

Abstract
Recognition of hepatitis C virus (HCV)-infected hepatocyes and interferon (IFN) induction are critical in antiviral immune response. We hypothesized that cell–cell contact between plasmacytoid dendritic cells (pDCs) and HCV-infected cells was required for IFNα induction via involvement of cell surface molecules. Co-culture of human peripheral blood mononuclear cells (PBMCs) with genotype 1a full length HCV genomic replicon cells (FL) or genotype 2a JFH-1 virus infected hepatoma cells (JFH-1), not with uninfected hepatoma cells (Huh7.5), induced IFNα production. Depletion of pDCs from PBMCs attenuated IFNα release and purified pDCs produced high levels of IFNα after co-culture with FL replicons or JFH-1 infected cells. IFNα induction by HCV-containing hepatoma cells required viral replication, direct cell–cell contact with pDCs, and receptor-mediated endocytosis. We determined that the tetraspanin proteins, CD81 and CD9 and not other HCV entry receptors were required for IFNα induction in pDCs by HCV infected hepatoma cells. Disruption of cholesterol-rich membrane microdomains, the localization site of CD81 or inhibition of CD81 downstream molecule, Rac GTPase, inhibited IFNα production. IFNα induction involved HCV RNA and Toll-like receptor 7 (TLR7). IFNα production by HCV infected hepatoma cells was decreased in pDCs from HCV infected patients compared to normal controls. We found that pre-exposure of normal PBMCs to HCV viral particles attenuated IFNα induction by HCV infected hepatoma cells or TLR ligands and this inhibitory effect could be prevented by an anti-HCV E2 blocking antibody. In conclusion, our novel data show that recognition of HCV-infected hepatoma cells by pDCs involves CD81/CD9-associated membrane microdomains and induces potent IFNα production.
Keywords: TEM, Rac GTPase, HCV E2, cell, cell contact, lipid rafts
Introduction
Hepatitis C virus (HCV), a positive-sense single-stranded RNA virus, infects human hepatocytes (1). The host immune defense determines successful HCV elimination or chronic infection that often leads to cirrhosis and hepatocellular carcinoma (2). Type I IFNs play a central role in anti-HCV immunity and peg-IFNα plus ribavirin is used clinically for HCV eradication (3). The process by which innate immune cells recognize HCV-infected hepatocytes is poorly understood. Although HCV infection activates IFNβ synthesis through the RIG-I and TLR3 pathways, this response is attenuated due to the serine protease activity of HCV NS3/4A that cleaves the adaptor proteins MAVS and TRIF, respectively (4, 5). Despite this, IFN-stimulated genes (ISGs) are expressed at high levels in the liver in chronic HCV infection raising the possibility that other cell types and/or IFNs may participate in induction of ISGs (6). Indeed, HCV infected livers are enriched with plasmacytoid dendritic cells (pDCs), a cell population with a high Type I IFN production capacity (7).
Dendritic cells (DCs) bridge innate and adaptive immunity and play a central role in anti-viral defense. Human mDCs express TLR3 which sense dsRNA and produce IFNβ, while pDCs are specialized to produce IFNα in amounts larger than any other cell types (8). Although TLR7 is highly expressed in pDCs and senses single-stranded RNA within the endosomal compartment (9), previous studies found that HCVcc (cell culture derived HCV particles) alone cannot or weakly induce mDC or pDC activation and HCVcc even inhibited pDC function upon TLR9 stimulation (10–12). A recent report showed that pDCs produced type I IFNs through a TLR7-dependent pathway in response to Huh7.5 cells containing replicating HCV RNA (13). However, it remains to be answered how pDCs recognize HCV hidden in infected hepatocytes to mount an interferon response.
HCV entry is a multi-step process where interactions between HCV E1–E2 envelope glycoproteins and glycosaminoglycans (GAGs) contribute to binding of the virus particles to host cells. The LDL receptor (LDLr) has been proposed as a capture molecule. Upon this initial engagement, the scavenger receptor B1 (SR-B1), a tetraspanin protein - CD81, together with the tight junction proteins, claudin-1 and occludin, synergistically contribute to HCV entry (14, 15). In this study, we evaluated the involvement of cell surface molecules and found a novel mechanism for recognition of HCV in infected hepatoma cells by pDCs which involves membrane microdomains formed by CD81- and CD9-associated tetraspanin webs and lipid rafts. We speculate that this novel pathway is critical for activation of pDCs in the liver tissue environment in HCV infected patients.
Materials and methods
Cells, replicons, and viruses
Huh7.5 cells and full length genomic replicon cells (FL) were kindly provided by C Rice, subgenomic replicons (BB7) by C Seeger and the JFH1/HCV by T. Wakita. All cells were maintained in low glucose DMEM medium containing 10% FBS, 10 μg/ml Ciprofloxacin and supplemented with non-essential amino acids. Additionally, both replicon cells were maintained in presence of 500 μg/ml G418. JFH1 genomic RNA was in vitro transcribed from the linearized pUC-vJFH1 plasmid as a template using an in vitro transcription kit (MEGAscript, Ambion). Transfection of in vitro synthesized JFH1 RNA constructs and production of JFH1 virus stock were performed as previously described (13).
Reagents
Neutralizing SR-B1 and DC-SIGN antibodies were from R&D, LDL-R antibody from Novartis, anti-E2 antibody (MBL-HCV1) was a generous gift from MassBiologics (16). Anti-CD81 (JS-81) was purchased from BD Biosciences, anti-CD9, anti-CD63 from Biolengend. See supplementary materials for information of other reagents.
Preparation of human PBMCs and pDCs and co-cultures
Human PBMCs and pDCs were isolated from peripheral blood from healthy adult human volunteers or HCV-infected patients after informed consent was obtained according to procedures approved by the Committee for Protection of Human Subjects in Research at the University of Massachusetts Medical School. Description of cell isolation, co-cultures, RNA extraction and analysis is described in the Supplementary materials.
RNAi knockdown
Two pre-designed siRNA for CD81 (S2722 and S2723) and one negative control siRNA (4390843) were used in this study. RNAi knockdown was performed using RNAiMAX system (invitrogen) according to the manufacturer’s instructions. Two days after siRNA transfection, cells were collected for flow cytometric analysis or co-culture experiments.
Statistical analysis
All data is from 3 or more than 3 independent experiments. Data presented as mean ± SD using the Student’s t test or Mann-Whitney test according to data distribution. P < 0.05 was considered to be statistically significant.
Results
Human PBMCs and purified pDCs produce IFNα in response to HCV-infected cells
Because immune cells can directly interact with hepatocytes in the liver, we evaluated whether co-culture of human PBMCs with HCV-infected hepatoma cells could induce IFNα production. PBMCs produced IFNα in response to HCV full-length (FL) replicon or JFH-1-infected Huh7.5 cells while uninfected Huh7.5 cells or subgenomic HCV-replicons (BB7) failed to induce IFNα production (Fig. 1A). There was no IFNα production in HCV-infected Huh7.5 cells or in HCV-exposed PBMCs in the absence of hepatoma cells (data not shown). PBMC stimulation with the TLR9 ligand (CpG-A), was the positive control for IFNα induction (Fig 1A). Plasmacytoid DCs produced large amounts of IFNα when co-cultured with HCV-infected hepatoma cells while depletion of pDCs significantly reduced IFNα production in PBMCs (Fig. 1B). Flow-cytometry analysis revealed intracellular IFNα increase in the pDC-gated populations in response to FL replicons, but not Huh7.5 cells or BB7 replicons (Fig 1C). TLR7/8 or TLR9 ligands induced strong intracellular IFNα expression in pDCs (Fig 1C).
Figure 1. IFNα is induced in human PBMCs by HCV-infected cells.
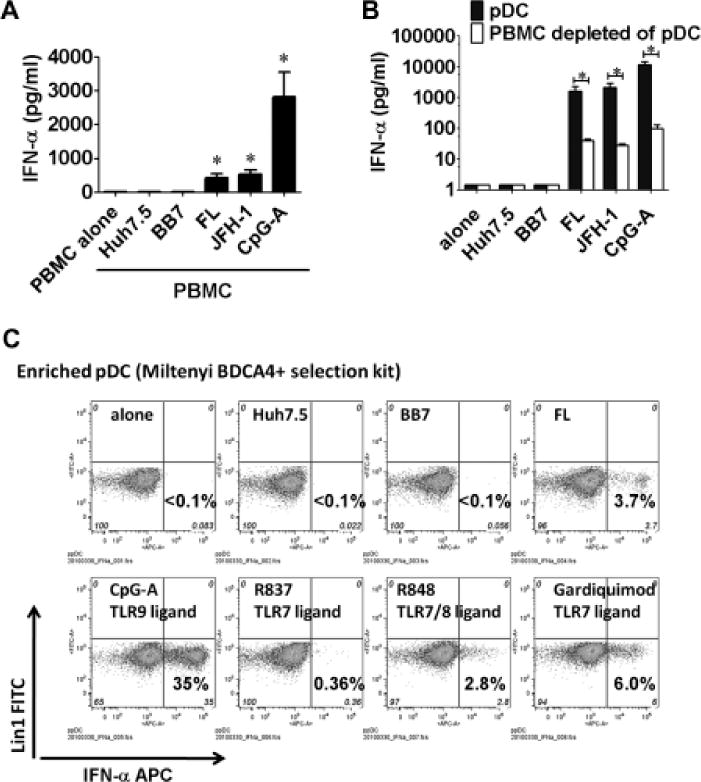
(A) Huh7.5, HCV replicon (BB7 or FL) or JFH-1-infected Huh7.5 cells were co-cultured with human PBMCs; CpG-A (2μM) was a positive control. (B) Huh7.5, HCV replicon or JHF-1-infected cells were co-cultured with pDCs or PBMCs depleted of pDC for 24 hours; IFNα in the supernatants was measured by ELISA. (C) Enriched pDCs were co-cultured with Huh7.5 or HCV replicon cells or TLR agonists (CpG-A 2μM, R837 2μg/ml, R848 2μg/ml or Garquimod 2μg/ml) and Golgistop was added after 6 hours. Ten hours later intracellular IFNα expression was determined by Flow cytometry. n=5, mean values ± SD (A, B); (*) indicates p<0.01 vs. PBMC alone, PBMC+Huh7.5 or BB7 group in (A); (*) indicates p<0.01 in (B).
Cell-to-cell contact between human PBMC and HCV-infected cells is required for IFNα induction
Induction of IFNα in pDCs by HCV-infected hepatoma cells may involve pathogen associated molecular patterns (PAMPs), secreted mediators, and/or cell–cell interactions. We found that in contrast to live cells, lysates of staurosporine-treated apoptotic FL replicons could not elicit IFNα induction suggesting that live, intact cells rather than their content induced IFNα in PBMCs (Fig. 2A and B). We determined a requirement for cell-to-cell contact for pDC activation by HCV-infected cells using transwell inserts separating PBMCs from HCV-infected cells and completely abolishing IFNα production (Fig. 2C). Transwell separation did not affect CpG-induced IFNα production (Fig 2C).
Figure 2. IFNα induction requires cell-to-cell contact between pDCs and live HCV-infected cells.
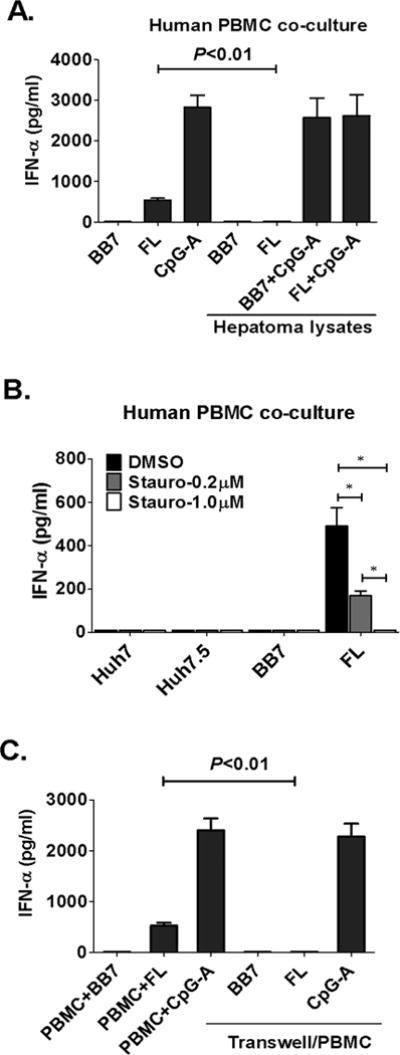
(A) Huh7.5, HCV replicon cells, or lysates from equivalent number of cells were co-cultured with human PBMCs for 24 hours; CpG-A (2μM) was a positive control. (B) staurosporine was added to Huh7.5 or HCV replicon cells for 8 hours, cells were washed and co-cultured with human PBMCs for 24 hours (C) transwell insert was introduced to co-cultures where indicated; n=4, mean values ± SD (A–C), (*) indicates p<0.01 in (B). IFNα in the supernatants was measured by ELISA.
CD81 and CD9 tetraspanins are involved in recognition of HCV-infected hepatoma cells by pDCs
Because only HCV-FL or JFH-1-infected and not subgenomic replicons induced IFNα in PBMCs requiring cell-to-cell contact, we hypothesized that cell surface molecules were involved in recognition of HCV structural proteins. HCV envelope glycoproteins (E1/E2) and selective host membrane receptors mediate viral attachment and entry (14, 15). We found that neutralizing anti-SR-B1, -LDL-R, -DC-SIGN and -HCV E2 antibodies did not prevent IFNα induction in PBMC by HCV-infected hepatoma cells (Fig. 3A and 3B). In contrast, addition of an anti-CD81 antibody significantly inhibited IFNα production in PBMCs in response to HCV-infected cells (Fig 3B). IFNα induction by TLR7/8- or TLR9-ligands was not affected by the anti-CD81 blocking antibody (Fig. 3B). Timing of anti-CD81 administration relative to co-culture was critical in inhibition of IFNα production. Introduction of anti-CD81 up to 3 hours after the initial cell–cell contact prevented IFNα induction but addition at later time points (>8 hours) failed to inhibit pDC activation (Fig. 3C). The inhibitory effect of anti-CD81 was dose-dependent (Fig. 3D).
Figure 3. Blocking CD81 inhibits pDC IFNα induction by HCV replicons or JFH-1 infected cells.
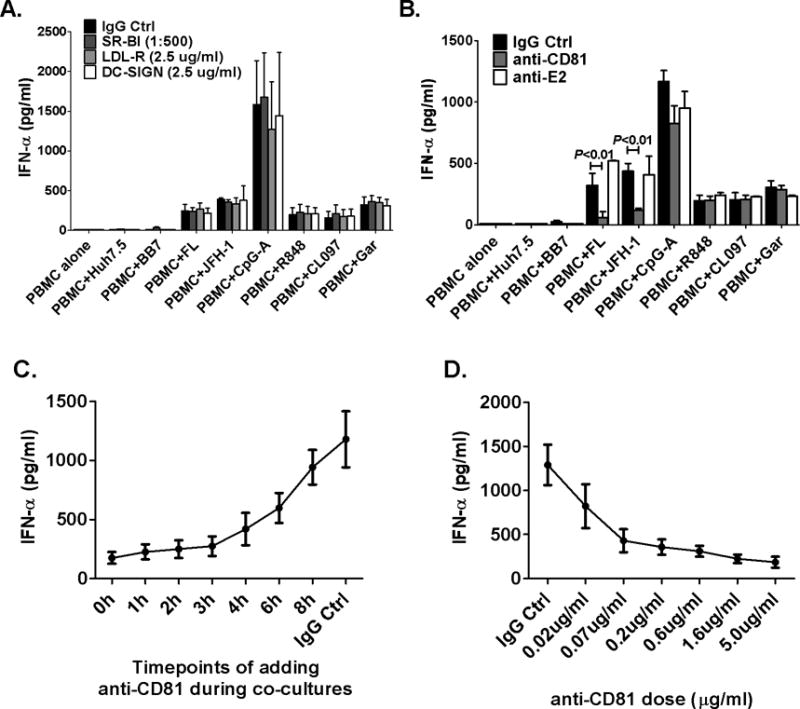
During co-culture, (A) blocking anti- SR-B1, LDL-R and DC-SIGN antibodies or (B) anti-HCV E2 antibody (5μg/ml) or anti-CD81 antibody (5μg/ml) were added. CpG-A (2μM), R848 (2μg/ml), CL097 (2μg/ml) and Garquimod (1μg/ml) included as controls. The results shown in (C, D) were from co-cultures between isolated human pDC and FL cells. (C) Anti-CD81 blocking antibody was added at the indicated time points after co-cultures; IFNα was measured 24 hours later by ELISA (D) different concentration of anti-CD81 antibody was used; n=3, mean values ± SD.
Beyond serving as a receptor for HCV, CD81 has a role in cell communication through formation of a tetraspanin web that allows association with other protein partners (17). We found that both human pDCs and HCV-infected hepatoma cells expressed high levels of surface CD81 (Fig. S1). Forty-eight hours after CD81-siRNA transfection, CD81 expression was decreased in hepatoma cells compared to control siRNA and HCV-NS3 expression was not affected (Fig. S2); however, CD81-siRNA knockdown in hepatoma cells did not inhibit IFNα (Fig. 4A) or MxA induction (Fig. 4B) in PBMCs. Because siRNA knockdown could not fully prevent CD81 expression in hepatocytes, we utilized an anti-CD81 blocking antibody and found that pre-treatment of hepatoma cells or pDCs with anti-CD81 blocking antibody could prevent IFNα induction by HCV infected hepatoma cells (Fig 4C).
Figure 4. Inhibition of CD81 on hepatoma cells does not affect IFNα production by PBMCss.
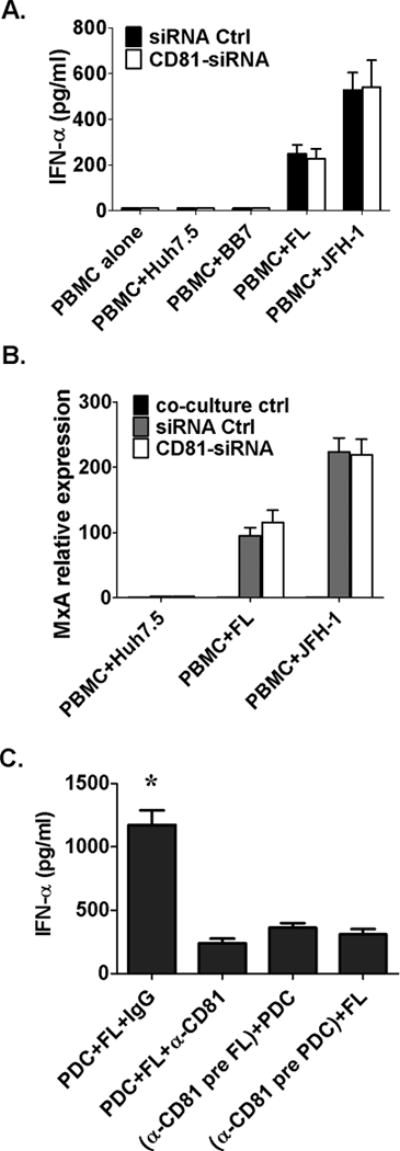
(A, B) Anti-CD81siRNA was transfected into Huh7.5, HCV replicon or JFH-1 infected cells 48 hours before their co-culture with PBMCs for 12 hours; RNA was extracted from cells for ISG detection and supernatants collected at 24 hours for IFNα measurement (ELISA), (C) pDC or FL cells were pretreated with anti-CD81 antibody separately for 1 hour, respectively, then washed and co-cultured for 24 hours; IFNα was measured by ELISA; n=3, mean values ± SD. (*) indicates p<0.01 vs. other groups in (C).
CD81, a member of the tetraspanin protein family, associates with other tetraspannin members (especially CD9) (18). Human pDCs highly expressed CD81 and CD9 and not CD63 and JFH-1 infected hepatoma cells had high CD81 and low CD63 and CD9 expression (Fig 5A). Both anti-CD9 and anti-CD81 antibodies significantly inhibited while anti-CD63 had no effect on IFNα production by pDC in response to HCV infected hepatoma cells (Fig. 5B and Fig. S4). Furthermore, CD9 and CD81 inhibition had additive negative effects on IFNα production (Fig. 5C) indicating the importance of membrane structures formed by CD9 and CD81 in pDCs activation.
Figure 5. CD9 is expressed on pDCs and CD9 blocking inhibits IFNα induction by HCV-infected hepatoma cells.
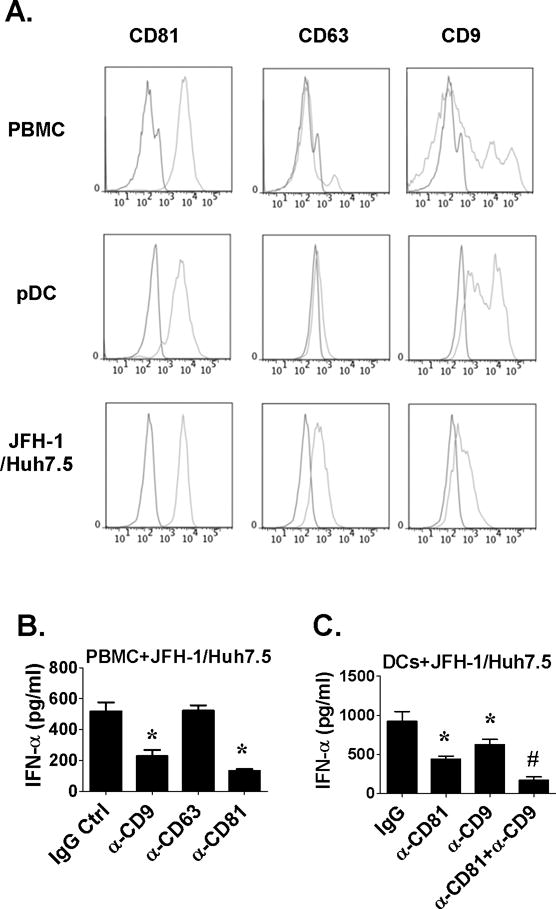
(A) PBMCs, purified pDCs or JFH-1-infected Huh7.5 cells were stained with FITC-conjugated anti-CD9, anti-CD63 and anti-CD9 or control antibodies and analyzed by FACS. (B) PBMCs or enriched DCs were co-cultured with JFH-1-infected Huh7.5 cells in the presence of anti-tetraspanin protein or control antibodies. Supernatants were collected 24 hours later for IFNα measurement. n=5, mean values ± SD. (*) indicates p<0.05 vs. control groups in (C, D) and (#) indicates p<0.05 vs. anti-CD81 and anti-CD9 group in (D).
CD81 molecule associated lipid rafts expressed in pDCs play a role in acquiring ligands from HCV-infected cells through endocytosis
Tetraspanin enriched micro-domains (TEMs) are platforms for interaction of membrane associated proteins and their signal transduction (19). CD81 and CD9 are heavily palmitoylated and directly bind to cholesterol (20). Treatment of cells with MβCD depletes cholesterol, reduces CD81 oligomerization (18), disrupts lipid rafts which are the platforms for CD81 interactions and TEM function. We found that disruption of lipid rafts in PBMCs with MβCD pretreatment significantly decreased IFNα production in response to HCV-FL replicons or JFH-1-infected cells, while pretreatment of hepatoma cells with MβCD had no effect (Fig. 6A). The reduction in IFNα was not due to cytotoxicity by MβCD because disruption of lipid rafts did not inhibit IFNα induction by TLR7/8 or TLR9 ligands (Fig. 6C). To dissect the inhibitory effect of anti-CD81 treatment in hepatoma cells and pDCs, CD81 knock-down hepatoma cells (CD81null cells) were co-cultured with PBMCs in the presence of anti-CD81 or control antibody. The inhibitory effect of anti-CD81 was not influenced by CD81 expression on hepatoma cells indicating that CD81 on pDCs played the role in recognition of HCV-infected cells (Fig. 6B).
Figure 6. CD81 protein, lipid rafts and endocytosis pathways are required for IFNα induction in PBMCs and hepatoma co-cultures.
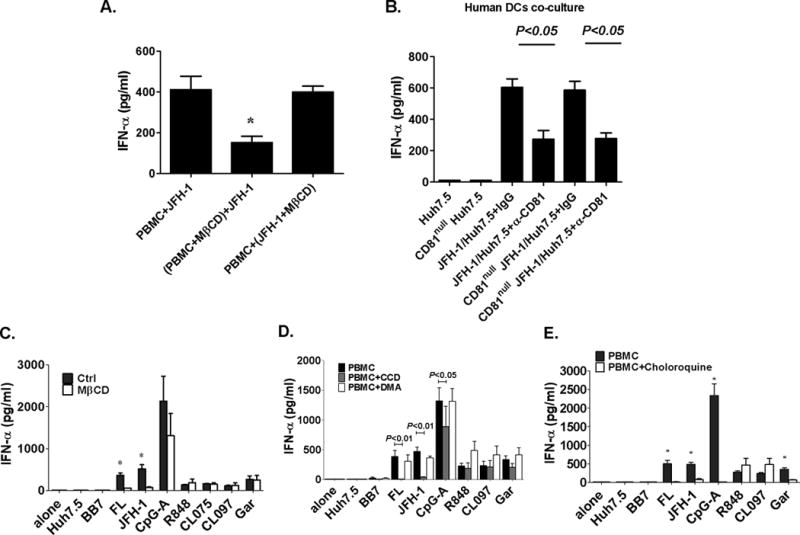
(A) PBMCs or JFH-1-infected Huh7.5 cells were pretreated with MβCD (10mM), respectively, and co-cultured as indicated. (B) Hepatoma cells were transfected with anti-CD81-siRNA or control siRNA for 48 hours then co-cultured with human PBMCs in the presence of anti-CD81 or control antibody. IFNα was measured in the supernatants. (C-E) Human PBMCs were treated with MβCD (10mM), CCD (10μM), DMA (50μM) or Choloroquine (10μM) for 30 minutes at 37°C where indicated and after washing co-cultured with HCV replicon or JFH-1-infected cells. CpG-A (5μM), R848 (2μg/ml), CL097 (2μg/ml) and Garquimod (1μg/ml) were included as controls. 24 hours later IFNα was measured in the supernatants. n=3, mean values ± SD. (*) indicates p<0.05 vs. other groups in (A), p<0.01 vs. MβCD pretreatment groups in (C) and vs. chrloroquine pretreatment groups in (E).
We defined that pretreatment of human PBMCs with CCD, an inhibitor of endocytosis, prevented while inhibition of macropinocytosis by DMA had no effect on IFNα induction in pDCs (Fig. 6D). These data suggested that pDCs utilize a CD81-dependent mechanism to acquire ligands from HCV-infected cells through endocytosis. We confirmed the requirement for endosomal acidification as chloroquine treatment of PBMCs prevented IFNα induction by FL replicons and JFH-1 infected cells (Fig. 6E).
Rac GTPase is involved in pDC-IFNα induction by HCV-infected hepatoma cells
Recent studies suggest that CD81 regulates cellular movements through modulating downstream cytoskeleton regulators, such as Syk or small GTPases, Rac and Rho (21–23). Using specific inhibitors of Syk, Rac and Rho, we found that only the Rac inhibitor (NSC-23766) prevented pDC IFNα production while inhibition of Syk (BAY61-3606) and Rho (CT04) had no effect (Fig. 7A). The Rac, but not Syk and Rho, inhibitor also prevented PBMC IFNα induction by Poly I:C, R848 and CpG-A (Fig. 7A). The inhibitory effect of NSC-23766 on pDC activation was dose-dependent. Low concentration of NSC-23766 pretreatment (20μm and 60μm) resulted in specific inhibition of PBMC IFNα induction by HCV-infected hepatoma cells and showed a minimal effect on IFNα induction by Poly I:C, R848 and CpG-A stimulation (Fig. 7B).
Figure 7. Rac GTPase inhibition diminishes IFNα induction in pDCs by HCV-infected hepatoma cells.
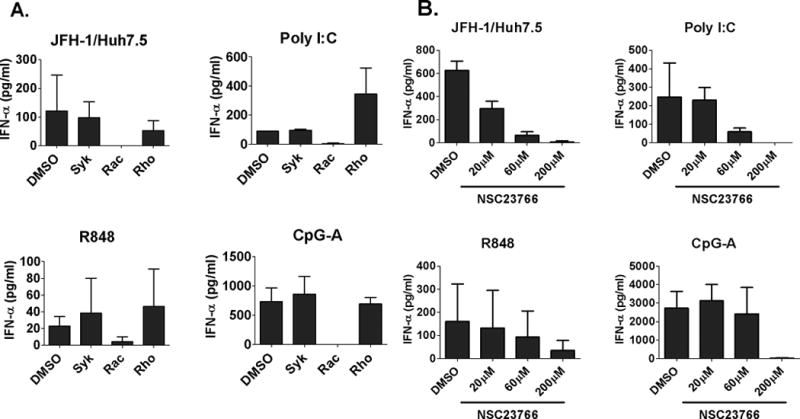
(A) PBMCs were pretreated with Bay61-3606 (20nM), NSC-23766 (200μM) or CT04 (1μg/ml) for 2 hours before co-culture with JFH-1 infected Huh7.5 cells or stimulation with Poly I:C (20μg/ml), R848 (2μg/ml) and CpG-A (2μM). (B) PBMC were pretreated with different doses of NSC-23766 for 2 hours before co-culture with JFH-1-infected Huh7.5 cells or stimulation with Poly I:C (20μg/ml), R848 (2μg/ml) and CpG-A (2μM). IFNα was determined by ELISA.
IFNα production by PBMCs of HCV-infected patients in response to HCV-infected hepatoma cells is decreased
We and others have reported that pDCs from chronic HCV infected patients have impaired IFNα production in response to viral PAMPs (11, 12). Here we show that compared to healthy controls, PBMCs from treatment-naïve patients with chronic HCV infection had significantly decreased IFNα production in response to HCV-infected hepatoma cells (Fig. 8A). As previous studies found that HCV viral particles could inhibit CpG-induced IFNα production in pDCs (11, 12), we hypothesized that HCV viral particles might inhibit pDC activation. Pre-treatment of normal PBMCs with infectious HCV particles significantly decreased IFNα production in response to HCV-infected hepatoma cells or CpG-A stimulation compared to HCV-naïve PBMCs (Fig. 8B).
Figure 8. Free HCV virus inhibits pDC IFNα induction in human PBMCs and hepatoma co-cultures.
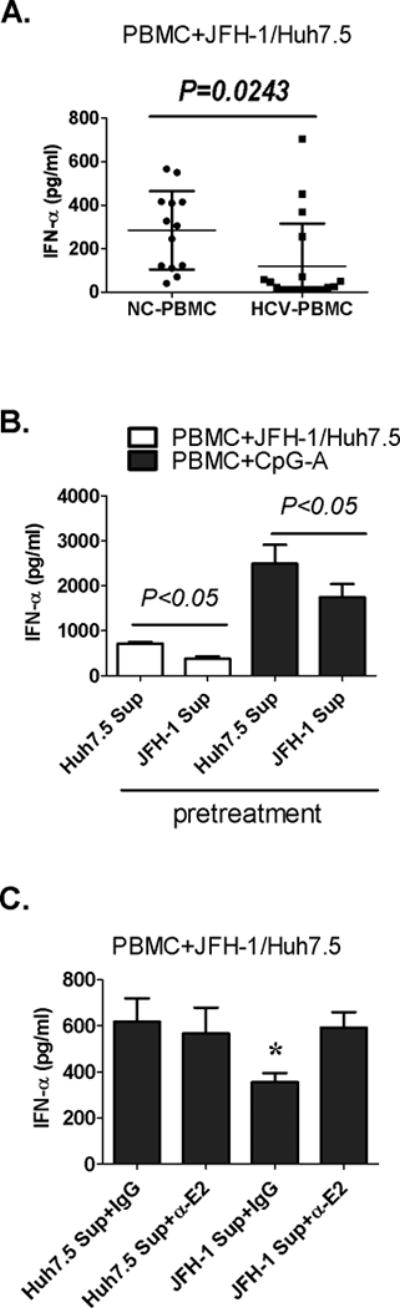
(A) PBMCs from normal controls (NC-PBMC) or non-treated HCV-infected patients (HCV-PBMC) were co-cultured with JFH-1 infected huh7.5 cells. (B–C) Normal PBMCs were pretreated with non-infectious (Huh7.5 Sup) or infectious supernatants (JFH-1 Sup) for 2 hours before co-culture with JFH-1-infected Huh7.5 cells. Neutralizing anti-HCV E2 antibody (α-E2) was added during pretreatment (C). IFNα was measure in the supernatants; n=3, mean values ± SD. (*) indicates p<0.05 vs. other groups in (C).
Because HCV glycoprotein E1–E2 proteins interact with CD81, we wondered whether the inhibitory effect of HCV pretreatment was due to interference with CD81 function. We found that a neutralizing anti-E2 antibody that prevents binding of HCV to CD81 reversed the inhibitory effect of the infectious HCV containing supernatants on PBMC IFNα production in response to HCV-infected hepatoma cells (Fig. 8C).
Discussion
Type I IFNs play a critical role in the host response that determines the outcome of HCV infection (6). The mechanisms that define immune recognition of HCV infected hepatocytes in the liver are still poorly understood. Here we report that human pDCs interact with HCV-infected hepatoma cells and in response, produce IFNα. We present novel evidence for an intimate crosstalk between the pDC and infected hepatocma cell that requires direct cell–cell contact and endocytosis. We show for the first time that IFNα induction in pDCs by HCV-infected cells depends on CD81/CD9 tetraspanins and the integrity of membrane lipid-rafts in pDCs. Our results demonstrate that the CD81-mediated pDC and hepatoma cell interaction and IFNα induction requires Rac-GTPase activity. Our novel data demonstrate that exposure of normal PBMCs to HCV virus can attenuate their IFNα production in response to HCV-infected hepatoma cells or TLR ligands suggesting a mechanism that may inhibit pDCs in vivo in a HCV infected host. These data indicate that pDC can sample HCV-infected cells to recognize infection and mount an anti-viral immune response with robust IFNα production but, likely, this process is attenuated in pDCs during acute and/or chronic HCV infection.
Plasmacytoid DCs produce large amounts of IFNα and capture and cross-present antigens to trigger adaptive immune responses (24). Studies showed that HIV-infected cells triggered more IFNα production in pDCs than HIV particles (25). Here we present new evidence that pDCs sample live HCV-infected cells through cell–cell contact and endocytic pathways involving CD81/CD9-associated cell surface structures but soluble HCV antigen, dying cells, or secreted HCV viral particles had no IFNα–inducing capacity. We found that HCV-FL cells but notsubgenomic replicon induced significant IFNα production from pDCs suggesting that HCV structural protein(s) played a role in this process. A recent report showed that HIV-1 transfected cells carrying defective envelope or fusion proteins, but not nucleocapsid protein, induced pDC activation (26). This together with our observations may suggest that the requirement for viral envelope proteins is a common element in sensing infected cells harboring RNA virus, while the proper assembly of the virus is not necessary. We suspect that envelope proteins might affect innate sensing though modulating viral-PAMP structure or transportation of PMAPs in the infected cells. However, investigation of the exact mechanisms of HCV envelope or other hepatocyte proteins in DC activation is beyond this study.
Considering the large size of hepatoma cells, it is unlikely that pDCs can take up an entire infected hepatoma cell during their close interaction but rather capture viral components through the endocytosis pathway. Our results suggested that the viral RNA causes pDC activation. Consistent with a previous report (13), we confirmed that a TLR7 inhibitory oligodeoxynucleotide (ODN, IRS661) strongly inhibited pDC-IFNα production while the DNA sensing receptor, TLR9 pathway, remained unaffected (Fig. S3). In vitro-synthesized HCV RNA segments or whole JFH-1 RNA triggered strong pDC-IFNα induction while extracted whole RNA from Huh7.5 cells failed to induce IFNα production (Fig. S5). Finally, the magnitude of IFNα induction in pDCs depended on the intracellular HCV-RNA levels in infected cells and eradication of HCV prevented pDC-IFNα production (Fig. S6).
CD81 was expressed on both HCV-infected hepatoma cells and human pDCs. We found that siRNA knock-down of CD81 in hepatocytes failed to prevent pDC activation, while pretreatment of pDC with anti-CD81 antibody significantly inhibited IFNα induction by HCV-infected hepatoma cells, suggesting a specific role of CD81 on pDCs and not on HCV-infected cells in the recognition process. CD81 is a member of the tetraspanin family (19). Tetraspanin proteins interact with one another, membrane receptors and signaling proteins to form functional structures in cell membranes called “tetraspanin microdomains” (TEM) which play important roles in migration, proliferation, differentiation and infectious disease (17). We determined that CD9, a CD81 partner protein, was expressed on pDCs and contributed to the interactions between pDCs and HCV-infected hepatoma cells as anti-CD9 antibody prevented IFNα induction. We found evidence for an additive co-operation between CD9 and CD81 in IFNα induction by HCV-infected hepatoma cells. Cholesterol physically and functionally associates with TEMs (18, 20) and MβCD, that depletes cholesterol, disrupts CD81 oligomerization and interferes with TEM function (18). Similar to anti-CD81 antibody treatment, pretreatment of pDCs with MβCD prevented IFNα induction by HCV-infected hepatoma cells indicating that the intact function of the CD81-associated cell membrane structures in pDCs was necessary for recognition of HCV-infected cells.
Due to its short cytosolic tail, CD81 cannot conduct signal transduction by itself and it interacts with partner proteins; however the molecular mechanisms downstream of CD81 are yet to be defined (15) (17). CD81 is important in cellular movement in human dendritic cells through regulating small GTPase, such as Rho and Rac (21–23). We hypothesized that CD81 mediated cell–cell interactions between pDCs and hepatoma cells involved Rho or Rac activities in pDCs and found that inhibition of Rac activity prevented IFNα production by pDCs. The effect of Rac inhibition was similar to CD81 blocking as they both specifically inhibited IFNα induction in pDCs by HCV-infected hepatoma cells and not by TLR agonists. Thus, we speculate that pDC and HCV-infected hepatoma interact through CD81 that signals by involving Rac activity in pDCs to induce IFNα production.
While impaired IFNα induction by TLR-agonists in pDCs from HCV-infected patients have been demonstrated before (27, 28), our novel data show impaired IFNα production by pDCs of patients with chronic HCV infection in response to HCV-infected hepatoma cells. Previous studies revealed several possible explanations for the decreased IFNα production capacity in pDCs of HCV infected patients including decreased number of circulating pDCs and functional impairment of the IFNα synthesis pathway (29–31). In this study, we reproduced the decreased IFNα production seen in patients by pre-incubating normal pDCs with infectious HCV virions and found that anti-HCV E2 antibodies prevented this inhibition. It is tempting to speculate that HCV virion interaction between E2 and CD81 on pDCs may interfere with subsequent CD81-mediated cell–cell interaction leading to attenuated IFNα induction by HCV-infected cells. This finding also implicates that large amount of released HCV virions or paticles during acute HCV infection might interfere with the innate sensing mechanisms of pDCs and lead to inefficient viral control and promote chronic infection. Consistent with this hypothesis, previous reports showed that HCV virions inhibited TLR9 and not TLR7 induced pDC activation (11) and HCV-E2 and CD81 interaction was associated with altered trafficking of dendritic cells (32) and inhibition of NK cell activation (33, 34).
In conclusions, we identified a novel mechanism for recognition of HCV-infected hepatoma cells by pDCs involving membrane microdomains formed by CD81-associated tetraspanin webs and lipid rafts. We speculate that this novel pathway is critical in activation of pDCs and the type I IFN response in the liver environment in HCV infected patients. Our data also suggest an intimate contact between immune cells and infected hepatocytes allowing “sampling” of the pathogenic signals to maintain host integrity.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R37AA014372 (GS). The authors thank Drs Charles M.Rice, Takaji Wakita, Christoph Seeger and Kui Li for kindly providing reagents.
Abbreviations used in this paper
- DC
dendritic cell
- ELISA
enzyme-linked immunosorbent assay
- HCV
hepatitis C virus
- IFN
interferon
- ISG
interferon stimulated gene
- MAVS
mitochondrial antiviral-signaling protein
- mDC
myeloid dendritic cell
- PAMPs
pathogen associated molecular patterns
- PBMC
peripheral blood mononuclear cell
- PCR
polymerase chain reaction
- pDC
plasmacytoid dendritic cell
- RLR
RIG-I like receptor
- siRNA
small interfering RNA
- TEM
tetraspanin-enriched microdomain
- TLR
toll-like receptor
- TRIF
TIR-domain-containing adapter-inducing interferon-β
References
- 1.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 2.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 3.Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 4.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 5.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, Heim MH. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A. 2008;105:7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lau DT, Fish PM, Sinha M, Owen DM, Lemon SM, Gale M., Jr Interferon regulatory factor-3 activation, hepatic interferon-stimulated gene expression, and immune cell infiltration in hepatitis C virus patients. Hepatology. 2008;47:799–809. doi: 10.1002/hep.22076. [DOI] [PubMed] [Google Scholar]
- 8.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 9.Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebihara T, Shingai M, Matsumoto M, Wakita T, Seya T. Hepatitis C virus-infected hepatocytes extrinsically modulate dendritic cell maturation to activate T cells and natural killer cells. Hepatology. 2008;48:48–58. doi: 10.1002/hep.22337. [DOI] [PubMed] [Google Scholar]
- 11.Shiina M, Rehermann B. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology. 2008;47:385–395. doi: 10.1002/hep.21996. [DOI] [PubMed] [Google Scholar]
- 12.Gondois-Rey F, Dental C, Halfon P, Baumert TF, Olive D, Hirsch I. Hepatitis C virus is a weak inducer of interferon alpha in plasmacytoid dendritic cells in comparison with influenza and human herpesvirus type-1. PLoS One. 2009;4:e4319. doi: 10.1371/journal.pone.0004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M, Chisari FV. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci U S A. 2010;107:7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Hahn T, Rice CM. Hepatitis C virus entry. J Biol Chem. 2008;283:3689–3693. doi: 10.1074/jbc.R700024200. [DOI] [PubMed] [Google Scholar]
- 15.Farquhar MJ, Harris HJ, McKeating JA. Hepatitis C virus entry and the tetraspanin CD81. Biochem Soc Trans. 2011;39:532–536. doi: 10.1042/BST0390532. [DOI] [PubMed] [Google Scholar]
- 16.Broering TJ, Garrity KA, Boatright NK, Sloan SE, Sandor F, Thomas WD, Jr, Szabo G, et al. Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. J Virol. 2009;83:12473–12482. doi: 10.1128/JVI.01138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yanez-Mo M, Barreiro O, Gordon-Alonso M, Sala-Valdes M, Sanchez-Madrid F. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol. 2009;19:434–446. doi: 10.1016/j.tcb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Silvie O, Charrin S, Billard M, Franetich JF, Clark KL, van Gemert GJ, Sauerwein RW, et al. Cholesterol contributes to the organization of tetraspanin-enriched microdomains and to CD81-dependent infection by malaria sporozoites. J Cell Sci. 2006;119:1992–2002. doi: 10.1242/jcs.02911. [DOI] [PubMed] [Google Scholar]
- 19.Levy S, Shoham T. The tetraspanin web modulates immune-signalling complexes. Nat Rev Immunol. 2005;5:136–148. doi: 10.1038/nri1548. [DOI] [PubMed] [Google Scholar]
- 20.Charrin S, Manie S, Thiele C, Billard M, Gerlier D, Boucheix C, Rubinstein E. A physical and functional link between cholesterol and tetraspanins. Eur J Immunol. 2003;33:2479–2489. doi: 10.1002/eji.200323884. [DOI] [PubMed] [Google Scholar]
- 21.Coffey GP, Rajapaksa R, Liu R, Sharpe O, Kuo CC, Krauss SW, Sagi Y, et al. Engagement of CD81 induces ezrin tyrosine phosphorylation and its cellular redistribution with filamentous actin. J Cell Sci. 2009;122:3137–3144. doi: 10.1242/jcs.045658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brazzoli M, Bianchi A, Filippini S, Weiner A, Zhu Q, Pizza M, Crotta S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J Virol. 2008;82:8316–8329. doi: 10.1128/JVI.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quast T, Eppler F, Semmling V, Schild C, Homsi Y, Levy S, Lang T, et al. CD81 is essential for the formation of membrane protrusions and regulates Rac1-activation in adhesion-dependent immune cell migration. Blood. 2011;118:1818–1827. doi: 10.1182/blood-2010-12-326595. [DOI] [PubMed] [Google Scholar]
- 24.Lui G, Manches O, Angel J, Molens JP, Chaperot L, Plumas J. Plasmacytoid dendritic cells capture and cross-present viral antigens from influenza-virus exposed cells. PLoS One. 2009;4:e7111. doi: 10.1371/journal.pone.0007111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology. 2005;343:256–266. doi: 10.1016/j.virol.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 26.Lepelley A, Louis S, Sourisseau M, Law HK, Pothlichet J, Schilte C, Chaperot L, et al. Innate sensing of HIV-infected cells. PLoS Pathog. 2011;7:e1001284. doi: 10.1371/journal.ppat.1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murakami H, Akbar SM, Matsui H, Horiike N, Onji M. Decreased interferon-alpha production and impaired T helper 1 polarization by dendritic cells from patients with chronic hepatitis C. Clin Exp Immunol. 2004;137:559–565. doi: 10.1111/j.1365-2249.2004.02550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mengshol JA, Golden-Mason L, Castelblanco N, Im KA, Dillon SM, Wilson CC, Rosen HR. Impaired plasmacytoid dendritic cell maturation and differential chemotaxis in chronic hepatitis C virus: associations with antiviral treatment outcomes. Gut. 2009;58:964–973. doi: 10.1136/gut.2008.168948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wertheimer AM, Bakke A, Rosen HR. Direct enumeration and functional assessment of circulating dendritic cells in patients with liver disease. Hepatology. 2004;40:335–345. doi: 10.1002/hep.20306. [DOI] [PubMed] [Google Scholar]
- 30.Szabo G, Dolganiuc A. Subversion of plasmacytoid and myeloid dendritic cell functions in chronic HCV infection. Immunobiology. 2005;210:237–247. doi: 10.1016/j.imbio.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 31.Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J Exp Med. 2011;208:2367–2374. doi: 10.1084/jem.20110654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nattermann J, Zimmermann H, Iwan A, von Lilienfeld-Toal M, Leifeld L, Nischalke HD, Langhans B, et al. Hepatitis C virus E2 and CD81 interaction may be associated with altered trafficking of dendritic cells in chronic hepatitis C. Hepatology. 2006;44:945–954. doi: 10.1002/hep.21350. [DOI] [PubMed] [Google Scholar]
- 33.Crotta S, Stilla A, Wack A, D’Andrea A, Nuti S, D’Oro U, Mosca M, et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J Exp Med. 2002;195:35–41. doi: 10.1084/jem.20011124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tseng CT, Klimpel GR. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J Exp Med. 2002;195:43–49. doi: 10.1084/jem.20011145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.