

Staphylococcus aureus CC30 is distinguished by its ability initiate complicated infections. This ability is due to acquisition of unique genes, SNPs and metabolic changes that attenuate virulence until the conditions become favorable for bacteremia and subsequent hematogenous seeding.
Keywords: genome, genotype, hematogenous infections, Staphylococcus aureus, transcriptome
Abstract
Background. The contemporary Staphylococcus aureus clonal complex (CC) 30 lineage is associated with complicated infections, including endocarditis and osteomyelitis. This lineage diverged from the phage-type 80/81 S aureus clone responsible for a major bacterial epidemic of the 20th century. The genome and transcriptome features that contribute to complicated infections of the CC30 lineage are unknown.
Methods. Twenty-nine clinical methicillin-resistant S aureus (MRSA) strains (8 from CC30 and 21 from other major CCs were evaluated for virulence using murine and Galleria mellonella sepsis models. Genomic features of CC30 were identified by comparative genome sequencing and RNA-Seq transcriptome analysis of the 29 strains and 31 previously sequenced S aureus genomes.
Results. The CC30 isolates displayed lower virulence in the sepsis models compared with other CCs [P < .0001]. Comparisons of orthologous proteins and transcriptome analysis identified genes (eg, nitric oxide reductase) and changes in metabolic pathways (eg, pyrimidine metabolism) that contribute to the distinct CC30 phenotype. Previously reported nonsynonymous single-nucleotide polymorphisms (SNPs) were found in accessory gene regulator C (agrC) and α-hemolysin (hla), molecules important for virulence. Additional nonsynonymous SNPs conserved across clinical CC30 isolates when compared with the first sequenced contemporary CC30 clone, MRSA-16, were identified in multiple genes, suggesting continuing evolutionary divergence in this lineage.
Conclusions. Genomic and transcriptional analyses suggest that the CC30 lineage has acquired metabolic features that contribute to persistent and complicated infections. Absence of sepsis-induced mortality in animal models may be due in part to its unique genomic profile and suggests that specific genotypes of S aureus elicit distinct types of infection types.
Staphylococcus aureus, a commensal of the skin and nares, is the cause of infections ranging from uncomplicated skin infections to serious complicated infections such as pneumonia, osteomyelitis, endocarditis, and sepsis [1–3]. The ability of S aureus to cause severe infections is attributed to its repertoire of virulence factors, many of which are transferred horizontally through the S aureus community [4, 5]. Staphylococcus aureus isolates are classified into different genetic lineages or clonal complexes (CCs) based on genome content, with a close association between bacterial genetic characteristics and the distinct clinical manifestations [6–12]. There are over 100 CCs of S aureus, and CC5, CC8, CC22, CC30, and CC45 are more frequently associated with hospital-acquired infections. Staphylococcus aureus CC30 is the major nasal carriage lineage, and it is associated with the majority of autologous infections [13]. Previous work from our group [8, 14, 15] demonstrated that the CC30 lineage is associated with hematogenous complications, including endocarditis, septic arthritis, and vertebral osteomyelitis. In addition, CC30 is more frequently associated with persistent versus resolving bacteremia, and it shows increased adhesion to endothelial cells, elevated resistance to human neutrophil peptide hNP-1, and increased membrane fluidity compared with resolving strains [16]. These attributes of CC30 isolates may potentially be responsible for invasion of endocardial surfaces and contribute to persistent infection. The genomic basis for these CC30 characteristics is unknown.
In the current investigation, we used in vivo sepsis models, comparative genomics, and RNA-Seq transcriptome analysis of CC30 and other CCs to (1) evaluate virulence and severity of infections and (2) identify genomic features that contribute to CC30 persistence and complicated infections. Staphylococcus aureus CC30 isolates were significantly less virulent compared with other CCs. The attenuated virulence of CC30 isolates in sepsis models is attributed to its distinct genomic architecture, single-nucleotide polymorphisms (SNPs) that inactivate key virulence genes, and differential regulation of metabolic and adhesin genes.
MATERIALS AND METHODS
All animal research was approved by Duke University Institutional Review Board and Institutional Animal Care & Use Committee, as appropriate. Genome sequence and assembly, assembly validation using Opgen optical maps, genome annotation, whole-chromosome phylogenetic analysis, Jaccard Orthologous Clustering (JOC) analysis, and SNP discovery and analysis in CC30 strains are described in Supplementary Materials and Methods.
Staphylococcus aureus Clinical Isolates
The 379 S aureus isolates (125 methicillin-resistant S aureus [MRSA] and 254 methicillin-sensitive S aureus [MSSA] isolates from CC1, CC5, CC8, CC5, CC30, and CC45) used for our initial genotypic multilocus sequence typing [MLST]/Spa typing were selected using strict definitions to identify 3 clinical groups that represent a progression from healthy individuals to those who are severely infected: (1) nasal carriage only (healthy controls), (2) uncomplicated infection, and (3) bacteremia with hematogenous complications [8]. Multiple levels of array-comparative genomic hybridization analyses identified differences in gene content relative to CC, MRSA, and MSSA status as well as clinical outcome. Two separate analyses, one dependent on CC status (found in CC5 and CC30) and a second independent of CC status, identified an identical set of 14 genes associated with complicated infections [8, 14]. The 29 MRSA isolates in the Complicated Infection Group (CIG) (Table 1) used in the current study were selected from the initial 379 S aureus isolates based on (1) carriage and clinical severity, (2) MRSA status, (3) presence and proportion of the 14 candidate virulence genes, and (4) CC. The CC30 CIG isolates belong to the previously described contemporary CC30 clone lineage that diverged from the phage-type 80/81 S aureus clone [17–19].
Table 1.
Methicillin-Resistant Staphylococcus aureus Strains in the CIG
| IDa | Clonal Complex | Group | B&J | Endo | Both | Outcomeb | Proportionc |
|---|---|---|---|---|---|---|---|
| 1057 | 5 | Complicated | X | 1 | 1 | ||
| 1096 | 5 | Complicated | X | 2 | 0.9 | ||
| 1150 | 5 | Complicated | X | 1 | 0.364 | ||
| 1165 | 5 | Complicated | X | 3 | 0.92 | ||
| 1213 | 5 | Complicated | X | 3 | 0.92 | ||
| 1750 | 5 | Complicated | X | 3 | 1 | ||
| 1769 | 5 | Complicated | X | 3 | 0.7 | ||
| 1612 | 8 | Complicated | X | 1 | 0.8 | ||
| 1114 | 8 | Complicated | X | 1 | 0.55 | ||
| 547 | 8 | Complicated | X | 1 | 0.86 | ||
| 2018 | 8 | Complicated | X | 1 | 0.75 | ||
| 1524 | 45 | Complicated | X | 3 | 0.3 | ||
| 290 | 45 | Complicated | X | 2 | 0.56 | ||
| 1835 | 1 | Complicated | X | 3 | 0.88 | ||
| 1176 | 30 | Complicated | X | 1 | 0.93 | ||
| 1242 | 30 | Complicated | X | 1 | 0.93 | ||
| 1500 | 30 | Complicated | X | 3 | 1 | ||
| 1605 | 30 | Complicated | X | 2 | 0.92 | ||
| 1267 | 30 | Complicated | X | 1 | 1 | ||
| 1214 | 30 | Complicated | X | 3 | 0.9 | ||
| 149 | 30 | Complicated | X | 3 | 0.9 | ||
| 1233 | 30 | Complicated | X | 3 | 1 | ||
| 1770 | 8 | Complicated | X | 3 | 0.58 | ||
| C34D | 8 | Nasal carriage | 0.2 | ||||
| C34D | 5 | Nasal carriage | 0.2 | ||||
| C348 | 5 | Nasal carriage | 1 | ||||
| C34D | 30 | Nasal carriage | 1 | ||||
| C93 | 15 | Nasal carriage | 0.1 | ||||
| C128 | 1 | Nasal carriage | 0.6 |
Abbreviations: B&J, bone and joint infection; Both, both bone and joint and endocarditis; CIG, Complicated Infection Group; Endo, endocarditis; ID, identification.
a Sequence data for the CIG genomes has been deposited in GenBank under PID (Pathway Interaction Database):
b 1, cured; 2, recurrent S aureus infection; 3, death due to S aureus infection.
c Proportion of 14 potential virulence genes more frequently associated with strains causing complicated infections [14]. This value is a fraction determined by X/14, where X = the number of virulence genes in the isolate.
PRJNA60651, PRJNA60653, PRJNA60655, PRJNA60657, PRJNA60659, PRJNA60661, PRJNA60663, PRJNA60665, PRJNA60667, PRJNA60669, PRJNA60671, PRJNA60673, PRJNA60675, PRJNA60677, PRJNA60679, PRJNA60681, PRJNA60683, PRJNA60685, PRJNA60687, PRJNA60689, PRJNA60691, PRJNA60693, PRJNA60695, PRJNA60697, PRJNA60699, PRJNA60701, PRJNA60703, PRJNA60705, PRJNA60707.
Staphylococcus aureus Reference Strains for Comparative Genome and Transcriptome Analysis
Staphylococcus aureus MRSA252 (a reference strain for the contemporary CC30 MRSA-16 lineage [20]), N315 (a CC5 reference strain [21]), THC60 (an S aureus CC30 isolate sequenced in the Human Microbiome Project [PRJNA159859]), and UAMS-1 (a commonly used clinical CC30 strain from an osteomyelitis patient [22]) were obtained from Network on Antimicrobial Resistance in Staphylococcus aureus or Mark Smeltzer (University of Arkansas).
Preparation of Staphylococcus aureus Cells
To prepare S aureus cells for injection, an isolated colony from a fresh tryptic soy agar (TSA) plate was inoculated to 10 mL fresh tryptic soy broth and incubated at 37°C/220 revolutions per minute (rpm) for overnight culture. An appropriate amount of overnight bacterial culture was added to 100 mL fresh tryptic soy broth (TSB) in a 500 mL Erlenmeyer flask to normalize the initial optical density ([OD] 600) to approximately 0.1. The flasks were incubated at 37°C/220 rpm to log-phase growth (OD 600 ∼ 1.0). Staphylococcus aureus cells were harvested by centrifugation, washed twice with phosphate-buffered saline (PBS), resuspended in PBS containing 20% glycerol to a concentration of approximately 106 colony-forming units (CFUs)/µL, aliquoted into individual cryovials, and immediately stored at −80°C until further use. An aliquot of PBS-washed culture was serially diluted and plated on TSA plates to enumerate CFUs. Frozen stocks were titered for CFUs in triplicate on separate occasions before use in any experiment.
Mouse Model of Intraperitoneal Sepsis
Six- to 8-week-old C57BL/6J mice used in this study were purchased from The Jackson Laboratory (Bar Harbor, ME). Colony-forming units (108) of each S aureus strain in 200 µL suspension in PBS were injected intraperitoneally into 5 mice in each experimental group. Mice were given supplemental care and observed continuously for survival for 8 hours postinfection and then every day for 5 days.
Wax Moth Model of Sepsis
The wax moth model of sepsis (described previously [23, 24]) was used in this study with slight modifications. In brief, the larval form of greater wax moth (Galleria mellonella) was purchased from Best Bet Inc. (Blackduck, MN) and stored at 4°C in dark until used for the sepsis experiments. The larvae were incubated at room temperature overnight prior to injection with S aureus. Twenty healthy larvae of similar size (approximately 250–300 mg) were injected in each experimental group, using 50 µL Hamilton syringes fitted with 26-gauge needles (Hamilton Company, Reno, NV), along with 20 uninjected and PBS-injected controls. The frozen stock of each culture (prepared as described earlier) was thawed on ice and diluted in PBS to the desired number of S aureus cells in the inoculum (10 µL of inoculum containing either 105 or 106 CFUs of S aureus). The larvae were incubated at 37°C, observed 4 hours postinfection, and then every day for 5 days to monitor survival. Dead larvae were identified as those melanized and unresponsive to stimuli.
RNA-Seq Transcriptome Analysis of Complicated Infection Group Isolates
Whole transcriptome analysis of the 29 MRSA CIG isolates, MRSA252, N315, and UAMS-1 was determined by RNA-Seq. To prepare isolates for RNA extraction, an overnight culture of S aureus was inoculated into 100 mL fresh TSB in a 500 mL Erlenmeyer flask to an OD (600) to approximately 0.1. The flasks were incubated at 37°C/220 rpm to log-phase growth (OD 600 ∼ 0.6). Total RNA was extracted by mechanical lysis in acid phenol [25], and rRNA was depleted by Ribo-Zero (Illumina, San Diego, CA) and sequenced with Illumina Tru-Seq. Low complexity reads, adapter, and vector contamination were removed using Seqclean (http://sourceforge.net/projects/seqclean/) and the National Center for Biotechnology Information (NCBI) UniVec database. Processed reads were then mapped to the MRSA252 genome with SHRiMP version 2.2.3 [26]. Differential expression analysis was performed using Cufflinks (cuffDiff2) version 2.0.2 [27] of the general transfer format annotation file for the reference genome and a false discovery rate of 0.05. The RNA expression levels for each gene were expressed as fragments per kilobase per million reads mapped (FPKM). Genes in which all samples did not have at least an FPKM of 1 were removed from consideration before downstream analysis. A greater than log2-fold increase or decrease in expression level was used to identify genes that were significantly different between the CCs. A custom PERL program was used to determine overlap of significant differentially expressed genes between samples within and between the CCs at a variety of levels of agreement (50%, 80%). Heat maps and dendograms were generated by per gene z-score that was computed from log10 FPKM values or hierarchical clustering of log10 FPKM values. Comparison of whole transcriptome expression between CCs was determined by multidimensional scaling (MDS) of the normalized compression distance. All RNA-Seq data (raw sequence reads and primary analysis) are deposited at NCBI and Sequence Read Archive under BioProject PRJNA255909.
Statistical Analyses
For survival analysis in both animal models, the survival data were pooled together into 2 groups (CC30 vs others), and the Kaplan-Meier survival curve was generated using GraphPad Prism, version 5.0 (La Jolla, CA). The survival difference between strains was assessed using the log-rank: Mantel-Cox test. To identify genes uniquely associated with CC30 (Table 1), protein clusters of related function identified using JOC (Supplementary Table 1) were first sorted by a t test comparison of clusters present in CC30 vs all other CCs. All clusters with P < .05 were then evaluated across all 60 genomes to calculate proportion of genomes containing each cluster (expressed as percentage).
RESULTS
Attenuated Virulence of Clonal Complex 30 in Murine and Galleria mellonella Sepsis Models
When compared with all other CCs studied, CC30 isolates demonstrated significantly attenuated virulence in both the murine intraperitoneal sepsis (P < .0001; Figure 1A) and G mellonella sepsis models (P < .0001; Figure 1B). This attenuated virulence of CC30 persisted when alternate infective doses of S aureus (106 CFU instead of 105 CFU, data not shown) were used in G mellonella sepsis model.
Figure 1.
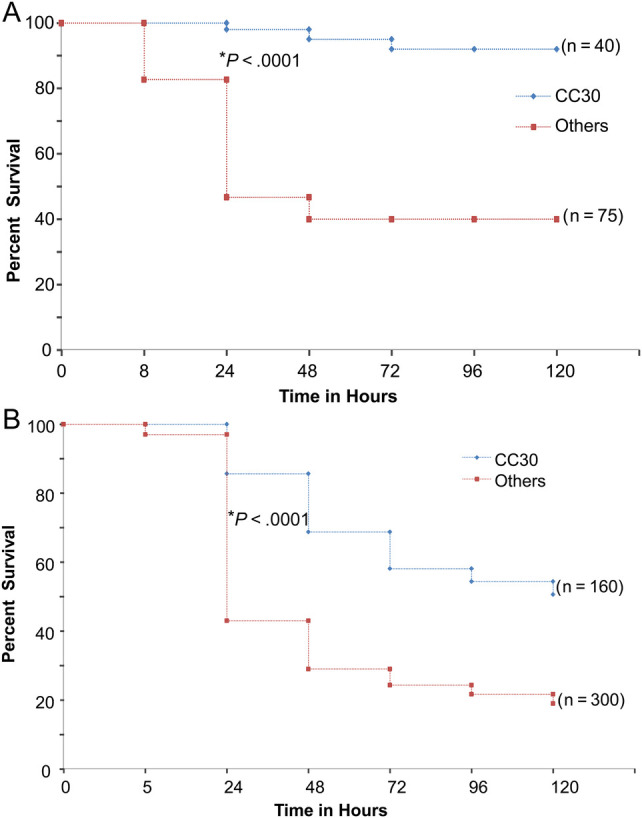
(A) Kaplan–Meier survival curve of C57BL/6J mice after infection with Staphylococcus aureus in the Complicated Infection Group (CIG). C57BL/6J mice were given intraperitoneal injections with 108 colony-forming units (CFUs) of each CIG S aureus isolates (n = 5 mice for each strain). Survival analysis was done using log-rank Mantel-Cox test in GraphPad prism. *, P value (log-rank: Mantel-Cox test); clonal complex (CC) 30 strains include 1176, 1242, 1500, 1605, 1267, 1233, and 1214. Others include CC1 (1835), CC5 (1057, 1096, 1150, 1165, 1213, 1750, 1769), CC8 (1612, 1114, 547, 2018, 1770), and CC45 (1524, 290). (B) Kaplan-Meier survival curve of greater wax moth (Galleria mellonella) after infection with S aureus in the CIG. The greater wax moth (G mellonella) were given injections with 105 CFU of each CIG S aureus isolates (n = 20 worms for each strain). Survival analysis was done using log-rank Mantel-Cox test in GraphPad prism. *P value (log-rank: Mantel-Cox test); strains used are identical to those in A.
Phylogenetic Relationships Between the Clonal Complexes
Phylogenetic information included in the core genome alignments of the 29 CIG isolates and 31 closed S aureus genomes available in GenBank were used to determine the genomic relatedness of the CIG isolates. There is significant congruence between the previously determined grouping by MLST into CCs and the current whole-genome phylogeny. As demonstrated by SNP whole-genome alignments (Figure 2) showing phylogenetic relationship between the CCs, all CC30 CIG isolates clustered into the same phylogroup, most closely related to the CC45 and CC398 lineages and most distant from CC5.
Figure 2.

Maximum likelihood single-nucleotide polymorphism (SNP) tree for 60 Staphylococcus aureus isolates, representing relationships between clonal complexes (CCs) in the Complicated Infection Group (CIG) and previously sequenced S aureus genomes. Single nucleotide polymorphisms were identified in pairwise genome comparisons between the predicted genes in all genomes. Single nucleotide polymorphisms for each genome were concatenated to form SNP pseudosequences and used to generate a phylogenetic tree using the HKY93 algorithm [28] with 500 bootstrap replicates.
Jaccard Orthologous Clustering Analysis to Distinguish Genome Features Unique to Clonal Complex 30 Isolates
Jaccard Orthologous Clustering analysis is typically used to group together highly similar proteins within a single genome/organism of interest and allows for one-to-many orthology. Comparisons of protein families between the 29 CIG genomes sequenced in this study (Table 1) and 31 additional complete S aureus genomes available in the GenBank were used to identify genomic features that distinguished the CC30 lineage from the other predominant lineages. A total of 2300 proteins were present among all tested CCs. The CC30 isolates possessed a distinct set of proteins, including 25 proteins encoded in all CC30 strains and absent in greater than 90% of all other CCs (Table 2 and Supplementary Table 1). Twenty-six proteins were uniquely associated with CIG CC30 isolates but were absent in CC30 isolates MRSA252 and THC60 (contemporary CC30 reference clones [20]). Functions of these 26 proteins suggested ongoing evolutionary changes among the CIG isolates. Twenty-eight proteins unique to CC30 were previously identified in islets of the MRSA252 genome [20].
Table 2.
Proteins Identified by Jaccard Orthologous Clustering as Associated with Staphylococcus aureus CC30 Isolates
| Representative Protein ID in Cluster (LOCUS Name) | Representative MRSA252 Protein in Cluster (LOCUS Name) | Common Name | CC30 Average Number of Proteins in Clustera | Present in All CC30 Strains and Absent in >90% of Other CC | Present in >50% of CC30 and Absent in >90% of Other CC | Present in CC30 CIG and Absent in MRSA252 or THC60 | Absent in >50% of CC30 and Present in >90% of Other CC | Absent in >90% of CC30 and Present in >90% of Other CC | t Test CC30 vs Others |
|---|---|---|---|---|---|---|---|---|---|
| CIG1176_2319 | SAR1587+b | PfkB Carbohydrate Kinase Family Protein | 1.0 | X | 1.97E–30 | ||||
| SAR0638 | SAR0638+ | Hypothetical (Putative Membrane Protein) | 1.0 | X | 1.97E–30 | ||||
| CIGC341D_1278 | SAR0717 | Bacterial Regulatory Helix-Turn-Helix, LysR Family Protein | 0.9 | X | 1.38E–29 | ||||
| CIG1500_1282 | SAR0721 | Multicopper Oxidase mco | 0.9 | X | 1.38E–29 | ||||
| CIG1267_1237 | SAR0715 | Glyoxalase | 0.9 | X | 1.38E–29 | ||||
| CIG1267_1239 | SAR0718 | Conserved Hypothetical 698 Family Protein | 0.9 | X | 1.38E–29 | ||||
| CIG1214_1043 | Ac | Pemk-Like Family Protein | 0.9 | X | 1.38E–29 | ||||
| CIG1214_2795 | A | Phosphoribosylformylglycinamidine Synthase Subunit PurS | 0.9 | X | 1.38E–29 | ||||
| CIG1233_343 | SAR2591+ | LysE Type Translocator Family Protein | 1.0 | X | 1.09E–22 | ||||
| SAR2592 | SAR2592+ | Fatty Acid Desaturase | 1.0 | X | 1.09E–22 | ||||
| HMPREF0772_10409 | SAR0091+ | Insertion Sequence Protein | 1.0 | X | 1.09E–22 | ||||
| CIG1500_1207 | A | Putative Inorganic Diphosphatase | 0.8 | X | 4.42E–21 | ||||
| CIGC341D_1257 | A | Putative Membrane Protein | 0.8 | X | 4.42E–21 | ||||
| CIG1267_2996 | A | Putative Membrane Protein | 0.8 | X | 4.42E–21 | ||||
| CIG1605_2630 | A | FRG Domain Protein | 0.8 | X | 4.42E–21 | ||||
| CIG149_1264 | A | HAD Hydrolase, IA, Variant 1 Family Protein | 0.8 | X | 4.42E–21 | ||||
| CIG1500_1272 | A | HTH Domain Protein | 0.8 | X | 4.42E–21 | ||||
| CIG1267_646 | SAR0087+ | 5-Methylcytosine-Specific Restriction Enzyme B Domain Protein | 1.0 | X | 1.70E–18 | ||||
| CIG1500_647 | SAR0088 | McrBC 5-Methylcytosine Restriction System Component Family Protein | 1.0 | X | 1.70E–18 | ||||
| SATW20_26470 | A | ABC Transporter ATP-Binding Protein | 0.0 | X | 1.70E–18 | ||||
| SAUSA300_0078 | SAR0720 | ATPase Copper Transport | 0.9 | X | 4.96E–17 | ||||
| CIG149_961 | SAR0368 | DNA Binding , Excisionase Family Domain Protein | 0.9 | X | 4.96E–17 | ||||
| SAB0348 | SAR0372 | Pathogenicity Island Protein | 1.8 | X | 5.58E–17 | ||||
| CIG1769_494 | A | ABC-2 Transporter Family Protein | 0.0 | X | 1.01E–15 | ||||
| CIGC348_980 | A | Beta-Grasp Domain Toxin Protein | 0.0 | X | 1.01E–15 | ||||
| SAA6008_02763 | SAR2788+ | Hypothetical (Putative Exported Protein) | 1.0 | X | 1.01E–15 | ||||
| CIG1176_934 | SAR2149+ | Hypothetical (Putative Exported Protein) | 1.0 | X | 1.01E–15 | ||||
| CIG149_981 | SAR0383 | Abi-Like Family Protein | 1.9 | X | 9.77E–15 | ||||
| CIG1176_1377 | SAR0704 | Bacteriophage CI Repressor Helix-Turn-Helix Domain Protein | 0.9 | X | 2.81E–14 | ||||
| CIG1524_2315 | SAR0697 | Bacteriocin Export ABC Transporter | 0.9 | X | 2.81E–14 | ||||
| ECTR2_1030 | A | Haemolytic Family Protein | 0.0 | X | 1.05E–13 | ||||
| CIG1165_915 | SAR0395 | Putative Membrane Protein | 0.4 | X | 8.92E–13 | ||||
| CIG1605_357 | A | Transposase | 1.3 | X | 8.92E–13 | ||||
| CIG149_1454 | SAR0838+ | Hypothetical (Putative Membrane Protein) | 1.0 | X | 1.05E–13 | ||||
| SAA6008_02764 | SAR2789+ | Subtilase Family Protease | 0.9 | X | 2.60E–12 | ||||
| CIG1605_650 | A | Acetyltransferase | 0.9 | X | 2.60E–12 | ||||
| CIG1605_1279 | SAR0723 | Copper-Translocating P-Type ATPase | 0.9 | X | 2.60E–12 | ||||
| CIG290_643 | A | Arylamine N-Acetyltransferase Family Protein | 0.9 | X | 2.60E–12 | ||||
| SAMSHR1132_23190 | A | HTH-Type Transcriptional Regulator SarU | 0.0 | X | 3.72E–12 | ||||
| CIG1605_203 | SAR2451+ | Bacterial Regulatory S, TetR Family Protein | 1.0 | X | 3.72E–12 | ||||
| HMPREF0772_10825 | SAR2453+ | Multidrug ABC Superfamily ATP Binding Cassette Transporter Permease Protein | 1.0 | X | 3.72E–12 | ||||
| CIG1214_1283 | SAR0696 | Conserved Hypothetical Family Protein | 0.8 | X | 2.96E–11 | ||||
| CIG290_224 | SAR2452+ | ABC Transporter Family Protein | 1.0 | X | 6.43E–11 | ||||
| SAEMRSA15_23960 | A | Putative Staphylococcal Accessory Regulator | 0.0 | X | 6.43E–11 | ||||
| SAR0097 | SAR0097+ | DNA-Binding Protein | 1.0 | X | 6.43E–11 | ||||
| SATW20_01030 | SAR0093+ | Hypothetical (Putative Membrane Protein) | 1.0 | X | 6.43E–11 | ||||
| CIG1233_404 | A | Putative Membrane Protein | 0.7 | X | 1.68E–10 | ||||
| M013TW_0379 | SAR2299 | Putative Transcriptional Regulator | 1.0 | X | 5.52E–10 | ||||
| CIG1176_846 | SAR0287 | Putative Transposase | 1.0 | X | 6.68E–10 | ||||
| CIG1165_437 | SAR2683 | Putative Membrane Protein | 0.2 | X | 9.32E–10 | ||||
| CIG1612_299 | A | Prevent-Host-Death Family Protein | 0.1 | X | 1.22E–09 | ||||
| SAB0782 | A | Leukocidin Chain lukM Precursor | 0.0 | X | 4.03E–09 | ||||
| CIG290_888 | A | Virulence Factor EsxB Family Protein | 0.0 | X | 4.76E–09 | ||||
| HMPREF0772_10238 | SAR0261+ | Nitric Oxide Reductase | 1.0 | X | 4.76E–09 | ||||
| CIG1267_2788 | A | Endodeoxyribonuclease RusA Family Protein | 0.6 | X | 6.03E–09 | ||||
| SAR0724 | SAR0724 | Cadmium Efflux System Accessory Protein | 0.9 | X | 1.11E–08 | ||||
| CIG1233_2758 | A | Helix-Turn-Helix Domain Protein | 0.8 | X | 1.35E–08 | ||||
| CIG149_1032 | A | Toxic Shock Syndrome Toxin-1 | 0.8 | X | 1.35E–08 | ||||
| CIG1214_1405 | SAR0824 | Malic Enzyme, NAD Binding Domain Protein | 1.0 | X | 2.54E–08 | ||||
| CIG1176_970 | A | Helix-Turn-Helix Family Protein | 0.9 | X | 7.02E–08 | ||||
| CIGC128_313 | A | Surface Protein G | 0.0 | X | X | 3.42E–07 | |||
| SAT0131_00223 | SAR0228+ | Glutamine Amidotransferase Class-I | 1.0 | X | 3.42E–07 | ||||
| CIG1176_657 | SAR0098 | Acetyltransferase Family Protein | 1.0 | X | 3.84E–07 | ||||
| CIG1267_946 | A | Phage Integrase Family Protein | 0.8 | X | 6.68E–07 | ||||
| CIG1214_965 | A | DNA Binding, Excisionase Family Domain Protein | 0.8 | X | 6.68E–07 | ||||
| CIG1835_198 | SAR2774 | Collagen Adhesin | 1.0 | X | 1.18E–06 | ||||
| CIGC341D_1691 | A | Seryl-tRNA Synthetase | 0.5 | X | 1.59E–06 | ||||
| SAA6008_00068 | SAR0092+ | Amidohydrolase | 1.0 | X | 2.23E–06 | ||||
| CIG1057_526 | A | Putative Membrane Protein | 0.0 | X | 3.19E–06 | ||||
| CIG1233_2421 | SAR1826 | ATP-Binding Protein | 0.9 | X | 4.11E–06 | ||||
| SAOV_2044 | A | Drug/Metabolite Transporter Permease | 0.0 | X | 7.12E–06 | ||||
| CIG290_550 | SAR2748 | Intracellular Adhesion Protein D | 0.2 | X | 1.03E–05 | ||||
| CIG1057_701 | A | Bacterial Transferase Hexapeptide Family Protein | 0.0 | X | X | 3.36E–05 | |||
| CIG1769_827 | A | O-Antigen Ligase Family Protein | 0.0 | X | X | 3.36E–05 | |||
| ECTR2_116 | A | Capsular Polysaccharide Synthesis Protein Cap5K | 0.0 | X | X | 3.36E–05 | |||
| SARLGA251_01310 | A | Capsular Polysaccharide Synthesis Enzyme | 0.0 | X | X | 3.36E–05 | |||
| CIGC348_1890 | A | MAP Domain Protein | 0.0 | X | 7.11E–05 |
Abbreviations: CIG, Complicated Infection Group; ID, identification.
a Average number of proteins in cluster was determined by X/11, where X = total number of proteins in clusters identified in all CC30 included in the comparison and 11 = total number of CC30 isolates included in the comparison (Supplementary Table 2).
b +, within genomic islets of MRSA252.
c A, not in MRSA252.
Single-Nucleotide Polymorphisms in Clonal Complex 30 Complicated Infection Group Isolates
The CIG genomes were compared with the contemporary CC30 reference strain, MRSA252, and the N315 (CC5) genomes (described in Materials and Methods) to identify specific SNPs potentially associated with unique phenotypic characteristics of the CC30 lineage. When compared with MRSA252, nonsynonymous SNPs were identified among 131 unique open reading frames (ORFs) in the 9 CC30 CIG isolates (Supplementary Table 2). Stop codons were identified in 5 unique ORFs, and 42 SNPs were identified in intergenic regions of the CC30 CIG genomes. Thirty-five synonymous and nonsynonymous SNPs were present in all CIG CC30 isolates when compared with MRSA252. Similar to the previous analysis of the historic and contemporary CC30 lineage [17–20, 29], SNPs were identified in the following: (1) agrC (G→A) that predicts a Gly-to-Arg substitution at residue 55 (agrCG55R); (2) hla, where a premature stop codon is introduced; and (3) PSMα3, which results in lower cytolytic and chemotactic activity. The ratio of nonsynonymous to synonymous SNPs (90:64 from a total of 154 SNPs) (Supplementary Table 2) across all CC30 isolates relative to MRSA252 through the entire genome suggests continuing evolutionary divergence or adaptation in this lineage. Further details of SNPs identified in the CC30 CIG genomes are listed in Supplementary Table 2.
Transcriptomic Analysis of the Complicated Infection Group Isolates
To test the hypothesis that transcriptome level differences in the CIG isolates were associated with differences in virulence and fitness, we conducted RNA-Seq whole transcriptome analysis of the 29 CIG isolates, MRSA252, N315, and UAMS-1 (a CC30 osteomyelitis isolate and a commonly used laboratory strain [22]). Our analysis included the following: (1) identification of strain-specific transcriptional differences within and between CCs (Figures 3 and 4) and (2) MDS (Figure 5) of all CIG isolates to evaluate overall transcriptional differences between the CCs. The CC30 CIG clinical isolates and MRSA252 share a common transcriptome, whereas the CC1, CC5, CC8, CC15, and CC45 randomly cluster into 3 distinct transcriptome groups. The transcriptome of UAMS-1 is distinct from the CC30 CIG strains and MRSA252. Genes with higher expression levels in CC30 relative to all other CC examined in our study include protein A (spa), multiple putative membrane proteins (SAR2274, SAR2275), and multiple putative exported proteins (SAR2016, SAR0437, and SAR0694). Genes with lower expression levels in CC30 relative to all other CC include the following (Figure 4): (1) several encoding metabolic functions, such as multiple members of the pyrimidine biosynthesis pathway [Carbamoyl-phosphate synthase large chain(carB), Dihydroorotase (pyrC), Bifunctional protein(pyrR), Orotate phosphoribosyltransferase(pyrE), and Orotidine 5′-phosphate decarboxylase(pyrF)]; (2) 3 genes [ABC transporter extracellular binding protein(SAR0641), ABC transporter permease protein(SAR0642), and ABC transporter ATP-binding protein(SAR0643)] encoding an operon similar to a Staphylococcus epidermidis iron repressible ABC transport system; and (3) an azoreductase (acpD). Comparison within the CC30 isolates identified 38 genes (excluding ribosomal proteins and transposases) that are differentially expressed within this CC (data not shown). These are primarily metabolic genes, including genes that participate in lactate metabolism (l-lactate dehydrogenase [ldh1] and l-lactate permease [lldP2]) and riboflavin biosynthesis (riboflavin biosynthesis protein [ribA], bifunctional riboflavin biosynthesis protein [ribD], riboflavin synthase alpha chain [ribE], and 6,7-dimethyl-8 ribityllumazine synthase [ribH]).
Figure 3.

Heat map of RNA-Seq transcriptome analysis for 1259 selected genes from the Staphylococcus aureus Complicated Infection Group (CIG) and reference strains. Total RNA was extracted from log-phase cultures (optical density, 600 ∼ 0.6), sequenced, and analyzed as described in Materials and Methods. Genes that were identified as significantly different between the clonal complexes (CCs) had fragments per kilobase per million reads mapped (FPKM) values of at least 1 and a greater than log2-fold increase or decrease in expression level. The heat map shows log10 FPKM values for 1259 selected genes (rows) and 32 samples (columns). Color corresponds to per-gene z-score that is computed from log10 FPKM (after adding 0.01). Genes were omitted from the original list of 2807 if the per-gene variance was less than 0.05 (low-variability genes) or if the FPKM expression was less than 10 for all 32 samples (uniformly, low-expressed genes). Based on these criteria, approximately 45% of the genes are shown. Genes and samples were hierarchically clustered based on Euclidean distance of z-score data and average linkage (dendrogram not shown for genes). Tick marks to the left of the heat map indicate genes in Figure 4. The figure on the right shows data expanded for 13 genes in cluster near top of heat map indicate FPKM expression of genes in CC30 relative to other CCs.
Figure 4.

Heat map of RNA-Seq transcriptome analysis for 52 selected genes from the Staphylococcus aureus Complicated Infection Group (CIG). The heat map shows log10 fragments per kilobase per million reads mapped (FPKM) values for 52 selected genes indicated by tick marks in Figure 3 (rows) and 32 samples (columns). Color corresponds to per-gene z-score that is computed from log10 FPKM (after adding 0.01). Genes and samples were hierarchically clustered based on Jensen-Shannon divergence distance using non-log FPKM data (after adding 1) and average linkage (dendrogram not shown for genes). Abbreviation: CC, clonal complex.
Figure 5.

Multidimensional scaling of transcriptome data for the Staphylococcus aureus Complicated Infection Group (CIG) isolates and reference strains. Multidimensional scaling was performed using a Jensen-Shannon divergence sample-sample distance matrix using fragments per kilobase per million reads mapped (+1) values for 2807 available genes. The first 2 coordinates are shown. Circles are CIG isolates from complicated infections and triangles are healthy controls (nasal carriage isolates). Clonal complex is indicated by color (see key). Reference S aureus strains (UAMS-1, MRSA252, and N315) are black asterisks.
DISCUSSION
Previous studies have shown that the S aureus CC30 lineage is associated with an increased risk for hematogenous complicated infections, including endocarditis, septic arthritis, vertebral osteomyelitis [8, 14, 15], and persistent bacteremia [30]. The current study sought to evaluate this association by genotype and severity of infections using in vivo infection models, comparative genome sequencing, and transcriptome analysis.
One intriguing finding from this study is the diminished virulence phenotype of the CC30 isolates in the animal sepsis models. This outcome is in variance with our previous results showing an association of CC30 with severe human infections [8], but it is in agreement with a recent report from DeLeo et al [17] where contemporary CC30 isolates displayed attenuated virulence in mouse sepsis model. The underlying basis for these differences can be attributed to either the host background or virulence potential of the CC30 isolates. Infections caused by CC30 occurred in a hospital setting, with individuals having existing risk factors for infection. The association of CC30 with bloodstream infections and persistent bacteremia suggests that they are adapted to long-term colonization and persistence, resulting in a high colonization burden in the human host [8, 31]. This is supported by recent data from Cheung et al [29], who demonstrates that the SNP in CC30 PSMα3 contributes to an attenuated proinflammatory potential in this lineage. The variance can also be attributed use of the mouse as a surrogate for human infections. The cardiovascular physiology of mice is distinct from humans, which compromises their use as models to assess pathogenesis and mechanisms of immune protection against S aureus infection. Future studies will use a rabbit model, whose immune and cardiovascular systems are more similar to humans [32].
The JOC analysis demonstrated that the CC30 isolates possess a distinct set of core proteins, with 25 proteins uniquely associated with CC30 isolates. Examples include a nitric oxide reductase (Nor) and fatty acid desaturase (Fad), which likely contribute to protection against production of radical nitric oxide (NO) by activated phagocytes and changes in membrane fluidity, respectively. CC30 Nor is a homolog of Neisseria meningitides MC58 norB, which is essential for persistence in the nasopharynx and evasion of macrophage response [33]. All lineages of S aureus are capable of metabolically adjusting to nitrosative stress by expressing an NO-inducible l-lactate dehydrogenase (ldh1) and a NO-detoxifying flavohemoglobin (hmp) and do not require Nor for this protective response [34]. However, recent work by Lewis et al [35] on CC30 Nor suggests that it contributes to NO-dependent respiration during nitrosative stress, potentially enhancing protection from NO in the CC30 lineage. The S aureus Fad is a homolog of a fatty acid desaturase from Bacillus subtilis, Des, which is involved in membrane adaptation during cold shock, decreased daptomycin susceptibility, and contributes to long-term survival in vitro [36]. In a similar manner, the S aureus Fad may play a role in maintaining membrane functions and contribute to the development of persistent infections. It is noteworthy that homologs of Fad are found in isolates of S epidermidis and Staphylococcus capitis, suggesting that fad was acquired through lateral gene transfer from these commensal staphylococcal species associated with endocarditis and bloodstream infections [37, 38]. Functional characterization of CC30 strains with targeted deletions of fad is needed to determine its role in multiple staphylococcal species.
The potential association between the transcriptome of CIG CC30 isolates to virulence and fitness were assessed by RNA-Seq transcriptome analysis of the 29 CIG isolates and reference strains MRSA252, N315, and UAMS-1. The CC30 isolates segregated into a transcriptome cluster that is distinct from all other CCs (Figure 5), which suggests that the transcriptome is a distinguishing feature of the CC30 that contributes to the attenuated virulence and persistence of this lineage. It is noteworthy that UAMS-1, a commonly used laboratory strain of CC30, has transcriptional features distinct from the CIG CC30 clinical isolates. A significant feature of the CC30 transcriptome is increased expression of protein A, a consequence of agrCG55R and defective Agr global regulatory system. Binding of protein A to different subclasses of immunoglobulins inhibits the B-cell responses during infection, interferes with the development of protective immunity [39–41], and leads to the overall immune suppression against staphylococcal infections [42]. A diminished immune response in healthcare-associated S aureus infections may contribute to persistence and survival of CC30, leading to chronic complicated infections [17].
Down-regulation of genes in the pyrimidine biosynthetic pathway (carB, pyrR, pyrE, and pyrF) (Figure 4) and subsequent decreases in levels of thymidine in the CC30 lineage are suggestive of thymidine-dependent small colony variants (SCVs) [43]. Other aspects of CC30 that are similar to thymidine-dependent SCVs include an Agr negative phenotype and increased levels of protein A. Thymidine-dependent SCVs also have decreased levels of hla transcription, whereas the hla in CC30 has a stop codon at amino acid (Glutamine 113: Q113Stop), making it nonfunctional. Our analysis suggests that the CC30 lineage shares some of the key metabolic features of SCVs and may be able to more quickly transition to a SCV-like bacterium.
CONCLUSIONS
In summary, we have previously demonstrated that the CC30 lineage is distinguished by its ability to persist in the human host where it can initiate complicated infections such as infectious endocarditis and osteomyelitis. The inability of CC30 to cause sepsis-induced mortality in animal models suggests that this lineage has acquired genetic features including increased expression of protein A, which, in concert with the accumulation of SNPs in several virulence genes and regulators (eg, agrC, hla, psmα3), may be of strategic significance for CC30 isolates to better persist in and colonize the human host by escaping the immune response and attenuating its virulence until the conditions become favorable for bacteremia and subsequent hematogenous seeding. In addition, genes unique to CC30 such as nor and fad and decreased expression of metabolic genes responsible for production of SCVs may contribute to immune evasion and long-term survival of these strains in the endocardial and osteoarticular host environments. Further analysis, including transcriptome profiling using in vivo models and functional analysis of the genes unique to CC30, is needed to validate the molecular mechanisms responsible for the persistent phenotype.
Supplementary Material
Supplementary material is available online at Open Forum Infectious Diseases (http://OpenForumInfectiousDiseases.oxfordjournals.org/).
Acknowledgments
We thank Ann L. Gill for technical and experimental support. RNA-Seq and transcriptome analysis of the Staphylococcus aureus strains in this study were completed by the University of Rochester Genomics Research Center.
Financial support. This work was supported by The National Institute of Allergy and Infectious Diseases at the National Institutes of Health (grant R01AI095111; to S. R. G.). V. G. F. was supported by a National Institute of Allergy and Infectious Diseases at the National Institutes of Health Mid-Career Mentoring Award K24-AI093969. Sequencing and annotation of the S aureus Complicated Infection Group strains in this study were supported by The National Institute of Allergy and Infectious Diseases at the National Institutes of Health (HHSN272200900007C to the Institute for Genome Sciences).
Potential conflicts of interest. V. G. F. served as Chair of V710 Scientific Advisory Committee (Merck), has received grant support from Cerexa, Pfizer, Advanced Liquid Logic, MedImmune, has been a paid consultant for Merck, Astellas, Affinium, Bayer, Theravance, Cubist, Cerexa, Durata, Pfizer, NovaDigm, Novartis, Medicines Company, Biosynexus, MedImmune, and Inimex, and has received honoraria from Merck, Astellas, Cubist, Pfizer, Theravance, and Novartis.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Chambers HF, Deleo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 2009; 7:629–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kallen AJ, Mu Y, Bulens S et al. Health care-associated invasive MRSA infections, 2005–2008. JAMA 2010; 304:641–8. [DOI] [PubMed] [Google Scholar]
- 3.Klevens RM, Morrison MA, Nadle J et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007; 298:1763–71. [DOI] [PubMed] [Google Scholar]
- 4.David MZ, Daum RS. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev 2010; 23:616–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerrish RS, Gill AL, Fowler VG, Gill SR. Development of pooled suppression subtractive hybridization to analyze the pangenome of Staphylococcus aureus. J Microbiol Methods 2010; 81:56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhattacharya D, Carleton H, Tsai CJ et al. Differences in clinical and molecular characteristics of skin and soft tissue methicillin-resistant Staphylococcus aureus isolates between two hospitals in Northern California. J Clin Microbiol 2007; 45:1798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell SJ, Deshmukh HS, Nelson CL et al. Genotypic characteristics of Staphylococcus aureus isolates from a multinational trial of complicated skin and skin structure infections. J Clin Microbiol 2008; 46:678–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fowler VG Jr, Nelson CL, McIntyre LM et al. Potential associations between hematogenous complications and bacterial genotype in Staphylococcus aureus infection. J Infect Dis 2007; 196:738–47. [DOI] [PubMed] [Google Scholar]
- 9.Herron-Olson L, Fitzgerald JR, Musser JM, Kapur V. Molecular correlates of host specialization in Staphylococcus aureus. PLoS One 2007; 2:e1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Highlander SK, Hulten KG, Qin X et al. Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol 2007; 7:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindsay JA, Moore CE, Day NP et al. Microarrays reveal that each of the ten dominant lineages of Staphylococcus aureus has a unique combination of surface-associated and regulatory genes. J Bacteriol 2006; 188:669–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Belkum A, Melles DC, Snijders SV et al. Clonal distribution and differential occurrence of the enterotoxin gene cluster, egc, in carriage- versus bacteremia-associated isolates of Staphylococcus aureus. J Clin Microbiol 2006; 44:1555–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wertheim HF, van Leeuwen WB, Snijders S et al. Associations between Staphylococcus aureus genotype, infection, and in-hospital mortality: a nested case-control study. J Infect Dis 2005; 192:1196–200. [DOI] [PubMed] [Google Scholar]
- 14.Gill SR, McIntyre LM, Nelson CL et al. Potential associations between severity of infection and the presence of virulence-associated genes in clinical strains of Staphylococcus aureus. PLoS One 2011; 6:e18673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nienaber JJ, Sharma Kuinkel BK, Clarke-Pearson M et al. Methicillin-susceptible Staphylococcus aureus endocarditis isolates are associated with clonal complex 30 genotype and a distinct repertoire of enterotoxins and adhesins. J Infect Dis 2011; 204:704–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiltshire MD, Foster SJ. Identification and analysis of Staphylococcus aureus components expressed by a model system of growth in serum. Infect Immun 2001; 69:5198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeLeo FR, Kennedy AD, Chen L et al. Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus. Proc Natl Acad Sci U S A 2011; 108:18091–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McAdam PR, Templeton KE, Edwards GF et al. Molecular tracing of the emergence, adaptation, and transmission of hospital-associated methicillin-resistant Staphylococcus aureus. Proc Natl Acad Sci U S A 2012; 109:9107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGavin MJ, Arsic B, Nickerson NN. Evolutionary blueprint for host- and niche-adaptation in Staphylococcus aureus clonal complex CC30. Front Cell Infect Microbiol 2012; 2:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holden MT, Feil EJ, Lindsay JA et al. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci U S A 2004; 101:9786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuroda M, Ohta T, Uchiyama I et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 2001; 357:1225–40. [DOI] [PubMed] [Google Scholar]
- 22.Gillaspy AF, Hickmon SG, Skinner RA et al. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun 1995; 63:3373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuchs BB, O'Brien E, Khoury JB, Mylonakis E. Methods for using Galleria mellonella as a model host to study fungal pathogenesis. Virulence 2010; 1:475–82. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Lara J, Needham AJ, Foster SJ. Invertebrates as animal models for Staphylococcus aureus pathogenesis: a window into host-pathogen interaction. FEMS Immunol Med Microbiol 2005; 43:311–23. [DOI] [PubMed] [Google Scholar]
- 25.Lasa I, Toledo-Arana A, Dobin A et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc Natl Acad Sci U S A 2011; 108:20172–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.David M, Dzamba M, Lister D et al. SHRiMP2: sensitive yet practical SHort Read Mapping. Bioinformatics 2011; 27:1011–2. [DOI] [PubMed] [Google Scholar]
- 27.Trapnell C, Roberts A, Goff L et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012; 7:562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 1985; 22:160–74. [DOI] [PubMed] [Google Scholar]
- 29.Cheung GY, Kretschmer D, Duong AC et al. Production of an attenuated phenol-soluble modulin variant unique to the MRSA clonal complex 30 increases severity of bloodstream infection. PLoS Pathog 2014; 10:e1004298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiong YQ, Fowler VG, Yeaman MR et al. Phenotypic and genotypic characteristics of persistent methicillin-resistant Staphylococcus aureus bacteremia in vitro and in an experimental endocarditis model. J Infect Dis 2009; 199:201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cox RA, Conquest C, Mallaghan C, Marples RR. A major outbreak of methicillin-resistant Staphylococcus aureus caused by a new phage-type (EMRSA-16). J Hosp Infect 1995; 29:87–106. [DOI] [PubMed] [Google Scholar]
- 32.Salgado-Pabon W, Schlievert PM. Models matter: the search for an effective Staphylococcus aureus vaccine. Nat Rev Microbiol 2014; 12:585–91. [DOI] [PubMed] [Google Scholar]
- 33.Stevanin TM, Moir JW, Read RC. Nitric oxide detoxification systems enhance survival of Neisseria meningitidis in human macrophages and in nasopharyngeal mucosa. Infect Immun 2005; 73:3322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richardson AR, Libby SJ, Fang FC. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 2008; 319:1672–6. [DOI] [PubMed] [Google Scholar]
- 35.Lewis AM, Rice KC. Quantitative real-time PCR (qPCR) workflow for analyzing Staphylococcus aureus gene expression [Epub ahead of print]. Methods Mol Biol 2015. PMID: 25646613. [DOI] [PubMed] [Google Scholar]
- 36.Aguilar PS, Lopez P, de Mendoza D. Transcriptional control of the low-temperature-inducible des gene, encoding the delta5 desaturase of Bacillus subtilis. J Bacteriol 1999; 181:7028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui B, Smooker PM, Rouch DA et al. Differences between two clinical Staphylococcus capitis subspecies as revealed by biofilm, antibiotic resistance, and pulsed-field gel electrophoresis profiling. J Clin Microbiol 2013; 51:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monk AB, Boundy S, Chu VH et al. Analysis of the genotype and virulence of Staphylococcus epidermidis isolates from patients with infective endocarditis. Infect Immun 2008; 76:5127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Falugi F, Kim HK, Missiakas DM, Schneewind O. Role of protein A in the evasion of host adaptive immune responses by Staphylococcus aureus. MBio 2013; 4:e00575–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graille M, Stura EA, Corper AL et al. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc Natl Acad Sci U S A 2000; 97:5399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romagnani S, Giudizi MG, del Prete G et al. Demonstration on protein A of two distinct immunoglobulin-binding sites and their role in the mitogenic activity of Staphylococcus aureus Cowan I on human B cells. J Immunol 1982; 129:596–602. [PubMed] [Google Scholar]
- 42.Kobayashi SD, DeLeo FR. Staphylococcus aureus protein A promotes immune suppression. MBio 2013; 4:e00764–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Proctor RA, von Eiff C, Kahl BC et al. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol 2006; 4:295–305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.