

Abstract
Proinsulin misfolding in the endoplasmic reticulum (ER) initiates a cell death response, although the mechanism(s) remains unknown. To provide insight into how protein misfolding may cause β-cell failure, we analyzed mice with the deletion of P58IPK/DnajC3, an ER luminal co-chaperone. P58IPK−/− mice become diabetic as a result of decreased β-cell function and mass accompanied by induction of oxidative stress and cell death. Treatment with a chemical chaperone, as well as deletion of Chop, improved β-cell function and ameliorated the diabetic phenotype in P58IPK−/− mice, suggesting P58IPK deletion causes β-cell death through ER stress. Significantly, a diet of chow supplemented with antioxidant dramatically and rapidly restored β-cell function in P58IPK−/− mice and corrected abnormal localization of MafA, a critical transcription factor for β-cell function. Antioxidant feeding also preserved β-cell function in Akita mice that express mutant misfolded proinsulin. Therefore defective protein folding in the β-cell causes oxidative stress as an essential proximal signal required for apoptosis in response to ER stress. Remarkably, these findings demonstrate that antioxidant feeding restores cell function upon deletion of an ER molecular chaperone. Therefore antioxidant or chemical chaperone treatment may be a promising therapeutic approach for type 2 diabetes.
Introduction
Type 2 diabetes (T2D) is a disease epidemic characterized by hyperglycemia in the context of insulin resistance. During the pathogenesis of T2D, insulin resistance pressures pancreatic β-cells to compensate by increasing insulin production. Unfortunately, one-third of insulin-resistant individuals develop β-cell failure and lose β-cell mass, therefore requiring insulin replacement therapy. Although extensively studied, the mechanisms leading to β-cell failure are poorly understood; however, several factors identified include genetic predisposition, hyperglycemia, hyperlipidemia, and inflammatory cytokines (1). Recent studies suggest that increased proinsulin synthesis overwhelms the capacity of the endoplasmic reticulum (ER) to properly fold, process, and secrete insulin (1,2). Under these conditions, termed ER stress, cells activate the adaptive unfolded protein response (UPR) to resolve the protein-folding defect. Increased expression of UPR genes is observed in islets from humans with T2D (3,4); therefore it was proposed that the UPR is activated in β-cells in response to insulin resistance as part of a compensatory mechanism to increase insulin production (5,6).
The UPR signals through three ER-localized transmembrane sensors, the double stranded RNA–activated protein kinase–like ER kinase (PERK), the inositol-requiring protein 1α, and the activating transcription factor 6α (7,8). When misfolded proteins accumulate, PERK phosphorylates the α-subunit of eukaryotic translation initiation factor 2 (eIF2α) to attenuate mRNA translation and reduce the protein-folding load. Paradoxically, eIF2α phosphorylation is required for translation of selective mRNAs, including activating transcription factor 4. However, if the UPR adaptive response is not sufficient to resolve the protein-folding defect, cells initiate apoptotic cell death, in part through induction of the proapoptotic gene Chop/Gadd153 (9).
Previous studies demonstrated a requirement for UPR signaling in β-cell function. Deletion of Xbp1 in mice or Perk in humans and mice causes β-cell failure (10–13). Notably, mice with a β-cell-specific Ser51 to Ala mutation at the PERK phosphorylation site in eIF2α also develop β-cell failure, similar to Perk deletion (14). The Ser51Ala eIF2α mutation releases the brake on mRNA translation, thereby increasing proinsulin synthesis in β-cells to cause accumulation of misfolded proinsulin in the ER, loss of insulin secretory granules, oxidative stress, and β-cell apoptosis (15,16). Importantly, these findings provide evidence that a modest increase in proinsulin synthesis (∼30%), similar to that which may occur in the insulin-resistant state but in the absence of hyperglycemia, hyperlipidemia, and inflammatory cytokines, is sufficient to initiate all the characteristics of β-cell failure observed in T2D. Because increased protein synthesis would also challenge the protein folding machinery in the ER of the β-cell, we analyzed whether protein misfolding in the ER causes oxidative stress.
Mechanistically, the regulation of proinsulin synthesis, folding, and processing in the β-cell is largely unknown. The most abundant molecular chaperone in the ER is a DnaK family member, the peptide-dependent ATPase glucose-regulated protein (GRP)78/BiP. P58IPK/DNAJc3 is a DnaJ family member that acts as a co-chaperone for GRP78 by promoting peptide binding and stimulating the GRP78 ATPase activity (17–19). P58IPK expression is increased in islets from individuals with T2D (4). P58IPK deletion causes a mild protein-folding defect in the ER in mouse embryonic fibroblasts (17,18). P58IPK deletion does not, however, cause a significant phenotype other than eventual β-cell failure with glucose intolerance (20,21). Here we show that the protein-folding defect in P58IPK−/− mice reduces β-cell mass and function that is associated with activation of the proapoptotic UPR and increased levels of oxidative stress. In addition, either deletion of Chop or treatments with chemical chaperones or antioxidants dramatically preserve β-cell mass and function in P58IPK−/− mice. The results identify three different interventions that preserve β-cells with defective chaperone function that may have relevance to therapy for T2D in humans.
Research Design and Methods
Mice
P58IPK−/− mice were backcrossed eight times into C57BL/6J mice (The Jackson Laboratory). P58IPK−/− mice were bred with Chop−/− mice in a C57BL/6J background. RIP1-Tag2 mice (22) were obtained from the National Cancer Institute. Studies were performed with littermate male mice that were housed with 12-h light/12-h dark cycles with free access to water and standard rodent chow (Laboratory Rodent Diet 5001; LabDiet). Chow supplemented with butylated hydroxyanisole (BHA; Sigma-Aldrich) was formulated by Research Diets Inc. (0.7% BHA in a base of Laboratory Rodent Diet 5001). Male mice were housed pairwise for the diet studies. 4-Phenylbutyric acid (PBA; Calbiochem) was administered through intraperitoneal injection. All animal care and procedures were conducted according to the protocols and guidelines approved by the University of Michigan Committee on the Use and Care of Animals or the Sanford-Burnham Medical Research Institute Institutional Animal Care and Use Committee. Blood glucose, glucose tolerance test (GTT), and insulin tolerance test measurements were performed as described previously (23).
Islet Isolation and Glucose-Stimulated Insulin Secretion
Islets were isolated by intraductal collagenase P (Roche) perfusion, histopaque-1077 gradient (Sigma-Aldrich) purification, and hand picking (16). Glucose-stimulated insulin secretion (GSIS) was performed as described previously (23).
Generation of β-Cell Lines and Cell Culture
RIP1-Tag2 mice were crossed with P58IPK−/− mice to generate heterozygous F1 mice, which were intercrossed to obtain RIP1-Tag2;P58IPK−/− or RIP1-Tag2;P58IPK+/+ mice. Islet tumors were excised and subjected to collagenase digestion, yielding P58IPK+/+ and P58IPK−/− mouse insulinoma (MI) cells. Established MI cell lines and INS1 cells were maintained in RPMI 1640 medium (11 mmol/L glucose) supplemented with 10% FBS, 2 mmol/L sodium pyruvate, 50 μmol/L β-mercaptoethanol, and penicillin/streptomycin. MIN6 cells were maintained in DMEM (27.5 mmol/L glucose) supplemented with 15% FBS and penicillin/streptomycin. A replication-defective adenovirus expressing Akita mutant proinsulin was generated using established protocols (Ad-Easy system; Stratagene) and was used with adenovirus encoding β-galactosidase (β-gal) as the control. MI cells were infected with adenoviruses for 24 h at 100 multiplicity of infection.
Islet Morphology and Immunohistochemistry
Pancreata were isolated, fixed with 10% buffered formalin, and embedded in paraffin. Sections were prepared and either stained with hematoxylin and eosin for visualization of islet morphology by light microscopy or subjected to immunohistochemistry. Nitrated proteins were detected with anti-nitrotyrosine antibody (Upstate) using the Histostatin-Plus Kit (Zymed Laboratories). Anti-insulin (1:200; Linco, 4011–01) and anti-glucagon (1:200; Linco, 4030–01F) antibodies were identified with anti–guinea pig Texas red (1:1000; Jackson ImmunoResearch, 106–075–003,) and with goat anti-rabbit Alexa Fluor 488 as the secondary antibody (1:1000; Invitrogen, A11008), respectively. Anti-PDX1 (1:200; Millipore, 07–696) and anti-4-hydroxynonenal (1:200; Abcam, ab46545) antibodies were identified with antirabbit Alexa Fluor 594 as the secondary antibody (1:1000; Invitrogen, A11037). Anti-MafA (1:100; Santa Cruz Biotechnology, sc-27140) antibody was identified with anti-goat Alexa Fluor 594 as the secondary antibody (1:1000; Invitrogen, A11058). In Fig. 6F, anti-proinsulin (1:200; HyTest Ltd., 2PR8) antibody was identified with anti-mouse Alexa Fluor 594 (1:1000; Invitrogen, A11005). In Fig. 7C and Supplementary Fig. 8, anti-insulin antibodies was identified with anti–guinea pig Alexa Fluor 488 (1:1000; Invitrogen, A11073).
Figure 6.

Antioxidant treatment prevents β-cell failure in P58IPK−/− mice. A and B: Mice with the indicated genotypes at 20–22 weeks of age were fed control chow or chow supplemented with BHA for up to 39 weeks (n = 3 or 4 mice per group). A: Blood glucose was measured either ad libitum or after a 5-h short fast. B: GTTs were performed on mice with the indicated genotypes before and 2 and 40 weeks after feeding with control or BHA-supplemented chow (n = 5–7 mice per group). Significance was shown between P58IPK−/− mice fed with control or BHA-supplemented chow. C: Pancreatic insulin contents were measured in mice fed control or BHA-supplemented chow for 40 weeks (n = 5 or 6 mice per group). D: Islet mass was measured in pancreatic sections from 16- to 20-week-old P58IPK−/− mice fed control or BHA-supplemented chow for 6 weeks. The percentage indicates the total islet mass over the pancreas area in each pancreata (n = 4 mice per group). E: GSIS (left panel) and insulin contents in total islet lysates (right panel) were measured in mice with the indicated genotypes fed control chow or BHA-supplemented chow for 40 weeks. Islets from two animals per condition were analyzed in triplicate. F: Insulin and proinsulin immunohistochemistry was performed on pancreatic sections from P58IPK+/+ and P58IPK−/− mice fed control or BHA-supplemented chow for 40 weeks. Representative images are shown. Scale bars represent 30 μm. G: Transmission electron microscopy was performed on pancreatic sections from P58IPK−/− mice fed control or BHA-supplemented chow for 22 weeks. Representative images are shown. Scale bar represents 0.5 μm. *P < 0.05; **P < 0.01.
Figure 7.

Antioxidant feeding improves β-cell function in P58IPK−/− mice. A: HODEs were measured in extracts from 50 islets isolated from mice aged 7–9 weeks that were fed control chow or BHA-supplemented chow for 3 weeks (n = 5–7 mice per group). B: Immunohistochemistry for 4-HNE was performed on pancreatic sections from P58IPK−/− mice fed control chow or BHA-supplemented chow for 6 weeks. Representative images are shown. Arrows indicate nuclei. Scale bar represents 100 μm. C: Immunohistochemistry for MafA and insulin was performed on pancreatic sections from P58IPK+/+ or P58IPK−/− mice fed control or BHA-supplemented chow for 6 weeks. Representative images are shown. Scale bar represents 100 μm. All data are mean ± SEM. **P < 0.01.
Islet Mass Measurement
For islet mass measurements, the entire pancreas was serially sectioned into 5-μm samples collected at 50-μm increments. We then chose 25–40 slides spanning each pancreas for hematoxylin and eosin histochemistry. Images covering the entire tissue sample were subsequently captured using an EVOS microscope (Invitrogen). The entire pancreas tissue area and islet area in each image were measured using ImageJ software (National Institutes of Health, Bethesda, MD). Relative islet mass (percentage) was defined as the sum of the islet area measured in all the images from each mouse divided by the sum of the total pancreas tissue area measured in all the images from each mouse, times 100. This technique was applied to 1) the P58IPK−/− and P58IPK+/+ mice at 16–20 weeks of age (n = 3 mice per group) (Fig. 1); 2) P58IPK−/−Chop+/− and P58IPK−/−Chop−/− at 16 weeks of age (n = 4 mice per group) (Fig. 3); 3) P58IPK−/−mice at 16–20 weeks of age were subsequently fed with control or BHA-supplemented chow for 6 weeks (n = 4 mice per group) (Fig. 4).
Figure 1.

Deletion of P58IPK in mice causes β-cell failure. A: Blood glucose was measured in P58IPK+/+ and P58IPK−/− mice at the indicated age after a 5-h fast (n = 5 or 6 mice per group). B: A GTT was performed (n = 5 or 6 mice [6–7 weeks of age] per group). C: An insulin tolerance test was performed (n = 5 or 6 mice [9–10 weeks of age] per group). D: Serum insulin after a 5-h fast was measured in 16- to 20-week-old mice after a 5-h fast. E: Western blot analysis was performed using 100 islets isolated from 10-week-old mice. F: Pancreatic insulin content was measured and normalized to protein content (n = 5 mice [10–12 weeks of age] per group). G: Islet mass was measured in pancreatic sections from 16- to 20-week-old mice. The percentage indicates the total islet mass over the pancreas area in each pancreata (n = 3 mice per group). H: Immunohistochemistry for insulin and glucagon was performed on pancreatic sections from 10- to 12-week-old mice. Representative images are shown. Scale bar represents 100 μm. I: Transmission electron microscopy was performed on pancreatic sections from 12-week-old mice. Representative images are shown. Scale bar represents 2 μm in upper panels and 0.5 μm in lower panels. ER, rough endoplasmic reticulum; M, mitochondria. *P < 0.05; **P < 0.01.
Figure 3.

P58IPK−/− β-cells are more susceptible to ER stress. A–C: Quantitative RT-PCR was performed using total RNA extracted from P58IPK+/+ and P58IPK−/− β-cell lines treated with Tm (2 μg/mL) for 0, 8, or 24 h: pancreatic β-cell-specific genes (A); cell death genes (B); and UPR genes (C). D: Cell viability was measured in P58IPK+/+ and P58IPK−/− β-cell lines treated with vehicle (Veh) (DMSO), Tm (2 μg/mL), or thapsigargin (Tg; 300 nmol/L) for 24 h and 0.6 mmol/L H2O2 for 8 h. E: Western blot analysis was performed using P58IPK+/+ and P58IPK−/− β-cell lines treated with vehicle (DMSO), Tg (300 nmol/L), and Tm (2 μg/mL) for 24 h. F: Western blot analysis was performed using P58IPK+/+ and P58IPK−/− β-cell lines treated with Tm (2 μg/mL) for 24 h. Cell lysates were harvested at the indicated time points. G and H: P58IPK+/+ and P58IPK−/− β-cell lines were infected with adenovirus-expressing β-gal (rAd-β-gal) or Akita proinsulin (rAd-Akita) for 24 h. G: Total RNA was isolated for quantitative RT-PCR. H: Cell viability was measured. All data are mean ± SEM of three independent experiments. *P < 0.05; **P < 0.01. hr, hours.
Figure 4.

Chemical chaperone treatment preserves β-cell function in P58IPK−/− mice. P58IPK+/+ and P58IPK−/− mice aged 22–26 weeks were treated intraperitoneally with saline or PBA (500 mg/kg body weight/day) for the indicated durations. Blood glucose was measured after a 5-h fast (A) and GTTs were performed before and after treatment with PBA for 42 days (B) (n = 4 or 5 mice per group). Significant differences between P58IPK−/− mice treated with saline or PBA are shown. *P < 0.05; **P < 0.01. Significance was shown between P58IPK−/− and P58IPK−/− with PBA treatment.
Insulin and Hydroxyoctadecadienoic Acid Measurements
Pancreata were homogenized in a cold solution of acid/ethanol containing 75% ethanol and 0.18 mol/L hydrochloric acid, followed by overnight incubation at 4°C. The extracts then were centrifuged, and supernatants were diluted into sample buffer. Insulin was measured by ELISA (ALPCO). Lipid peroxidation (hydroxyoctadecadienoic acids [HODEs]) was measured as previously described (16).
Cell Viability Assays
Relative cell viabilities were measured using Cell Counting Kit-8 (Dojindo, Rockville, MD) according to the manufacturer’s instructions.
Gene Expression Studies
Total RNA was extracted from cells or islets (at least 50) using RNAeasy (Qiagen). The relative amounts of mRNAs were measured by quantitative real-time PCR with SYBR green (Bio-Rad). Real-time primer sequences are shown in Supplementary Table 1.
Western Blot Analyses
Cells were collected in cell lysis buffer (50 mmol/L Tris-HCl [pH 7.4], 150 mmol/L NaCl, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate and protease inhibitors [Roche]). Primary antibodies were as follows: anti-PARP (1:1000; Cell Signaling Technology, 9542), anti-tubulin (1:5000; Sigma-Aldrich, T5168), anti-CHOP (1:200; Santa Cruz Biotechnology, sc-575), and anti-KDEL (1:1000; Abcam, ab12223), anti-insulin (1:1000; Sigma-Aldrich, I2018), and anti-proinsulin (1:1000; HyTest Ltd., 2PR8). Anti-P58IPK antibody was obtained from one of the authors (M.G.K.). Anti-rabbit antibody conjugated with horseradish peroxidase (1:5000; Millipore, AQ132P) for CHOP and PARP, and anti-mouse antibody (1:5000; Millipore, AP192P) for tubulin, KDEL, insulin, and proinsulin, were used as secondary antibodies.
Transmission Electron Microscopy
Transmission electron microscopy was performed on pancreas tissue as previously described (16).
Statistical Analysis
Data are presented as means ± SEMs. The statistical significance of differences between groups was evaluated using the Student t test. P values <0.05 were considered statistically significant.
Results
P58IPK−/− Mice Develop Diabetes as a Result of Pancreatic β-Cell Failure Associated With Oxidative Stress
To investigate the effect of P58IPK deletion on β-cell physiology, we first analyzed blood glucose concentrations (BGCs) and body weight of P58IPK+/+ and P58IPK−/− male mice fed a regular diet (Fig. 1A and Supplementary Fig. 1A). After 10 weeks of age, the fasting BGC in P58IPK−/− mice steadily increased and became significantly greater after 21 weeks of age compared with that of P58IPK+/+ mice, consistent with earlier observations (20). Although there was no significant difference in fasting BGCs between genotypes at 10–14 weeks, the P58IPK−/− mice were glucose intolerant as early as 6–7 weeks (Fig. 1B). Interestingly, there was no significant difference in islet morphology or mass 1 day after birth, indicating that the loss of β-cell mass and/or function starts after birth and likely does not originate from a developmental defect (Supplementary Fig. 1B). There were, however, no significant differences in insulin sensitivity (Fig. 1C) or liver morphology (Supplementary Fig. 1C) between genotypes.
Hyperglycemia in the P58IPK−/− mice correlated with reduced serum (Fig. 1D) and pancreatic insulin contents (Fig. 1E and F) and decreased islet size and mass (Fig. 1G and H and Supplementary Fig. 1D). Transmission electron microscopy demonstrated β-cells from P58IPK−/− mice had a marked decrease in insulin granules and a distended ER, indicating that P58IPK−/− β-cells experience ER stress (Fig. 1I). Although the number of mitochondria was not significantly different between genotypes (Supplementary Fig. 1E), mitochondria in P58IPK−/− cells were more swollen, suggesting mitochondrial damage (Fig. 1I). The ER luminal distension and mitochondrial swelling in P58IPK−/− β-cells suggested the β-cells experience ER stress and oxidative stress (16). Indeed, a specific antibody against nitrotyrosine, a product of tyrosine nitration mediated by reactive nitrogen species, intensively stained islets from P58IPK−/− mice compared with islets from P58IPK+/+ mice (Fig. 2A). In addition, lipid peroxidation (HODE) was significantly greater in islets from P58IPK−/− mice (Fig. 2B), indicating oxidative stress.
Figure 2.
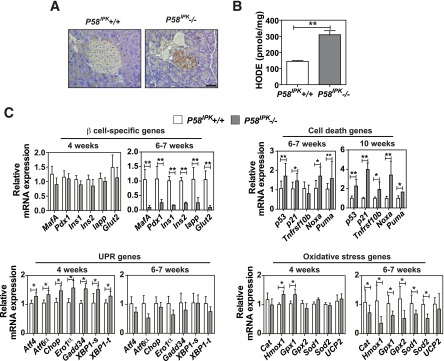
Deletion of P58IPK causes oxidative stress in β-cells. A: Immunohistochemistry for nitrotyrosine was performed on pancreatic sections from 12-week-old mice. Representative images are shown. Scale bar represents 100 μm. B: HODEs were measured in extracts from 50 islets isolated from 10- to 12-week-old mice (n = 6 mice per group). C: Quantitative RT-PCR was performed using total RNA extracted from islets from P58IPK+/+ and P58IPK−/− mice at 4, 6–7, or 10 weeks of age (n = 3–6 mice per group). All data are mean ± SEM. *P < 0.05; **P < 0.01.
To provide insight into the mechanism of β-cell failure in the absence of P58IPK, we analyzed gene expression in islets from mice at the age of 4 and 6–7 weeks. The expression of the β-cell-specific genes Pdx1 and MafA1 and their downstream transcriptional targets Ins1, Ins2, and Glut2 was comparable at 4 weeks; however, their expression was significantly reduced at 6–7 weeks in P58IPK−/− islets (Fig. 2C). Importantly, expression of Hmox1 and Gpx1 genes was increased at 4 weeks, whereas expression of Cat, Hmox1, Gpx1, Gpx2, Sod1, and Sod2 was significantly decreased in P58IPK−/− islets (Fig. 2C), suggesting oxidative stress occurs at 4 weeks in P58IPK−/− islets. It is noteworthy that the expression of cell death genes was increased at 6–7 weeks and further increased at 10 weeks of age in P58IPK−/− islets. In addition, the expression of UPR-related genes was significantly increased at 4 weeks in P58IPK−/− islets compared with P58IPK+/+ islets, indicating the presence of ER stress at this age (Fig. 2C).
P58IPK Deletion in β-Cells Increases Susceptibility to ER Stress–Induced Apoptosis
To investigate the role of the UPR in β-cell failure associated with P58IPK deletion, we generated SV40 T antigen-immortalized β-cell lines from P58IPK−/− and P58IPK+/+ mice. The expression of β-cell-specific genes in these lines was comparable to that in the well-characterized mouse insulinoma cell line MIN6 (Supplementary Fig. 2), indicating these cell lines represent a reasonable in vitro system to investigate β-cell physiology. In response to ER stress induced by tunicamycin (Tm), which inhibits N-linked glycosylation, the expression of the β-cell-specific genes MafA, Pdx1, Ins1, Ins2, and Glut2 was greatly decreased in P58IPK−/− insulinoma cells compared with P58IPK+/+ insulinoma cells, possibly suggesting dedifferentiation and/or dysfunction (24) (Fig. 3A). In addition, Tm induction of the proapoptotic genes P53, Tnfrsf10b, Tnfr, and Noxa (Fig. 3B), as well as the ER stress–induced proapoptotic genes Atf3 and Chop, was significantly greater in P58IPK−/− cells compared with P58IPK+/+ cells (Fig. 3C). Indeed, the viability of P58IPK−/− β-cells was significantly reduced compared with P58IPK+/+ cells in response to ER stress (Tm and thapsigargin) as well as oxidative stress (H2O2), with increased CHOP expression and PARP cleavage (Fig. 3D and E). Notably, CHOP expression peaked ∼8 h after Tm treatment in both cell lines, but CHOP expression was sustained in P58IPK−/− cells; this correlated with increased PARP cleavage 24 h after Tm treatment (Fig. 3F). This is consistent with a requirement for attenuated CHOP expression to survive ER stress (25).
To test whether P58IPK−/− β-cells are more susceptible to physiological conditions of ER stress, we expressed the insulin 2 Akita mutant containing a Cys96Tyr mutation in the A chain (26), which is known to misfold and cause ER stress–mediated β-cell death (27). Upon Akita mutant expression in the β-cell lines, induction of Chop and Atf3 was significantly greater in the P58IPK−/− β-cells (Fig. 3G), and this correlated with reduced viability (Fig. 3H). These results show that P58IPK−/− β-cells exhibit increased susceptibility to ER stress–mediated cell death, possibly associated with increased expression of CHOP.
Either Chemical Chaperone Treatment or Chop Deletion Prevents β-Cell Failure in P58IPK−/− Mice
To test whether ER stress causes the β-cell failure observed in P58IPK−/− mice, we treated P58IPK−/− mice with the chemical chaperone 4-PBA, which relieves ER stress (28–30). Six weeks after PBA treatment, fasting hyperglycemia (Fig. 4A) and glucose intolerance (Fig. 4B) were reduced, suggesting that the P58IPK−/− β-cell failure results from protein misfolding in the ER, consistent with a defect in chaperone function. The results support the notion that a chemical chaperone can complement the requirement for protein chaperone function to preserve β-cell function.
Previous studies demonstrated that Chop deletion protects β-cells in models of T2D and in Akita mice, which express a folding-defective mutant proinsulin (23,27). In particular, studies suggested that CHOP is required for oxidative stress that occurs in response to ER stress (23,31–33). In addition, we demonstrated that CHOP expression was significantly greater in P58IPK−/− cells compared with P58IPK+/+ cells. Therefore we investigated the requirement for CHOP in the cell death observed in P58IPK−/− β-cells. P58IPK−/− mice were crossed with Chop−/− mice to generate P58IPK−/−Chop−/− mice. At 16 weeks of age, Chop deletion reduced hyperglycemia, which was sustained up to 41 weeks of age (Fig. 5A). Significantly improved glucose tolerance was observed at 16 weeks of age, which was more prominent at 36 weeks of age (Fig. 5B). P58IPK−/−Chop−/− mice maintained postprandial BGCs within the normal range and increased insulin secretion upon fasting and refeeding (Supplementary Fig. 3A and B). The improved glucose homeostasis in P58IPK−/−Chop−/− mice was associated with increased insulin content (Fig. 5C) and islet mass (Fig. 5D and Supplementary Fig. 3C). Strikingly, Chop deletion preserved intracellular organelle integrity and insulin granule content in the P58IPK−/− β-cells (Fig. 5E). These results demonstrate that Chop deletion protects β-cells from ER stress caused by the deletion of a protein chaperone in the ER.
Figure 5.

Chop deletion preserves β-cell function in P58IPK−/− mice. A: Blood glucose was measured at the indicated times, either ad libitum or after a 5-h fast, in mice with the indicated genotypes up to 41 weeks of age (n = 7–13 mice per group). B: GTTs were performed in mice with the indicated genotypes. Glucose doses were 2 mg/kg body weight for mice 14 and 16 weeks of age and 1 mg/kg body weight for mice 36 weeks of age (n = 7–13 mice per group). Significance was shown between P58IPK−/− Chop+/− and P58IPK−/− Chop−/−. C: Pancreatic insulin was measured in mice with the indicated genotypes at 16 weeks of age (n = 4–6 mice per group). D: Islet mass was measured in pancreatic sections from 16-week-old P58IPK−/− Chop+/− or P58IPK−/− Chop−/− mice and their littermates. The percentage indicates the total islet mass over the pancreas area in each pancreata (n = 4 mice per group). E: Transmission electron microscopy was performed on pancreatic sections from P58IPK+/−Chop+/− mice and P58IPK−/−Chop−/− mice at 38–41 weeks of age. Representative images are shown. Scale bar represents 0.5 μm. All data are mean ± SEM. *P < 0.05; **P < 0.01.
Antioxidant Treatment Restores β-Cell Function in P58IPK−/− Mice and Akita Mice
The oxidative damage observed in P58IPK−/− islets (Fig. 2), as well as the previously suggested relationship between protein misfolding in the ER and reactive oxygen species (ROS) production (34), prompted us to assess the contribution of oxidative stress to β-cell failure by analyzing the effect of the antioxidants BHA, sodium 4,5-dihydroxybenzene-1,3-disulfonate (Tiron), 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPOL), and manganese [III] tetrakis (4-benzoic acid) porphyrin (MnTBAP). Among these treatments, Tiron (Supplementary Fig. 4) and MnTBAP (Supplementary Fig. 5) did not alter the abnormal BGCs or GTT results in P58IPK−/− mice, whereas treatment with TEMPOL improved the GTT results in P58IPK−/− mice after 42 days (Supplementary Fig. 6). Of all these treatments, feeding P58IPK−/− mice with a BHA-supplemented diet for as little as 1 week normalized the BGCs after ad libitum feeding and short fasts; these were maintained up to 38 weeks (Fig. 6A), with no significant difference in body weight (Supplementary Fig. 7A). Strikingly, 2 weeks after feeding of the BHA-supplemented diet, the GTT results were completely normalized in the P58IPK−/− mice compared with P58IPK+/+ mice (Fig. 6B), with no significant difference in insulin sensitivity (Supplementary Fig. 7B). The normalized GTT result was maintained with up to 40 weeks of feeding of the BHA-supplemented diet (Fig. 6B). In addition, feeding BHA-supplemented chow to P58IPK−/− mice also improved blood glucose homeostasis and insulin secretion upon fasting and subsequent refeeding (Supplementary Fig. 7C and D). The increased insulin secretion correlated with increased insulin content (Fig. 6C) and islet mass (Fig. 6D and Supplementary Fig. 7E). The improved β-cell function also was observed by GSIS assays using islets isolated from mice that were fed with or without BHA-supplemented chow (Fig. 6E). Immunofluorescence microscopy demonstrated BHA-supplemented chow increased insulin and proinsulin content in the P58IPK−/− mice (Fig. 6F). Because deletion of P58IPK was engineered with insertion of a green fluorescent protein cassette, green fluorescent protein fluorescence is observed in acinar cells, although it is not detectably expressed in the islets. In addition, feeding P58IPK−/− mice with BHA-supplemented chow preserved organelle integrity and insulin granule content (Fig. 6G). BHA feeding reduced oxidative damage in P58IPK−/− islets, as indicated by significantly reduced HODE concentrations, an indication of oxidative stress, in the islets from mice fed BHA-supplemented chow (Fig. 7A). In addition, feeding P58IPK−/− mice with BHA-supplemented chow for 6 weeks significantly reduced the nuclear localization of the lipid peroxidation product 4-hydroxynonenal (HNE) (Fig. 7B). 4-HNE was previously noted for nuclear localization under conditions of oxidative stress (35,36). The reduced nuclear localization of 4-HNE suggests decreased oxidative damage in P58IPK−/− islets fed BHA-supplemented chow. The disappearance of MafA, but not PDX1, from the nucleus was previously associated with oxidative stress–mediated β-cell dysfunction (37); therefore we analyzed the localization of MafA and PDX1 in the islets. Nuclear localization of MafA, but not PDX1, was diminished in islets from P58IPK−/− mice and was restored in islets from P58IPK−/− mice fed BHA-supplemented chow (Fig. 7C and Supplementary Fig. 8). These results demonstrate that feeding of BHA-supplemented chow not only preserves but also restores β-cell function and mass in islets of P58IPK−/− mice by reducing oxidative stress.
The dramatic effect of BHA feeding on preserving β-cell function in P58IPK−/− mice prompted us to test its potential to improve β-cell function in a more severe model of ER protein misfolding in the β-cell using the Akita mouse, which has a Cys96Tyr mutation in the insulin gene; this mutation disrupts disulfide bond formation, resulting in misfolded proinsulin and oxidative stress in β-cells (38). Indeed, BHA feeding improved glucose homeostasis in heterozygous Akita mice (Fig. 8A and B).
Figure 8.

BHA feeding improves glucose homeostasis in Akita mice. Wild-type and heterozygous Akita+ male littermate mice 10–20 weeks of age were fed control chow or BHA-supplemented chow for up to 36 weeks. A: Blood glucose concentrations were measured between 9 and 10 a.m. ad libitum (n = 3 or 4 mice per group). B: GTTs were performed before and 8 weeks after feeding control chow or BHA-supplemented chow. Glucose (1 g/kg body weight) was administered intraperitoneally to mice fasted for 5–6 h (n = 3 or 4 mice per group). C: Model depicting β-cell demise caused by ER protein misfolding, subsequent ROS production, and interventions demonstrated to preserve β-cell function. All data are mean ± SEM. **P < 0.01.
Discussion
In this study, we showed that deletion of the ER co-chaperone protein P58IPK causes β-cell failure through ER stress– and oxidative stress–mediated cell death. P58IPK−/− mice become hyperglycemic with reduced glucose tolerance as a result of decreased islet mass and β-cell insulin content and granules. At the ultrastructural level, the defect in chaperone function disrupted intracellular architecture, including distension of the ER and mitochondria. We demonstrated that Chop deletion, treatment with the chemical chaperone PBA, and antioxidant BHA feeding all improve β-cell function and mass in P58IPK−/−mice, indicating that P58IPK deletion causes β-cell failure because of protein misfolding in the ER and oxidative stress.
One of the unique features of pancreatic β-cells is their relatively low expression and the activity of several antioxidant enzymes (39). The expression of catalase and glutathione peroxidase is ∼5%, and that of superoxide dismutase is ∼30%, compared with the amount of liver (40). As a consequence, β-cells are susceptible to oxidative stress. Indeed, antioxidant treatment improved β-cell function in immortalized cell lines and isolated islets (41), as well as in rodent (37,42–44) and human (45) islets in vivo. Here we demonstrated that antioxidant feeding prevents β-cell failure and even restores ER function in β-cells that have a primary chaperone defect that would disrupt protein folding in the ER, adding further support to the notion that protein misfolding in the ER causes oxidative stress.
A fundamental question is where the ROS are generated in β-cells during the progression to T2D. Several reports have demonstrated an association between protein misfolding in the ER and ROS production. An increase in protein synthesis was sufficient to cause protein misfolding in the ER and oxidative stress (15,16,33). In addition, expression of Akita mutant proinsulin (38; see also the results reported here), high urokinase expression (46), and expression of clotting factor VIII that is prone to misfolding (31) also caused oxidative stress. Although mechanisms have been proposed, including cysteine oxidation during disulfide bond formation to transfer electrons to molecular oxygen to produce H2O2 (47), induction of NADPH oxidase (48), and mitochondrial dysfunction (49), this is a poorly studied subject (34). Our results indicate that a primary defect in the ER protein-folding machinery is sufficient to initiate ROS production, resulting in β-cell failure typical of that observed in T2D (50), which could be rescued by antioxidant treatment (Fig. 8C). In total, these findings suggest that oxidative stress and ER stress occur concomitantly and are principal factors that contribute to β-cell death during T2D.
Of all the antioxidants tested (Tiron, MnTBAP, TEMPOL, and BHA), BHA—and to a lesser extent TEMPOL—most significantly protected β-cell function. It is noteworthy that Tiron forms stable complexes with iron, suggesting the Fenton reaction may not cause β-cell toxicity. MnTBAP selectively neutralizes peroxynitrate over superoxide, suggesting peroxynitrate may not play a primary role in the β-cell failure described here. TEMPOL, which did protect P58IPK−/− islets to some extent, is a membrane-permeable superoxide scavenger. BHA most significantly improved β-cell function. BHA is a lipophilic free radical scavenger and may protect β-cells by preventing lipid peroxidation. Of all four antioxidants tested, BHA and TEMPOL are more related structurally. Further studies are required to characterize the unique ability of BHA to preserve the function of β-cells that have P58IPK deleted or that express misfolded mutant Akita proinsulin.
In conclusion, our findings show that maintenance of the protein-folding capacity of β-cells is essential to maintain β-cell function and minimize the deleterious effects of oxidative stress. Importantly, antioxidant feeding can preserve cell function in the absence of an ER protein chaperone. Therefore antioxidant or chemical chaperone treatments may provide a promising therapeutic approach for T2D.
Article Information
Acknowledgments. The authors thank Taryn Goode of the Sanford-Burnham Medical Research Institute for assistance with manuscript preparation and the members of the Kaufman laboratory for critical input. Electron, confocal, and light microscopy were performed at the University of Michigan Microscope and Image Analysis Laboratory.
Funding. This work was supported in part by National Institutes of Health grants R37-DK-042394, R01-DK-088227, and R01-HL-052173 (to R.J.K.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. Ja.H., B.S., and R.J.K. designed the research, analyzed data, and wrote the manuscript. Ja.H., B.S., J.K., V.K.K., A.P., M.W., Ju.H., S.W., S.P., and S.H.B. performed the research. M.G.K. contributed reagents. R.J.K. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-1357/-/DC1.
Ja.H. and B.S. contributed equally to this work.
References
- 1.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem 2012;81:767–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemaire K, Schuit F. Integrating insulin secretion and ER stress in pancreatic β-cells. Nat Cell Biol 2012;14:979–981 [DOI] [PubMed] [Google Scholar]
- 3.Huang CJ, Lin CY, Haataja L, et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007;56:2016–2027 [DOI] [PubMed] [Google Scholar]
- 4.Laybutt DR, Preston AM, Akerfeldt MC, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007;50:752–763 [DOI] [PubMed] [Google Scholar]
- 5.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev 2008;29:317–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eizirik DL, Cnop M. ER stress in pancreatic beta cells: the thin red line between adaptation and failure. Sci Signal 2010;3:pe7. [DOI] [PubMed] [Google Scholar]
- 7.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011;334:1081–1086 [DOI] [PubMed] [Google Scholar]
- 8.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol 2012;197:857–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011;13:184–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delépine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet 2000;25:406–409 [DOI] [PubMed] [Google Scholar]
- 11.Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell 2001;7:1153–1163 [DOI] [PubMed] [Google Scholar]
- 12.Lee AH, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci U S A 2011;108:8885–8890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab 2006;4:491–497 [DOI] [PubMed] [Google Scholar]
- 14.Scheuner D, Song B, McEwen E, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 2001;7:1165–1176 [DOI] [PubMed] [Google Scholar]
- 15.Scheuner D, Vander Mierde D, Song B, et al. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med 2005;11:757–764 [DOI] [PubMed] [Google Scholar]
- 16.Back SH, Scheuner D, Han J, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab 2009;10:13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rutkowski DT, Kang SW, Goodman AG, et al. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol Biol Cell 2007;18:3681–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrova K, Oyadomari S, Hendershot LM, Ron D. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J 2008;27:2862–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tao J, Petrova K, Ron D, Sha B. Crystal structure of P58(IPK) TPR fragment reveals the mechanism for its molecular chaperone activity in UPR. J Mol Biol 2010;397:1307–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ladiges WC, Knoblaugh SE, Morton JF, et al. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 2005;54:1074–1081 [DOI] [PubMed] [Google Scholar]
- 21.Oyadomari S, Yun C, Fisher EA, et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell 2006;126:727–739 [DOI] [PubMed] [Google Scholar]
- 22.Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 1985;315:115–122 [DOI] [PubMed] [Google Scholar]
- 23.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 2008;118:3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012;150:1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rutkowski DT, Arnold SM, Miller CN, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol 2006;4:e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 1997;46:887–894 [DOI] [PubMed] [Google Scholar]
- 27.Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 2002;109:525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A 2000;97:1796–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh OV, Pollard HB, Zeitlin PL. Chemical rescue of deltaF508-CFTR mimics genetic repair in cystic fibrosis bronchial epithelial cells. Mol Cell Proteomics 2008;7:1099–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao SS, Zimmermann EM, Chuang BM, et al. . The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 2013;144:989–1000.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malhotra JD, Miao H, Zhang K, et al. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci U S A 2008;105:18525–18530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004;18:3066–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han J, Back SH, Hur J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 2013;15:481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 2007;9:2277–2293 [DOI] [PubMed] [Google Scholar]
- 35.Chiarpotto E, Allasia C, Biasi F, et al. Down-modulation of nuclear localisation and pro-fibrogenic effect of 4-hydroxy-2,3-nonenal by thiol- and carbonyl-reagents. Biochim Biophys Acta 2002;1584:1–8 [DOI] [PubMed] [Google Scholar]
- 36.Yan J, Hales BF. Depletion of glutathione induces 4-hydroxynonenal protein adducts and hydroxyurea teratogenicity in the organogenesis stage mouse embryo. J Pharmacol Exp Ther 2006;319:613–621 [DOI] [PubMed] [Google Scholar]
- 37.Harmon JS, Bogdani M, Parazzoli SD, et al. β-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 2009;150:4855–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan Q, Tang W, Zhang X, et al. Proinsulin atypical maturation and disposal induces extensive defects in mouse Ins2+/Akita β-cells. PLoS One 2012;7:e35098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grankvist K, Marklund SL, Täljedal IB. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J 1981;199:393–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997;46:1733–1742 [DOI] [PubMed] [Google Scholar]
- 41.Lortz S, Tiedge M. Sequential inactivation of reactive oxygen species by combined overexpression of SOD isoforms and catalase in insulin-producing cells. Free Radic Biol Med 2003;34:683–688 [DOI] [PubMed] [Google Scholar]
- 42.Kaneto H, Kajimoto Y, Miyagawa J, et al. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999;48:2398–2406 [DOI] [PubMed] [Google Scholar]
- 43.Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U S A 1999;96:10857–10862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamamoto M, Yamato E, Toyoda S, et al. Transgenic expression of antioxidant protein thioredoxin in pancreatic beta cells prevents progression of type 2 diabetes mellitus. Antioxid Redox Signal 2008;10:43–49 [DOI] [PubMed] [Google Scholar]
- 45.Hussain SA. Silymarin as an adjunct to glibenclamide therapy improves long-term and postprandial glycemic control and body mass index in type 2 diabetes. J Med Food 2007;10:543–547 [DOI] [PubMed] [Google Scholar]
- 46.Nakagawa H, Umemura A, Taniguchi K, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014;26:331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 2004;164:341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol 2010;191:1113–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaufman RJ, Malhotra JD. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim Biophys Acta 2014;1843:2233–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robertson RP. Beta-cell deterioration during diabetes: what’s in the gun? Trends Endocrinol Metab 2009;20:388–393 [DOI] [PMC free article] [PubMed] [Google Scholar]