

Abstract
NKX3.1 is a prostate specific homeodomain protein and tumor suppressor whose expression is reduced in the earliest phases of prostatic neoplasia. NKX3.1 expression is not only diminished by genetic loss and methylation, but the protein itself is a target for accelerated degradation caused by inflammation that is common in the aging prostate gland. NKX3.1 degradation is activated by phosphorylation at C-terminal serine residues that mediate ubiquitination and protein turnover. Since NKX3.1 is haploinsufficient, strategies to increase its protein stability could lead to new therapies. Here, a high throughput screen was developed using a siRNA library for kinases that mediate NKX3.1 degradation. This approach identified several candidates of which DYRK1B, a kinase that is subject to gene amplification and overexpression in other cancers, had the greatest impact on NKX3.1 half-life. Mechanistically, NKX3.1 and DYRK1B were shown to interact via the DYRK1B kinase domain. In addition, an in vitro kinase assay showed that DYRK1B phosphorylated NKX3.1 at serine185, a residue critical for NKX3.1 steady state turnover. Lastly, small molecule inhibitors of DYRK1B prolonged NKX3.1 half-life. Thus, DYRK1B is a target for enzymatic inhibition in order to increase cellular NKX3.1.
Implications
DYRK1B is a promising and novel kinase target for prostate cancer treatment mediated by enhancing NKX3.1 levels.
Introduction
Genetic studies commencing decades ago with karyotype analyses and currently represented by whole genome sequencing have repeatedly and consistently implicated 8p21, the locus of NKX3.1, as the most commonly disrupted genetic target of prostate cancer (1-4). NKX3.1 is a prostate-specific homeobox gene that controls differentiation (5), epithelial cell growth (6), and stem cell maintenance (7). In early prostate cancer, reduced levels of the haploinsufficient NKX3.1 protein are seen in the majority of prostate cancers (8). The degree of protein loss in primary prostate cancer cells is related to Gleason grade, suggesting that prostate cancer phenotype is, to some degree, under control of NKX3.1. During prostate cancer progression there is selective pressure to decrease NKX3.1 expression further (9). NKX3.1 protein loss is mediated by several mechanisms including genetic loss, gene methylation, and posttranslational modification (8). For example, phosphorylation at serine 185 controls NKX3.1 steady state turnover by signaling ubiquitination and subsequent degradation in the proteasome (10). However, NKX3.1 protein is further destabilized by inflammation that releases cytokines causing phosphorylation at serine 196 and markedly decreasing protein half-life. Thus, inhibition of the kinases that trigger protein degradation can increase intracellular NKX3.1 levels and may mediate growth inhibition and differentiation of prostate cancer.
Hypothesizing that kinase inhibition could affect NKX3.1 levels and thereby impact prostate cancer therapy, a kinase siRNA library was screened to select kinases that could affect cellular levels of NKX3.1. Of 720 kinase candidates, seven were found after two rounds of library screening consistently to elevate NKX3.1 levels. As a confirmation of on-target effects, the siRNAs were shown actually to knock down their cognate targets. Among the final candidates, DYRK1B a developmentally important kinase (11), that has been implicated in cell cycle control (12, 13) and cancer (14-16), had the greatest effect on NKX3.1. DYRK1B interacted directly with the prostate suppressor protein and phosphorylated a key residue that signaled protein degradation. Moreover, pharmacologic inhibition of DYRK1B stabilized NKX3.1 protein in cultured cells demonstrating an interaction that should be explored by preclinical development.
Materials and Methods
Cells and culture
The human prostate cell lines LNCaP and HEK (human embryonic kidney)-293T cells were cultured in modified IMEM (improved minimum essential medium; Invitrogen) supplemented with 5% FBS (fetal bovine serum) at 5% CO2 and 37°C. For establishment of stable sublines of LNCaP Cells expressing PL-N-NKX3.1, LNCaP cells were transfected with PL-N-NKX3.1 and selected in the presence of 500 μg/ml G418. Individual clones were picked up and cultured for expansion. Expression of PL-N-NKX3.1 was verified by western blotting analysis with anti-NKX3.1 antibody. Clone 7 (PLNKX7) was used for this experiment.
Kinase Inhibitors
Compounds NCGC00185981, NCGC00185963, NCGC00185976, and NCGC00185984 were provided by Craig Thomas, National Cancer Institute, Bethesda, MD. Compound A (16) was purchased from Calbiochem. Compound 39621, a MARK inhibitor, was purchased from EMD Millipore.
Plasmids
Mammalian expression vector encoding full-length human NKX3.1 (pCMV-MYC-NKX3.1) with MYC tag has been reported previously. Flag-tagged murine DYRK1B cloned in p3XFlag-CMV7.1 was provided by Dr. Miyata of Kyoto University, Japan (17). The ProLabel-NKX3.1 fusion construct (PL-N-NKX3.1) was produced by cloning the PCR-amplified full-length NKX3.1 in-frame with the ProLabel-N vector (Clontech). The NKX3.1 fragment was obtained by PCR amplification with pCMV-MYC-NKX3.1 as template using PCR primers that introduce an EcoRI and BamHI sites. PCR products were digested, purified and subcloned into the EcoRI/BamHI sites of ProLabel-N. The construct was confirmed by direct sequencing.
ProLabel is a small (about 6 kDa) tag that is capable of producing a strong chemiluminescent signal via an enzyme complementation assay. The ProLabel tag allows direct detection of tagged protein of interest in a high throughput screening.
Chemiluminescent assays
Chemiluminescent detection high throughput 96-well homogeneous assays were performed with ProLabel Detection Kit II (Clontech) according to the protocol provided by manufacturer. Briefly, cells in 96-well plates were lysed in their culture medium by adding Lysis/CL Working Solution. 20 μl of EA reagent were added to each sample and incubated at room temperature for 1 hour. Chemiluminescent signals were measured with a plate reader.
High Throughput siRNA Screening
The human kinase siRNA Set V4.1, in a 96-well microtiter plate format, which targets 720 human kinases genes, was purchased from Qiagen. siRNA and transfection reagent concentrations, the transfection method and efficiency in LNCaP cells were optimized prior to the high throughput screen (Fig. 2, for example). For first-round screening, the master plate collections consisted of four non-overlapping duplex siRNAs to each target gene; aliquots from the four sets were pooled into one master plate. Each plate consisted of three negative-universal siRNA control came together with the original master plates. These negative controls were AllStars negative control (Cat 1027280), negative siRNA control (Cat 1022076) and siRNA control against GFP. As a positive control 20 nM NKX3.1 siRNA was included. In addition, eight wells per plate were used as mock transfection control. For siRNA screening, 1 μL of combined siRNAs were aliquoted into 96-well plates, mixed with 2 μL of Lipofectamine RNAiMAX (Invitrogen) which has been diluted by 10-fold with Opti-MEM right before the experiments. 18 μL of Opt-MEM were then added to each well and mixed gently and incubated at room temperature for about 20 min. Reverse transfection was performed by dispensing PLNKX7 cells (8000 cells in 80 μL) into each well. The final volume per well was 100 μL and contained a final concentration of 25 nM for each siRNA from the four duplexes for each target. Each plate was transfected in duplicate at different times to deduce reproducible inhibition. After 72 hours cells were exposed to 100 μg/ml cycloheximide for 60 min. Half of mock transfections (four wells each plate) and NKX3.1. siRNA controls were treated with DMSO.
Figure 2.
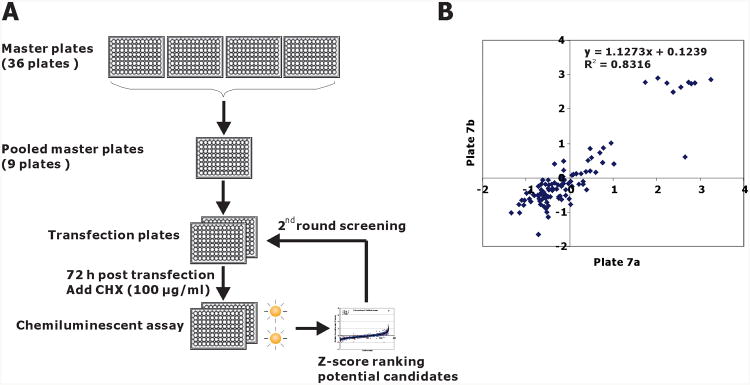
Chemiluminescence assay of PL-NKX3.1 degradation in 96-well plates. A. Schematic diagram of the siRNA strategy for kinome siRNA screening. From master plates, four target-specific siRNAs were pooled into working transfection plates, siRNA pools from each transfection plate were reverse transfected into LNCaP-PLNKX7 cells. After 72 h of transfection, the plates were treated with cycloheximide for 1 hr. Chemiluminescent assay was performed with Prolabel Detection Kit II. B. Plate-to-plate reproducibility is shown in a pairwise plot of Z-score values from replicate plates 7a and 7b that were the master plates used for first round screening. Cells were plated in a replicate manner, but the transfections were performed separately at different times. Linear regression indices of the pairwise comparison are shown.
The second round screening was limited to 43 candidate siRNAs based on Z-score ranking. Each four specific siRNAs of these target genes were separately transfected into LNCaP-PLNKX7 cells in 96-well plates as stated above and treated with cycloheximide for 1 hour before chemiluminescent assay. From the second-round screening, we selected 10 candidate siRNAs for which we designed four custom individual siRNAs per gene (Dharmacon, SMARTpool) that were used for final validation in LNCaP cells by directly assaying endogenous NKX3.1 by immunoblot.
The screening data were normalized using the standard Z-score method by correcting the raw data of chemiluminescent signals for plate row variation, and then normalizing and pooling data from all assay plates. The assumption of the screening is that the majority of the siRNAs are non-hits and the null distribution is normal (18). All the cells transfected with kinase siRNAs and half of the mock transfected cells were treated with cycloheximide for 60 min and half of the mock transfected cells (three wells) were treated with DMSO control. Since the chemiluminescent signals in DMSO-treated cells were about 2 fold higher than that in cycloheximide-treated cells, signal readings in DMSO-treated cells (three wells) were excluded when we calculated the averages of the readings. A Z-score cutoff of 1.42, corresponding to a p ≤ 0.05, was used to select the candidates for our second-round screening. In addition, to utilize the capacity of the microtiter plate, the total number of samples subjected to secondary screening was increased to 43.
In vitro DYRK1B kinase assay
GST–DYRK1B was purchased from Life Technologies (Carlsbad, CA). His-NKX3.1 was prepared as described previously. In vitro kinase assay was performed according to the assay conditions reported previously (19, 20). Briefly, 200 ng GST-DYRK1B was incubated with 300 ng His-NKX3.1 for 30 min at 30°C in 10 μl kinase buffer (25 mM Hepes pH 7.4, 5 mM MgCl2, 0.5 mM dithiothreitol) in the presence of 250 μM ATP. For dynamic and DYRK1B dose-dependent analyses, in vitro kinase assays were also performed with increasing concentrations of GST-DYRK1B (up to 200 ng per reaction) for different times (from 5 min to 120 min). Reactions were separated on SDS-PAGE containing 25 μM Phos-tag and 50 μM MnCl2 and transferred onto nitrocellulose membrane. Western blot analysis was performed with rabbit NKX3.1 antibody.
In vivo DYRK1B kinase assay
293T cells were plated onto 100 mm dishes and cotransfected with pcDNA3.1-NKX3.1 with either Flag-mDYRK1B or pcDNA3.1. Cells were harvested and cell lysates were fractionated on SDS-PAGE containing 25 μM Phos-tag and 50 μM MnCl2 and transferred onto nitrocellulose membrane as described above. NKX3.1 was detected by immunoblotting.
Coimmunoprecipitation
HEK-293T cells were engineered to express MYC–NKX3.1 and Flag-mDYRK1B and the cell lysates were prepared 48 hr after transfection. Cell lysates were used to perform the coimmunoprecipitation assays with monoclonal antibody against either Flag (M2, Sigma-Aldrich) or goat NKX3.1 antiserum (Santa Cruz). After extensive washing, the pellets were fractionated by SDS/PAGE (4–20% gel) and subjected to immunoblotting with anti-Flag antibody or NKX3.1 antiserum.
Identification of NKX3.1 phosphorylation sites by mass spectrometry
The in vitro kinase assay was performed as described above and the phosphorylated NKX3.1 was run on SDS-PAGE and the gel piece containing NKX3.1 was excised for LC-MS/MS analysis. Dried gel slices were rehydrated and digested in 80 μL 12.5 ng/μL trypsin Gold/50 mM, ammonium bicarbonate overnight at 37°C. After the digestion was complete, condensed evaporated water was collected from tube walls by brief centrifugation a bench-top microcentrifuge (Eppendorf, Hauppauge, NY). The gel pieces and digestion reaction were mixed with 50 μL 2.5% trifluoroacetic acid and rigorously mixed for 15 min. The solution with extracted peptides was transferred into a fresh tube. The remaining peptides were extracted with 80 μl 70% acetonitrile/5% trifluoroacetic acid mixture using rigorous mixing for 15 min. The extracts were pooled and dried to completion (1.5–2 h) in a SpeedVac. The dried peptides were reconstituted in 30 μL 0.1% TFA by mixing for 5 min and stored in ice or at −20 °C prior to analysis.
The concentrated peptide mix was reconstituted in a solution of 2% acetonitrile, 2 % formic acid for MS analysis. Peptides were loaded with the autosampler directly onto a 2 cm C18 pre-column and were eluted from the column using a Thermo Easy-nLC1000 UHPLC with a 10 min gradient from 2% buffer B to 35 % buffer B (100% acetonitrile, 0.1% formic acid). The gradient was switched from 35% to 85% buffer B over 1 min and held constant for 2 min. Finally, the gradient was changed from 85% buffer B to 98% buffer A (100% water, 0.1% formic acid) over 1 min, and then held constant at 98% buffer A for 5 more minutes. The application of a 2.0 kV distal voltage electrosprayed the eluting peptides directly into the mass spectrometer equipped with an Easy-spray source (Thermo Finnigan). The mass spectrometer was set to a targeted analysis method to only acquire CID MS/MS of the expected unphosphorylated (m/z 673.83) and phosphorylated (m/z 713.84) peptides. These MS/MS scans were acquired in the Orbitrap at 15,000 resolution with a scan range of m/z 150-1500. Mass spectrometer-scanning functions and HPLC gradients were controlled by the Xcalibur data system (Thermo Finnigan, San Jose, CA). The acquired MS data were analyzed manually to confirm the precursor mass, fragmentation ions, and serine phosphorylation in the targeted peptide.
Results
Development and validation of a cell-based assay to monitor NKX3.1 stabilization
We developed an assay for screening a kinome siRNA library to identify kinases that caused the degradation of endogenous NKX3.1 in LNCaP cells. To monitor rapidly NKX3.1 levels in cells, ProLabel, a 6 kDa tag that produces a strong chemiluminescent signal in an enzyme complementation reaction, was fused in-frame to the C-terminus of NKX3.1. The screening was carried out with knockdown reagents applied to LNCaP cells transiently transfected with the PL-N-NKX3.1 plasmid. After application of siRNAs, cells were lysed in the culture wells for chemiluminescence assay (Figure 1A).
Figure 1.

A. LNCaP-PLNX7 cells were transfected with a plasmid, PL-N-NKX3.1, allowing the expression of a chimeric NKX3.1 protein fused to the ProLabel tag at its C-terminal end. Upon transcription and translation, the PL-N-NKX3.1 fusion protein was phosphorylated and subjected to degradation through the ubiquitin-proteasome pathway. As the enzyme donor, the ProLabel tag in PL-N-NKX3.1 fusion protein can associate with enzyme acceptor (EA) to form an active enzyme that cleaves the chemiluminescent substrate; the resulting signal can be detected with a standard luminometer. Therefore, PL-N-NKX3.1 protein levels could be easily determined by cell-based high-throughput screening assays. B. PL-N-NKX3.1 was transiently transfected into 293T cells and the fusion protein was stained with NKX3.1 antiserum. C. Immunoblots from LNCaP cells transiently transfected with PL-N-NKX3.1 and 48 hr after transfection, cells were treated with cycloheximide for the indicated times. D. Immunoblot of extracts from a derivative LNCaP cell line expressing PL-N-NKX3.1 (clone #7, or PLNKX7). Cells were treated with cycloheximide and harvested at the indicated times for detection with NKX3.1 antiserum. E. Quantification of the immunoblot in D by densitometric scanning. F. LNCaP-PLNKX7 cells were transfected with NKX3.1 siRNA by reverse transfection. Immunoblotting was performed with NKX3.1 antiserum for both PL-NKX3.1 and endogenous NKX3.1. G. Sensitivity of the LNCaP-PLNKX7 detection assay was demonstrated in 96-well plates at an LNCaP cell density of 7000 cells/well. Cells were lysed directly in 96 well plate 4 days after transfection and chemiluminescence was determined.
The tagged NKX3.1 fusion protein was expressed well in both 293T and LNCaP cells and, like the native protein, localized to the nucleus (Figure 1B). LNCaP cells transiently transfected with PL-N-NKX3.1 expressed high levels of the fusion protein and LNCaP cells engineered for stable expression of the fusion protein showed levels comparable to the endogenous protein (Figures 1C and 1D). Importantly, the fusion protein and the endogenous native protein showed similar half-lives in LNCaP cells (Figures 1D and 1E). A single LNCaP clone (LNCaP-PLNKX7), selected for high expression of the fusion protein, was shown to undergo knockdown of both endogenous and fusion protein by NKX3.1 siRNA, thus demonstrating its utility for library screening (Figures 1F and 1G).
Kinome siRNA library screening
Initial kinome library screening was done by plating LNCaP-PLNKX7 into 96-well plates preloaded with the transfection reagent/siRNA mixture that included four separate siRNA duplexes for each kinase. After 72 hr 100 μg/ml cycloheximide was added and cells were lysed and assayed after an additional 60 min. Each plate contained mock transfections and NKX3.1 siRNA controls (Figure 2A). Luminescence data was normalized and analyzed using the Z-score method as described in Materials and Methods. Pairwise plots showed plate-to-plate reproducibility of the assay (Figure 2B). Forty-three genes with average Z scores above 1.415 were selected for secondary screening in order to fill a full microtiter plate. Genes with Z scores of 1.65 or higher were significantly different from control with a p ≤ 0.05. In the second-round screening, the protocol was the same as that used in the primary screening except that each of four specific siRNAs of these target genes were separately transfected into cells and PL-N-NKX3.1 fusion protein expression was monitored after cycloheximide treatment.
We previously showed that mutation of NKX3.1 N-terminal serines individually to alanine changed the NKX3.1 protein half-life 40-100% (10). Since we did not expect complete kinase inhibition in our siRNA screening we first set the screening threshold for NKX3.1 stabilization to 35% increase in residual NKX3.1 compared to mock-transfected cells exposed to cycloheximide. The results of the secondary screenings are shown in Figure 3A. Ten candidate kinases were selected by the secondary screening. Nine of these, all except BLK, were shown to be expressed in LNCaP cells (Figure 3B). To validate the effect of the kinases on NKX3.1 we generated a pool of four specific siRNA molecules for each enzyme (SMARTpool, Dharmacon). The validation was performed in the parent LNCaP cells and the level of endogenous NKX3.1 was assessed by immunoblotting. The expression of each candidate kinase was effectively knocked down by the corresponding siRNAs obtained from the kinome siRNA library, indicating these siRNA sequences could be used for our final validation assays (Figure 3C).
Figure 3.

A. NKX3.1 expression levels in duplicate assays of the second round screening. Independent result are shown in blue and red, representing the mean of four individual siRNA transfections. For mock transfection (Ctrl), cells were treated with or without 100 μg/ml cycloheximide for 60 min. Gene symbols in red are those chosen for final validations. B. Expression of endogenous kinase expression in LNCaP cells. Except BLK (B lymphoid tyrosine kinase), the expression of all other kinase candidate mRNAs was easily detected with semi-quantitative RT-PCR. C. Custom siRNAs were used to knock down kinase expression in LNCaP cells. RT-PCR was performed. For semiquantitative RT-PCR reaction cycles were as follows: PRKAR1A: 25 cycles; PIK3R1: 26 cycles; DYRK1B: 28 cycles; MARK2 and WNK3: 30 cycles; MAP4K1: 31 cycles; CDKN2A: 32 cycles; GRK7: 34 cycles.
As a final step we analyzed the effect of each kinase siRNA on endogenous NKX3.1 in LNCaP cells. Specific siRNAs from the library were transfected into LNCaP cells exposed to cycloheximide for up to 60 minutes. The immunoblots in Figure 4A show the effects of kinase inhibition on endogenous NKX3.1. DYRK1B had the most profound effect of the group on NKX3.1 half-life (Figure 4B). Knockdown of RPS6KA5 or PRKAR1A did not affect the stability of NKX3.1 (data not shown). It should be noted that in the primary and secondary screening assays, knockdown of PRKAR1A always led to a dramatic increase in the level of fusion protein PL-N-NKX3.1 with the highest Z score (average Z score = 14.43). This was also true in LNCaP-PLNKX7 cells when it was knocked down with the custom siRNA we synthesized. However, neither of these siRNAs affected the level of endogenous NKX3.1. We did not study the interaction of PRKAR1A with the PL-N-NKX3.1 fusion protein, but made the preliminary conclusion that the effect seen in the screening was an artifact of the fusion protein and not related to native NKX3.1. PRKAR1A would not have been a likely candidate for NKX3.1 suppression as the kinase subunit itself is a suppressor protein whose inactivation is associated with a syndrome of benign neoplasia, Carney's triad (21).
Figure 4.

Kinase knockdown of NKX3.1 expression. A. LNCaP cells were transfected with control or kinase siRNA. Cycloheximide 100 μg/ml was added at 72 h after and cell extracts were prepared at the indicated times for immunoblots. B. Immunoblots were quantitated with Image J and NKX3.1 half-life was determined using Graph Pad Prism.
Physical association of NKX3.1 with DYRK1B
Knockdown of DYRK1B by siRNA was consistently found to have the most pronounced effect on NKX3.1 half-life. We were able to demonstrate physical association between the two proteins we used Flag-tagged DYRK1B and exogenous NKX3.1 expression in 293T cells (Figure 5A). Kinase-dead DYRK1B that was inactivated by missense mutation (DYR1B-KD) was still able to associate with NKX3.1. However, the DYRK1B N-terminus alone (DYRK1B-ΔKD) did not bind to NKX3.1(Figure 5B). Thus the kinase domain is the likely site of interaction with NKX3.1.
Figure 5.

Interaction between NKX3.1 and DYRK1B. A. 293T cells transfected with MYC-NKX3.1 and Flag-tagged mDYRK1B were lysed and subjected to immunoprecipitation with α-Flag (M2) or mouse IgG. Immunoprecipitates were analyzed by immunoblotting with either MYC or Flag antibodies. The arrows indicate specific protein bands. B.293T cells were engineered as in A to express NKX3.1 and murine DYRK1B constructs as shown below the gels. C. Dose and time-dependence of NKX3.1 association with DYRK1B. GST-DYRK1B (0-200 ng/reaction) and His-NKX3.1 were combined in an in vitro kinase assay in the presence of ATP as described in Materials and Methods. Reactions were separated on SDS-PAGE containing 25 μM Phos-tag and 50 μM MnCl2. NKX3.1 was detected by immunoblotting with NKX3.1 antiserum. D. Upper panel: 293T cells expressing MYC-NKX3.1 and transfected with either pcDNA3.1 or Flag-mDYRK1B were analyzed for phosphorylation or diluted about 200 fold with phage λ phosphatase buffer followed by concentrating with Amicon Centricon YM-10 and Amicon Ultra-0.5 ml (10K). The cleared and concentrated lysates were then treated with λ phosphatase for 60 min at 30°C and analyzed with 25 μM Phos-tag. Lower panel: 293T cells expressing MYC-NKX3.1 and transfected with either pcDNA3.1 or Flag-mDYRK1B were treated with or without 1 μM 185981 for 16 hr. NKX3.1 phosphorylation was determined by immunoblotting a gel run with 20 μM Phos-Tag.
DYRK1B phosphorylates NKX3.1 in vitro
We demonstrated in vitro phosphorylation of polyhistidyl-NKX3.1 by purified GST-DYRK1B (22). The reaction products were separated on SDS-PAGE containing 25 μM Phos-tag and 50 μM MnCl2 and transferred onto nitrocellulose membrane. Both phosphorylated and native NKX3.1 were visualized by immunoblotting with NKX3.1 antibody. Retardation of phosphorylated NKX3.1 was seen to be dependent on DYRK1B concentration and time (Figure 5C). In addition, Flag-DYRK1B expression in 293T cells resulted in phosphorylation of exogenously expressed MYC-NKX3.1 in vivo. MYC-NKX3.1 was cotransfected with either Flag-mDYRK1B or pcDNA3.1 vector control into 293T cells and NKX3.1 phosphorylation was seen and reversed by addition of phage-λ phosphatase (Figure 5D). Moreover, 185981 inhibited phosphorylation of MYC-NKX3.1 by DYRK1B.
To map the phosphorylation sites in NKX3.1 by DYRK1B, in vitro kinase reaction products were separated by conventional SDS-PAGE, excised, digested with trypsin and analyzed by mass spectrometry (MS/MS). We used LC-MS/MS analysis to detect the tryptic peptide containing S185 from in vitro phosphorylated NKX3.1. Correct precursor mass and fragment ion masses of the targeted peptide were manually verified. Neutral loss of S185 was identified and manually verified in the acquired tandem mass spectra as shown in Figure 6A. Serine 185, a key residue in signaling NKX3.1 turnover, is highly conserved across species (Figure 6B).
Figure 6.

A. HPLC/MS/MS spectrum of phosphopeptide KQLpSSELGDLEK. The arrow pointed at corresponding bn ions minus H3PO4, which serve to confirm the phosphopeptide. Mass accuracy: ±10ppm (monoisotopic mass). B. NKX3.1 from various species aligned to show sequence conservation across the C-terminal domains.
DYRK1B inhibitors extend NKX3.1 half-life
Since our long-term goal is the pharmacologic increase of intracellular NKX3.1 in prostate cancer, we asked whether small molecule DYRK1B inhibitors affected the half-life of NKX3.1. Using the same assay of endogenous NKX3.1 half-life shown in Figure 4 we treated LNCaP cells with DYRK1B inhibitors after the cells had been exposed to cycloheximide for one hour. The compounds listed in Table 1 all prolonged NKX3.1 half-life. The most active compound was NCGC00185981 that has specificity against both DYRK and CLK family kinases with high affinity for DYRK1B (23). A second compound with higher IC50 for DYRK1B but with no specificity for CLK kinases NCGC00185984 was not as active in blocking NKX3.1 degradation. A MARK2 inhibitor, 39621, was used because MARK2 was one of the kinases identified in the second round kinome library screening (24). Compound 39621 was substantially less active as an inhibitor of NKX3.1 degradation than the DYRK1B inhibitors.
Table 1. Effect of Kinase Inhibitors on NKX3.1 Half-Life.
| IC50-nM | Target | μM | T1/2 | |
|---|---|---|---|---|
| DMSO | 24.7 | |||
| Compound A | 68 | DYRK1B | 0.10 | 31.6 |
| 0.50 | 46.9 | |||
|
| ||||
| DMSO | 24.7 | |||
| NCGC00185981 | 25 | DYRK1B | 0.10 | 51.0 |
| 0.50 | ≫50 | |||
|
| ||||
| DMSO | 28.0 | |||
| NCGC00185963 | 1510 | DYRK1B | 0.50 | >50 |
| 2.00 | 40.0 | |||
|
| ||||
| DMSO | 28.0 | |||
| NCGC00185976 | 103 | DYRK1B | 0.50 | 66.0 |
| 2.00 | 34.8 | |||
|
| ||||
| DMSO | 28 | |||
| 39621 | 3600 | MARK2 | 5.00 | 33.5 |
| 10.00 | 60.5 | |||
| 20.00 | 42.0 | |||
|
| ||||
| DMSO | 26.0 | |||
| NCGC00185984 | 72 | DYRK1B | 0.01 | 39.5 |
| 0.05 | 29.5 | |||
| 0.10 | 36.5 | |||
| 0.25 | 41.0 | |||
| 0.50 | 40.5 | |||
| 1.00 | 65.5 | |||
Discussion
Both pathologic and physiologic processes contribute to the erosion of NKX3.1 expression in the aging prostate gland. Loss of this protein results in increased propensity for proliferation, susceptibility to DNA damage, and increased frequency of TMPRSS2-ERG gene rearrangement (8, 25, 26). In mouse models heterozygosity of Nkx3.1 has minimal effect on prostate function as shown by effects on prostatic fluid, but more profound effects on cell proliferation (5). In fact, Nkx3.1 heterozygosity predisposes to prostatic hyperplasia and dysplasia not dissimilar from complete loss of Nkx3.1, but with longer latency. Nkx3.1 heterozygosity also potentiates the effect of Pten heterozygosity on prostatic neoplasia (27). With the ultimate goal of inhibiting NKX3.1 loss in the prostate we have identified at least one kinase that targets NKX3.1 for phosphorylation important for steady-state protein turnover. DYRK1B was shown to phosphorylate NKX3.1 at serine 185, a key signal for ubiquitination subsequent proteasomal degradation (10).
Dual-specificity tyrosine-regulated kinase 1B (DYRK1B) or minibrain-related kinase (Mirk) is a nuclear serine-threonine kinase activated by tyrosine autophosphorylation (11). DYRK1B is a member of the conserved DYRK/minibrain family of tyrosine-regulated, arginine-directed serine/threonine protein kinases (28). DYRK1B is highly expressed in the testis and skeletal muscle and plays an important role in muscle differentiation, cell survival, and cell migration. More recently, it has been revealed that DYRK1B phosphorylates and stabilizes the cyclin-dependent kinase (CDK) inhibitor p27 in nontransformed cells thus contributing to maintenance of cell quiescence (29). DYRK1B also phosphorylates and destabilizes cyclins D1 and D3 and therefore blocks quiescent cells from traversing G1 phase of the cell cycle (30, 31).
DYRK1B has paradoxically also been shown to be pro-oncogenic and its inhibition results in growth inhibition of several cancer cell lines (14-16). DYRK1B effect on cell proliferation may be due in part to activation of cyclin D1 (12) and suppression of p27Kip1 (13). In addition a number of small molecule inhibitors of DYRK1B have been developed and (23, 32). Thus DYRK1B is already a target of investigational cancer therapy. DYRK1B inhibition retards the growth of cancer cells in model systems. Inhibition of DYRK1B in prostate cancer will have the tissue specific effect of increasing levels of the NKX3.1 suppressor protein besides its effect on cell cycle regulators. For these reasons DYRK1B is a novel and potentially important therapeutic target with unique relevance to prostate cancer.
Acknowledgments
Supported in part by Department of Defense Prostate Cancer Research Program W81XWH-11-1-0177, by NCI grant P01 CA154293, and by CCSG P30 CA013696-36 via the Bioinformatics Shared Resource and 5 P30 CA013696-39S3 to the Proteomics Shared Resource.
Footnotes
The authors have no competing interests and no conflicts to disclose.
Contributor Information
Liang-Nian Song, Email: ls2584@columbia.edu.
Jose Silva, Email: jose.silva@mssm.edu, jms2263@columbia.edu.
Antonius Koller, Email: ak3705@columbia.edu.
Andrew Rosenthal, Email: andrew.s.rosenthal@gmail.com.
Emily I. Chen, Email: ec2974@columbia.edu.
References
- 1.Vocke CD, Pozzatti RO, Bostwick DG, Florence CD, Jennings SB, Strup SE, et al. Analysis of 99 microdissected prostate carcinomas reveals a high frequency of allelic loss on chromosome 8p21-22. Cancer Res. 1996;56:2411–6. [PubMed] [Google Scholar]
- 2.Swalwell JI, Vocke CD, Yang Y, Walker JR, Grouse L, Myers SH, et al. Determination of a minimal deletion interval on chromosome band 8p21 in sporadic prostate cancer. Genes Chromosomes Cancer. 2002;33:201–5. doi: 10.1002/gcc.10015. [DOI] [PubMed] [Google Scholar]
- 3.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, Kim M, Desai N, Norton CR, et al. Roles for Nkx3.1 in prostate development and cancer. Genes and Development. 1999;13:966–77. doi: 10.1101/gad.13.8.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim MJ, Bhatia-Gaur R, Banach-Petrosky WA, Desai N, Wang Y, Hayward SW, et al. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer Res. 2002;62:2999–3004. [PubMed] [Google Scholar]
- 7.Wang X, Kruithof-de JM, Economides KD, Walker D, Yu H, Halili MV, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asatiani E, Huang WX, Wang A, Rodriguez OE, Cavalli LR, Haddad BR, et al. Deletion, methylation, and expression of the NKX3.1 suppressor gene in primary human prostate cancer. Cancer Res. 2005;65:1164–73. doi: 10.1158/0008-5472.CAN-04-2688. [DOI] [PubMed] [Google Scholar]
- 9.Bowen C, Bubendorf L, Voeller HJ, Slack R, Willi N, Sauter G, et al. Loss of NKX3.1 expression in human prostate cancers correlates with tumor progression. Cancer Res. 2000;60:6111–5. [PubMed] [Google Scholar]
- 10.Markowski MC, Bowen C, Gelmann EP. Inflammatory cytokines induce phosphorylation and ubiquitination of prostate suppressor protein NKX3.1. Cancer Res. 2008;68:6896–901. doi: 10.1158/0008-5472.CAN-08-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost HG. Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J Biol Chem. 1998;273:25893–902. doi: 10.1074/jbc.273.40.25893. [DOI] [PubMed] [Google Scholar]
- 12.Zou Y, Ewton DZ, Deng X, Mercer SE, Friedman E. Mirk/dyrk1B kinase destabilizes cyclin D1 by phosphorylation at threonine 288. J Biol Chem. 2004;279:27790–8. doi: 10.1074/jbc.M403042200. [DOI] [PubMed] [Google Scholar]
- 13.Jin K, Ewton DZ, Park S, Hu J, Friedman E. Mirk regulates the exit of colon cancer cells from quiescence. J Biol Chem. 2009;284:22916–25. doi: 10.1074/jbc.M109.035519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao J, Zhao Y, Lv Y, Chen Y, Wei B, Tian J, et al. Mirk/Dyrk1B mediates G0/G1 to S phase cell cycle progression and cell survival involving MAPK/ERK signaling in human cancer cells. Cancer Cell Int. 2013;13:2. doi: 10.1186/1475-2867-13-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu J, Deng H, Friedman EA. Ovarian cancer cells, not normal cells, are damaged by Mirk/Dyrk1B kinase inhibition. Int J Cancer. 2013;132:2258–69. doi: 10.1002/ijc.27917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewton DZ, Hu J, Vilenchik M, Deng X, Luk KC, Polonskaia A, et al. Inactivation of mirk/dyrk1b kinase targets quiescent pancreatic cancer cells. Mol Cancer Ther. 2011;10:2104–14. doi: 10.1158/1535-7163.MCT-11-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyata Y, Nishida E. DYRK1A binds to an evolutionarily conserved WD40-repeat protein WDR68 and induces its nuclear translocation. Biochim Biophys Acta. 2011;1813:1728–39. doi: 10.1016/j.bbamcr.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 18.Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods. 2009;6:569–75. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soppa U, Schumacher J, Florencio O V, Pasqualon T, Tejedor FJ, Becker W. The Down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle. 2014;13:2084–100. doi: 10.4161/cc.29104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gockler N, Jofre G, Papadopoulos C, Soppa U, Tejedor FJ, Becker W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009;276:6324–37. doi: 10.1111/j.1742-4658.2009.07346.x. [DOI] [PubMed] [Google Scholar]
- 21.Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 22.Ju JH, Maeng JS, Zemedkun M, Ahronovitz N, Mack JW, Ferretti JA, et al. Physical and functional interactions between the prostate suppressor homeoprotein NKX3.1 and serum response factor. J Mol Biol. 2006;360:989–99. doi: 10.1016/j.jmb.2006.05.064. [DOI] [PubMed] [Google Scholar]
- 23.Rosenthal AS, Tanega C, Shen M, Mott BT, Bougie JM, Nguyen DT, et al. Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk) Bioorg Med Chem Lett. 2011;21:3152–8. doi: 10.1016/j.bmcl.2011.02.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Timm T, von Kries JP, Li X, Zempel H, Mandelkow E, Mandelkow EM. Microtubule affinity regulating kinase activity in living neurons was examined by a genetically encoded fluorescence resonance energy transfer/fluorescence lifetime imaging-based biosensor: inhibitors with therapeutic potential. J Biol Chem. 2011;286:41711–22. doi: 10.1074/jbc.M111.257865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowen C, Gelmann EP. NKX3.1 activates cellular response to DNA damage. Cancer Res. 2010;70:3089–97. doi: 10.1158/0008-5472.CAN-09-3138. [DOI] [PubMed] [Google Scholar]
- 26.Bowen C, Ju JH, Lee JH, Paull TT, Gelmann EP. Functional activation of ATM by the prostate cancer suppressor NKX3.1. Cell Rep. 2013;4:516–29. doi: 10.1016/j.celrep.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim MJ, Cardiff RD, Desai N, Banach-Petrosky WA, Parsons R, Shen MM, et al. Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc Natl Acad Sci U S A. 2002;99:2884–9. doi: 10.1073/pnas.042688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedman E. Mirk/Dyrk1B in cancer. J Cell Biochem. 2007;102:274–9. doi: 10.1002/jcb.21451. [DOI] [PubMed] [Google Scholar]
- 29.Deng X, Mercer SE, Shah S, Ewton DZ, Friedman E. The cyclin-dependent kinase inhibitor p27Kip1 is stabilized in G(0) by Mirk/dyrk1B kinase. J Biol Chem. 2004;279:22498–504. doi: 10.1074/jbc.M400479200. [DOI] [PubMed] [Google Scholar]
- 30.Deng X, Ewton DZ, Friedman E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 2009;69:3317–24. doi: 10.1158/0008-5472.CAN-08-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Becker W. Emerging role of DYRK family protein kinases as regulators of protein stability in cell cycle control. Cell Cycle. 2012;11:3389–94. doi: 10.4161/cc.21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coombs TC, Tanega C, Shen M, Wang JL, Auld DS, Gerritz SW, et al. Small-molecule pyrimidine inhibitors of the cdc2-like (Clk) and dual specificity tyrosine phosphorylation-regulated (Dyrk) kinases: Development of chemical probe ML315. Bioorg Med Chem Lett. 2013;23:3654–61. doi: 10.1016/j.bmcl.2013.02.096. [DOI] [PMC free article] [PubMed] [Google Scholar]