition, this classification was suggested to be erroneous [11]. The need for reclassification was primarily based on the lack of 16S rRNA gene relatedness of the M. perideroedes and other members of the Methylobacteriaceae family (see Figure 1), which was originally transcribed based solely on 16S rRNA based phylogeny [12]. Kelly and others [11] noted that M. perideroedes had no closely related species (none > 90% 16S rRNA gene similarity), with no phenotypic traits that specifically associate it with other described bacterial families. As such, the authors suggested that Meganema be classified as a novel family, designated “Meganemaceae”, within the order Rhizobiales, or alternatively transferred to the family Caulobacteraceae within the order Caulobacterales based on Greengenes taxonomy [13]. However, the latest release of the Greengenes taxonomy (October 2012) no longer classifies Meganema as such, and phylogenetic analysis does not appear to support its inclusion in either the Methylobacteriaceae or Caulobacteriaceae families (Figure 1). Therefore, we propose that the genus be classified to the novel family “Meganemaceae”.
General features of M. perideroedes Gr1T are summarized in Table 1. The strain exhibits a filamentous morphology with irregular disc shaped cells that are approximately 1.5-2 μm in diameter and Gram stain negative (Figure 2). They are non-motile and oxidase and catalase positive. Growth is observed in the presence of NaCl up to 2% [w/v] and between 15–35°C, with an optimum growth temperature of 25–30°C. In pure culture they produce off-white cohesive colonies that are difficult to separate. Cells are Nile Blue and Neisser stain positive, indicating intracellular lipid and polyphosphate inclusions, respectively. They have a demonstrated aerobic organoheterotrophic metabolism and are unable to utilize nitrate as an electron acceptor [1]. Starch or tributyrin are not hydrolysed. Carbon sources supporting growth of the Gr1T isolate are unknown, although in situ strains of the genus were observed in activated sludge, with FISH-MAR, to assimilate acetate, propionate, butyrate, oleic acid, glucose, galactose, mannose, glycine and leucine, but not formate, pyruvate or ethanol [9].
Table 1.
Classification and general features of M. perideroedes Gr1 T [14]
| MIGS ID | Property | Term | Evidence code a |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [15] | |
| Phylum Proteobacteria | TAS [16] | ||
| Class Alphaproteobacteria | TAS [17] | ||
| Order Rhizobiales | TAS [18] | ||
| Family “Meganemaceae” | TAS [11] | ||
| Genus Meganema | TAS [1] | ||
| Species M. perideroedes | TAS [1] | ||
| Type strain Gr1 | TAS [1] | ||
| Gram stain | Negative | TAS [1] | |
| Cell shape | Irregular disc-shaped in filaments | TAS [1] | |
| Motility | Non-motile | TAS [1] | |
| Sporulation | Non-sporulating | NAS | |
| Temperature range | 15-35°C | TAS [1] | |
| Optimum temperature | 25-30°C | TAS [1] | |
| pH range; Optimum | Not reported | NAS | |
| Carbon source | Varied | NAS | |
| MIGS-6 | Habitat | Activated sludge | TAS [1,2,9] |
| MIGS-6.3 | Salinity | Not reported | NAS |
| MIGS-22 | Oxygen requirement | Aerobic | TAS [1] |
| MIGS-15 | Biotic relationship | Free-living | TAS [1] |
| MIGS-14 | Pathogenicity | Non-pathogen | NAS |
| MIGS-4 | Geographic location | Grindsted, Denmark | TAS [1] |
| MIGS-5 | Sample collection time | Not reported | NAS |
| MIGS-4.1 | Latitude | 55.758 | NAS |
| MIGS-4.2 | Longitude | 8.924 | NAS |
| MIGS-4.4 | Altitude | 41 m | NAS |
aEvidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence) [19].
Figure 2.
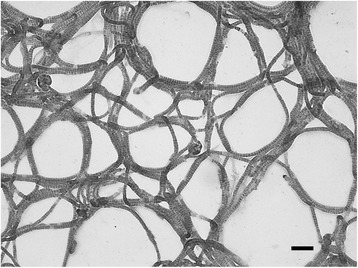
Grey-scale brightfield micrograph of M. perideroedes Gr1 T stained with safranin O. Scale bar represents 10 μm.
The main respiratory quinone is Q-10 and the fatty acid profile is dominated by C18:1ω7c (86.4%) with smaller amounts of C18:0 (3.8%), C16:0 (2.9%), summed feature 2 (C14:0 3-OH, C16:1 iso I)(2.4%), C18:0 3-OH (2.3%) and C19:0 10-methyl (1.1%) [1].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [20,21]. Sequencing strain Gr1T (DSM 15528T) is part of the Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes KMG project [22], a follow-up of the GEBA project [23], which aims to increase the sequencing coverage of key reference microbial genomes. The genome project is deposited in the Genomes OnLine Database [24] and the permanent draft genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE JGI using state of the art sequencing technology [25]. A summary of the project information is shown in Table 2.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Level 2: High-Quality Draft |
| MIGS-28 | Libraries used | Illumina Std. shotgun library |
| MIGS-29 | Sequencing platforms | Illumina HiSeq 2000, |
| MIGS-31.2 | Fold coverage | 620.1 × |
| MIGS-30 | Assemblers | Velvet v. 1.1.04; ALLPATHS v. r41043 |
| MIGS-32 | Gene calling method | Prodigal |
| Locus tag | B161DRAFT | |
| Genbank ID | ARFG00000000 | |
| Genbank Date of Release | December 12, 2013 | |
| GOLD ID | Gp0013029 | |
| BIOPROJECT | 169830 | |
| Project relevance | Tree of Life, GEBA-KMG | |
| MIGS-13 | Source material identifier | DSM 15528 |
Growth conditions and genomic DNA preparation
M. perideroedes Gr1T, DSM 15528, was grown in R2A medium (DSMZ medium 830) at 25°C [26]. DNA was isolated from 0.5-1.0 g of cell paste using Jetflex DNA purification kit (GENOMED 600100) following the standard protocol provided by the manufacturer but modified by an incubation time of 60 min, incubation on ice overnight on a shaker, the use of an additional 50 μl proteinase K, and the addition of 100 μl protein precipitation buffer. DNA is available through the DNA Bank Network [27].
Genome sequencing and assembly
The draft genome sequence was generated using the Illumina technology [28]. An Illumina Standard shotgun library was constructed and sequenced using the Illumina HiSeq 2000 platform which generated 14,100,926 reads totaling 2,115.1 Mbp. All general aspects of library construction and sequencing performed at the JGI can be found at [29]. All raw Illumina sequence data was passed through DUK, a filtering program developed at JGI, which removes known Illumina sequencing and library preparation artifacts (Mingkun L, Copeland A, Han J. DUK. 2011, in preparation). The following steps were then performed for assembly: (1) filtered Illumina reads were assembled using Velvet [30], (2) 1–3 kbp simulated paired end reads were created from Velvet contigs using wgsim [31], (3) Illumina reads were assembled with simulated read pairs using Allpaths–LG [23]. Parameters for assembly steps were: 1) Velvet (velveth: 63 –shortPaired and velvetg: −very clean yes –export-Filtered yes –min contig lgth 500 –scaffolding no –cov cutoff 10) 2) wgsim (−e 0 –1 100 –2 100 –r 0 –R 0 –X 0) 3) Allpaths–LG (PrepareAllpathsInputs: PHRED 64 = 1 PLOIDY = 1 FRAG COVERAGE = 125 JUMP COVERAGE = 25 LONG JUMP COV = 50, RunAllpathsLG: THREADS = 8 RUN = std shredpairs TARGETS = standard VAPI WARN ONLY = True OVERWRITE = True). The final draft assembly contained 22 contigs in 22 scaffolds. The total size of the genome is 3.4 Mbp and the final assembly is based on 368.8 Mbp of Illumina data, which provides an average 108.2 × coverage of the genome.
Genome annotation
Genes were identified using Prodigal [32] as part of the DOE-JGI genome annotation pipeline [33], followed by a round of manual curation using the JGI GenePRIMP pipeline [34]. The predicted CDSs were translated and used to search the NCBI non-redundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro database. These data sources were combined to assert a product description for each predicted protein. Additional gene prediction analysis and functional annotation was performed within the IMG-ER platform [35]. Pathway assessment for genomic insights also utilized the ‘MicroScope’ pipeline [36].
Genome properties
The assembly of the draft genome sequence consists of 22 scaffolds amounting to 3,409,949 bp, and the G + C content is 67.2% (Table 3). Of the 3,092 genes predicted, 3,033 were protein-coding genes, and 59 RNAs; No pseudogenes were identified. The majority of the protein-coding genes (82.0%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Table 3.
Genome statistics
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) | 3,409,949 | 100.00% |
| DNA coding (bp) | 3,058,064 | 89.68% |
| DNA G + C (bp) | 2,291,117 | 67.19% |
| DNA scaffolds | 22 | |
| Total genes | 3,092 | 100.00% |
| Protein-coding genes | 3,033 | 98.09% |
| RNA genes | 59 | 3.26% |
| Pseudogenes | 0 | 0% |
| Genes in internal clusters | Unknown | |
| Genes with function prediction | 2,536 | 82.02% |
| Genes assigned to COGs | 2,549 | 82.44% |
| Genes assigned Pfam domains | 2,617 | 84.67% |
| Genes with signal peptides | 403 | 13.03% |
| Genes with transmembrane helices | 675 | 21.83% |
| CRISPR repeats | 4 |
Table 4.
Number of genes associated with the 25 general COG functional categories
| Code | Value | % age | Description |
|---|---|---|---|
| J | 158 | 5.64 | Translation, ribosomal structure and biogenesis |
| A | 5 | 0.18 | RNA processing and modification |
| K | 147 | 5.25 | Transcription |
| L | 98 | 3.50 | Replication, recombination and repair |
| B | 1 | 0.04 | Chromatin structure and dynamics |
| D | 36 | 1.28 | Cell cycle control, cell division, chromosome partitioning |
| V | 37 | 1.32 | Defense mechanisms |
| T | 93 | 3.32 | Signal transduction mechanisms |
| M | 145 | 5.17 | Cell wall/membrane/envelope biogenesis |
| N | 17 | 0.61 | Cell motility |
| U | 69 | 2.46 | Intracellular trafficking, secretion, and vesicular transport |
| O | 124 | 4.43 | Posttranslational modification, protein turnover, chaperones |
| C | 184 | 6.57 | Energy production and conversion |
| G | 172 | 6.14 | Carbohydrate transport and metabolism |
| E | 325 | 11.60 | Amino acid transport and metabolism |
| F | 68 | 2.43 | Nucleotide transport and metabolism |
| H | 133 | 4.75 | Coenzyme transport and metabolism |
| I | 122 | 4.35 | Lipid transport and metabolism |
| P | 159 | 5.67 | Inorganic ion transport and metabolism |
| Q | 91 | 3.25 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 343 | 12.24 | General function prediction only |
| S | 274 | 9.78 | Function unknown |
| - | 543 | 17.56 | Not in COGs |
The total is based on the total number of protein coding genes in the genome.
Insights from the genome sequence
Analysis of the genome of M. perideroedes Gr1T indicates the potential for storage of polyphosphate, PHAs and glycogen, with the former two polymers supported by selective stains in axenic culture and in situ strains in activated sludge [1,9]. Storage of such polymers is common in Bacteria, and shown to be key to the metabolic strategies of several activated sludge organisms, such as the PAO and GAO phenotypes [37]. The PAO utilize aerobically stored polyphosphate to energize anaerobic carbon uptake. Whilst Meganema spp. appear to be able to store polyphosphates, they are unable to assimilate carbon anaerobically [9]. This may in part be due to the absence of the low affinity phosphate Pit transport gene, suggested to be key to the use of polyphosphate for energizing anaerobic carbon uptake in the PAO [38,39]. Distribution of Meganema spp. appears somewhat restricted to industrial WWTPs, without the anaerobic tanks implemented in EBPR plants. Thus the ability to assimilate PHA likely provides advantage during intermittent periods of carbon starvation or under the unbalanced growth conditions (i.e. high COD to N:P ratio) that often characterize industrial waste streams. Such an explanation was suggested for members of the alphaproteobacterial genus Amaricoccus, which also assimilate relatively high PHA reserves and only appear in high abundance in aerated systems treating industrial wastes [40-42].
A novel finding with analysis of the Gr1T genome was the apparent potential for methylotrophic growth. Putative genes for a methanol dehydrogenase (EC. 1.1.2.7), the formaldehyde oxidation pathway (glutathione-dependent), and a formate dehydrogenase (EC 1.2.1.2), were collocated on a putative operon in the genome. These together catalyze the oxidation of methanol to carbon dioxide via formaldehyde [43]. Analysis of potential assimilatory pathways for C1 compounds [43] revealed that key genes were missing for the described serine and ribulose monophosphate pathways, but present for the CBB cycle. Therefore, methanol may be assimilated, via oxidation to CO2, through the CBB carbon fixation pathway. Such a phenotype, sometimes referred to as “pseudomethylotrophy” or “autotrophic methylotrophy” [44], has previously been demonstrated for other related members of the order Rhizobiales [43,45]. Given the annotated potential for facultative methylotrophy, experimental validation of the ability was assessed in pure culture and for in situ community strains present in an environmental sample (for details see Additional file 1). Attempts to grow strain Gr1T on media with methanol as the sole carbon source were unsuccessful. More comprehensive experimental work is required to assess the ability for, and nature of, methylotrophic growth of the Gr1T strain. Methanol assimilation was also not detected for probe-defined in situ strains of the genus in the Grindsted WWTP (Additional file 1). The same negative result was obtained for formate assimilation in previous FISH-MAR investigations of the genus [9]. Thus, the ability for methylotrophy is yet to be empirically demonstrated for the genus and the importance of methanol metabolism remains to be resolved. In the case of the activated sludge environment, utilisation of other carbon sources in situ, including stored PHA, would be more energetically favorable. Methanol and/or formate oxidation to CO2 may supplement energy derived from other sources, or may not be important substrates for these organisms in activated sludge. This is consistent with previous observations that microorganisms in environmental systems are demonstrated to have more specialized physiologies and niches despite metabolic potentials for more diverse activities [46].
Putative denitrification genes were not located in the genome, supporting axenic characterisation of the Gr1 strain, which was unable to grow with nitrate as electron acceptor [1]. In situ strains of the genus have been demonstrated to assimilate some substrates anoxically in the presence of nitrate or nitrite, indicating an ability for denitrification [9]. Thus, members of the genus appear to vary in their potential for denitrification. The capacity to fix atmospheric nitrogen is common for other methylotrophic bacteria [43], but the absence of a nitrogenase (EC 1.18.6.1) indicates that this is not the case for M. perideroedes Gr1T.
Taxonomic proposals
Description of Meganemaceae fam. nov.
Meganemaceae (Me.ga.nem.a'ce.ae. N.L. neut. n. Meganema, type genus of the family; suff. -aceae ending denoting a family; N.L. fem. pl. n. Meganemaceae, the family of Meganema).
Filamentous morphology with irregular disc shaped cells. Cells stain Gram-negative and, Nile Blue and Neisser positive. The major quinone is Q-10. Fatty acid profiles are dominated by C18:1ω7c; characteristic hydroxy acids are C14:0 3-OH and C18:0 3-OH. Meganemaceae belongs to the order Rhizobiales and the type genus is Meganema.
Conclusions
The draft genome sequence of M. perideroedes Gr1T is 3.4 Mbp, consisting of 22 scaffolds with 3,033 protein-coding genes. The annotated ability of the organism for facultative methylotrophy could not be demonstrated in either pure culture or for in situ strains. Further work is required to elucidate the role of these pathways for the organism, and why they are maintained in the genome. The genome sequence presented here provides a resource for more detailed investigations of this biotechnologically important organism. Based on 16S rRNA gene analysis we formally propose the novel family Meganemaceae to include the genus Meganema.
Acknowledgements
The authors gratefully acknowledge the assistance of J Ildal and M Stevenson at the Center for Microbial Communities for technical assistance, Anja Frühling for growing M. perideroedes cultures and Evelyne-Marie Brambilla for DNA extraction and quality control (both at the DSMZ). We also acknowledge the LABGeM team at Genoscope for providing access to their automated annotation pipeline as well as for database maintenance and technical assistance. This work was performed under the auspices of the US Department of Energy's Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344. A.L. was supported in part by Russian Ministry of Science Mega-grant no.11.G34.31.0068 (PI. Dr Stephen J O'Brien).
Abbreviations
- CBB
Calvin-Bensen-Bassham
- COD
Chemical oxygen demand
- EBPR
Enhanced biological phosphorus removal
- FISH
Fluorescence in situ hybridization
- GAO
Glycogen accumulating organism
- IMG-ER
Integrated Microbial Genomes-Expert Review
- MAR
Microautoradiography
- MIGS
Minimal information about a genome sequence
- PAO
Polyphosphate accumulating organism
- PHA
Polyhydroxyalkanoate
- WWTP
Wastewater treatment plant
Additional file
Experimental validation of methylotrophy for the Gr1 T and in situ strains of the genus Meganema.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HPK supplied the DNA to the JGI for sequencing. SJM, PHN and HPK drafted the manuscript. All other authors were involved in sequencing, annotation and analysis of the genome and/or editing the final paper. All authors read and approved the final manuscript.
Contributor Information
Simon J McIlroy, Email: sjm@bio.aau.dk.
Alla Lapidus, Email: piterlabs@gmail.com.
Trine R Thomsen, Email: trt@bio.aau.dk.
James Han, Email: JKHan@lbl.gov.
Matthew Haynes, Email: mrhaynes@lbl.gov.
Elizabeth Lobos, Email: ealobos@lbl.gov.
Marcel Huntemann, Email: MHuntemann@lbl.gov.
Amrita Pati, Email: APati@lbl.gov.
Natalia N Ivanova, Email: NNIvanova@lbl.gov.
Victor Markowitz, Email: VMMarkovitz@lbl.gov.
Susanne Verbarg, Email: susanne.verbarg@dsmz.de.
Tanja Woyke, Email: twoyke@lbl.gov.
Hans-Peter Klenk, Email: hans-peter.klenk@newcastle.ac.uk.
Nikos Kyrpides, Email: nckyrpides@lbl.gov.
Per H Nielsen, Email: phn@bio.aau.dk.
References
- 1.Thomsen TR, Blackall LL, de Muro M, Nielsen JL, Nielsen PH. Meganema perideroedes gen. nov., sp. nov., a filamentous alphaproteobacterium from activated sludge. Int J Syst Evol Microbiol. 2006;56:1865–1868. doi: 10.1099/ijs.0.02916-0. [DOI] [PubMed] [Google Scholar]
- 2.Levantesi C, Beimfohr C, Geurkink B, Rossetti S, Thelen K, Krooneman J, Snaidr J, van der Waarde J, Tandoi V. Filamentous Alphaproteobacteria associated with bulking in industrial wastewater treatment plants. Syst Appl Microbiol. 2004;27:716–727. doi: 10.1078/0723202042369974. [DOI] [PubMed] [Google Scholar]
- 3.Dionisi D, Beccari M, Di Gregorio S, Majone M, Papini MP, Vallini G. Storage of biodegradable polymers by an enriched microbial community in a sequencing batch reactor operated at high organic load rate. J Chem Technol Biotechnol. 2005;80:1306–1318. doi: 10.1002/jctb.1331. [DOI] [Google Scholar]
- 4.Dionisi D, Majone M, Vallini G, Di Gregorio S, Beccari M. Effect of the applied organic load rate on biodegradable polymer production by mixed microbial cultures in a sequencing batch reactor. Biotechnol Bioeng. 2006;93:76–88. doi: 10.1002/bit.20683. [DOI] [PubMed] [Google Scholar]
- 5.Majone M, Beccari M, Di Gregorio S, Dionisi D, Vallini G. Enrichment of activated sludge in a sequencing batch reactor for polyhydroxyalkanoate production. Water Sci Technol. 2006;54(1):119–128. doi: 10.2166/wst.2006.379. [DOI] [PubMed] [Google Scholar]
- 6.Beccari M, Bertin L, Dionisi D, Fava F, Lampis S, Majone M, Valentino F, Vallini G, Villano V. Exploiting olive oil mill effluents as a renewable resource for production of biodegradable polymers through a combined anaerobic-aerobic process. J Chem Technol Biotechnol. 2009;84:901–908. doi: 10.1002/jctb.2173. [DOI] [Google Scholar]
- 7.Werker AG, Bengtsson SOH, Morgan-Sagastume F, Karlsson CAB, Blanchet EM. Method of treating wastewater and producing an activated sludge having a high biopolymer production potential. 2012. Patent No. US20120305478.
- 8.Valentino F, Beccari M, Fraraccio S, Zanaroli G, Majone M. Feed frequency in a sequencing batch reactor strongly affects the production of polyhydroxyalkanoates (PHAs) from volatile fatty acids. N Biotechnol. 2014;31:264–275. doi: 10.1016/j.nbt.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Kragelund C, Nielsen JL, Thomsen TR, Nielsen PH. Ecophysiology of the filamentous alphaproteobacterium Meganema perideroedes in activated sludge. FEMS Microbiol Ecol. 2005;54:111–122. doi: 10.1016/j.femsec.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Yarza P, Ludwig W, Euzeby J, Amann R, Schleifer KH, Glockner FO, Rossello-Mora R. Update of the All-Species Living Tree Project based on 16S and 23S rRNA sequence analyses. Syst Appl Microbiol. 2010;33:291–299. doi: 10.1016/j.syapm.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Kelly DP, McDonald IR, Wood AP. Family Methylobacteriaceae. In: Rosenberg E, Delong EF, Lory S, Stackebrandt E, Thompson F, editors. The Prokaryotes – Alphaproteobacteria and Betaproteobacteria. Heidelberg: Springer-Verlag; 2014. [Google Scholar]
- 12.Garrity G, Bell J, Lilburn T. Family IX. Methylobacteriaceae fam. nov. In: Brenner D, Krieg N, Staley J, Garrity G, editors. Bergey’s Manual of Systematic Bacteriology Part C: The Alpha- Beta-, Delta- and Epsilonproteobacteria Vol. 2. New York: Springer; 2005. pp. 567–571. [Google Scholar]
- 13.Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, Buchner A, Lai T, Steppi S, Jobb G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüssmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, Dawyndt P, Guralnick R, Goldstein P, Hall N, Hirschman L, Kravitz S, Lister AL, Markowitz V, Thomson N, Whetzel T. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the do-mains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s Manual of Systematic Bacteriology, 2nd edition, vol. 2 (The Proteobacteria), part B (The Gammaproteobacteria) New York: Springer; 2005. p. 1. [Google Scholar]
- 17.Garrity GM, Bell JA, Lilburn T. Class I.Alphaproteobacteriaclass. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s Manual of Systematic Bacteriology, 2nd edition, vol. 2 (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria) New York: Springer; 2005. p. 1. [Google Scholar]
- 18.Kuykendall LD. Order VI.Rhizobialesord. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s Manual of Systematic Bacteriology, 2nd edn, vol. 2 (The Proteobacteria), part C (The Alpha-,Beta-,Delta-, and Epsilonproteobacteria) New York: Springer; 2005. p. 324. [Google Scholar]
- 19.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klenk HP, Göker M. En route to a genome - based classification of Archaea and Bacteria? Syst Appl Microbiol. 2010;33:175–182. doi: 10.1016/j.syapm.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Göker M, Klenk HP. Phylogeny-driven target selection for genome-sequencing (and other) projects. Stand Genomic Sci. 2013;8:360–374. doi: 10.4056/sigs.3446951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kyrpides NC, Woyke T, Eisen JA, Garrity G, Lilburn TG, Beck BJ, Whitman WB, Hugenholz P, Klenk HP. Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes (KMG-I) project. Stand Genomic Sci. 2013;9:628–634. doi: 10.4056/sigs.5068949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, Hooper SD, Pati A, Lykidis A, Spring S, Anderson IJ, D’haeseleer P, Zemla A, Singer M, Lapidus A, Nolan M, Copeland A, Han C, Chen F, Cheng JF, Lucas S, Kerfeld C, Lang E, Gronow S, Chain P, Bruce D, et al. A phylogeny-driven Genomic Encyclopaedia ofBacteriaandArchaea. Nature. 2009;462:1056–1060. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v. 4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012;40:D571–D579. doi: 10.1093/nar/gkr1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, Goodwin L, Woyke T, Lapidus A, Klenk HP, Cottingham RE, Kyrpides NC. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS One. 2012;7:e48837. doi: 10.1371/journal.pone.0048837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.List of growth media used at the DSMZhttp://www.dmsz.de/catalogues/cataloque-microorganisms/culture-technology/list-of-media-for-microorganisms.html.
- 27.Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch AM, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreserv Biobank. 2011;9:51–55. doi: 10.1089/bio.2010.0029. [DOI] [PubMed] [Google Scholar]
- 28.Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5:433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- 29.JGI web sitehttp://www.jgi.doe.gov.
- 30.Zerbino D, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Githubhttps://github.com/lh3/wgsim.
- 32.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiati on site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci. 2009;1:63–67. doi: 10.4056/sigs.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods. 2010;7:455–457. doi: 10.1038/nmeth.1457. [DOI] [PubMed] [Google Scholar]
- 35.Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 36.Vallenet D, Engelen S, Mornico D, Cruveiller S, Fleury L, Lajus A, Rouy Z, Roche D, Salvignol G, Scarpelli C, Medigue C. MicroScope: a platform for microbial genome annotation and comparative genomics. Database (Oxford) 2009;2009:bap021. doi: 10.1093/database/bap021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seviour RJ, Mino T, Onuki M. The microbiology of biological phosphorus removal in activated sludge systems. FEMS Microbiol Rev. 2003;27:99–127. doi: 10.1016/S0168-6445(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 38.García Martín H, Ivanova N, Kunin V, Warnecke F, Barry KW, McHardy AC, Yeates C, He S, Salamov AA, Szeto E, Dalin E, Putnam NH, Shapiro HJ, Pangilinan JL, Rigoutsos I, Kyrpides NC, Blackall LL, McMahon KD, Hugenholtz P. Metagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communities. Nat Biotechnol. 2006;24:1263–1269. doi: 10.1038/nbt1247. [DOI] [PubMed] [Google Scholar]
- 39.McIlroy SJ, Albertsen M, Andresen EK, Saunders AM, Kristiansen R, Stokholm-Bjerregaard M, Nielsen KL, Nielsen PH. “Candidatus Competibacter”-lineage genomes retrieved from metagenomes reveal functional metabolic diversity. ISME J. 2014;8:613–624. doi: 10.1038/ismej.2013.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falvo A, Levantesi C, Rossetti S, Seviour R, Tandoi V. Synthesis of intracellular storage polymers by Amaricoccus kaplicensis, a tetrad forming bacterium present in activated sludge. J Appl Microbiol. 2001;91:299–305. doi: 10.1046/j.1365-2672.2001.01384.x. [DOI] [PubMed] [Google Scholar]
- 41.McIlroy SJ, Speirs LB, Tucci J, Seviour RJ. In situ profiling of microbial communities in full-scale aerobic sequencing batch reactors treating winery waste in Australia. Environ Sci Technol. 2011;45:8794–8803. doi: 10.1021/es2018576. [DOI] [PubMed] [Google Scholar]
- 42.McIlroy SJ. Phylogenetic diversity and ecophysiology of alphaproteobacterial glycogen accumulating organisms in enhanced biological phosphorus removal activated sludge systems. In: PhD Thesis. La Trobe University, School of Pharmacy and Applied Science; 2010.
- 43.Lidstrom ME. Aerobic methylotrophic prokaryotes. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E, editors. The Prokaryotes, vol. 2. New York: Springer; 2006. pp. 618–634. [Google Scholar]
- 44.Anthony C. The Biochemistry of Methylotrophs. New York: Academic Press; 1982. [Google Scholar]
- 45.Dedysh SN, Smirnova KV, Khmelenina VN, Suzina NE, Liesack W, Trotsenko YA. Methylotrophic autotrophy in Beijerinckia mobilis. J Bacteriol. 2005;187:3884–3888. doi: 10.1128/JB.187.11.3884-3888.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kindaichi T, Nierychlo M, Kragelund C, Nielsen JL, Nielsen PH. High and stable substrate specificities of microorganisms in enhanced biological phosphorus removal plants. Environ Microbiol. 2013;15:1821–1831. doi: 10.1111/1462-2920.12074. [DOI] [PubMed] [Google Scholar]