

Abstract
During pancreas development, endocrine precursors and their progeny differentiate, migrate, and cluster to form nascent islets. The transcription factor Neurogenin 3 (Neurog3) is required for islet development in mice, but its role in these dynamic morphogenetic steps has been inferred from fixed tissues. Moreover, little is known about the molecular genetic functions of NEUROG3 in human islet development. We developed methods for gene transduction by viral microinjection in the epithelium of cultured Neurog3-null mutant fetal pancreas, permitting genetic complementation in a developmentally relevant context. In addition, we developed methods for quantitative assessment of live-cell phenotypes in single developing islet cells. Delivery of wild-type NEUROG3 rescued islet differentiation, morphogenesis, and live cell deformation, whereas the patient-derived NEUROG3R107S allele partially restored indicators of islet development. NEUROG3P39X, a previously unreported patient allele, failed to restore islet differentiation or morphogenesis and was indistinguishable from negative controls, suggesting that it is a null mutation. Our systems also permitted genetic suppression analysis and revealed that targets of NEUROG3, including NEUROD1 and RFX6, can partially restore islet development in Neurog3-null mutant mouse pancreata. Thus, advances described here permitted unprecedented assessment of gene functions in regulating crucial dynamic aspects of islet development in the fetal pancreas.
Introduction
Development of solid organs is a dynamic process, but classical investigations of mammalian organogenesis have been limited largely to static analysis of fixed tissues. In the pancreas, epithelial cells form branched tubes that develop into exocrine acinar and ductal cells required for digestion. From that branching epithelium, endocrine progenitors differentiate and initiate cell shape changes culminating in delamination and outward migration into overlying mesenchyme. Subsequently, migrating endocrine cells differentiate into hormone-producing cells, including insulin+ and glucagon+ cells, that cluster to form morphologically distinct, vital structures called the islets of Langerhans, which regulate metabolism (1–3). Methods to visualize single cells developing along this endocrine lineage would advance our understanding of islet development.
Pancreatic endocrine precursor development is regulated by the basic helix-loop-helix transcription factor Neurogenin3 (Neurog3). Mice without Neurog3 form a pancreas but have no islet cells, other than a small transient population of glucagon+ cells (4), and die perinatally with severe hyperglycemia and no detectable insulin production (5). Mice ectopically expressing Neurog3 in early pancreatic progenitors divert cells toward an endocrine fate (5–7). These and other findings indicate that Neurog3 is both necessary and sufficient for inducing pancreatic endocrine differentiation and establishing islet cell fate during mouse development (8–13). Humans with mutations in NEUROG3 suffer from congenital diarrhea, caused by a deficit of intestinal endocrine cells, that is accompanied by a varying severity of insulin deficiency and diabetes (14–16). A subset of patient-derived NEUROG3 alleles may be hypomorphic or null, based on assays in cell lines and nonmammalian systems (17). Puzzlingly, some patients with intestinal anendocrinosis and neonatal diabetes harboring suspected null NEUROG3 alleles have low but detectable serum C-peptide, an indicator of insulin secretion, suggesting that NEUROG3-independent β-cell development may occur in humans (14,15). Thus, developmentally relevant systems for testing gene function of human alleles of NEUROG3 are needed. In mice, Neurog3 directly induces the expression of multiple targets, including genes encoding the transcription factors, NeuroD1, Pax4, Insm1, and Rfx6 (18–23). Mouse mutants lacking each of those factors have distinct endocrine defects, suggesting that gene networks governed by Neurog3 have multiple distinct phenotypic outputs (24,25). Thus, simple methods for assessing the cellular and molecular functions of Neurog3 and its targets in the developing pancreas could prove useful for decoding regulatory networks governing islet development.
During islet formation, studies of fixed tissues have provided evidence that Neurog3 is required for initiation of islet formation, which includes epithelial cell deformation, delamination, outward movement, hormone expression, and clustering into polyclonal multicellular aggregates. However, control of these transient features of islet development by Neurog3 remains unclear, reflecting a lack of assays to quantify transient cellular phenotypes. Prior studies show that Neurog3+ cells lose polarized expression of markers including Mucin and aPKC; express Snail2, a transcriptional regulator of the epithelial-to-mesenchymal transition; and appear to exit the fetal pancreatic epithelium (9,26). Hormone-expressing progeny derived from Neurog3+ endocrine progenitor cells are detected in fixed tissues both as single cells and as small clusters of multiple islet cells throughout development. As development progresses, these islet clusters enlarge and remodel to form mature islet morphology (27–30). Overexpression of Neurog3 in an immortalized pancreatic ductal cell line altered expression of a subset of genes involved in cell migration and intercellular interactions, suggesting a role for Neurog3 in initiating transcriptional changes that promote migratory phenotypes (31). Prior important live-cell analysis in the developing mouse pancreas (32) permitted description on a multicellular scale of fetal epithelial branching and β-cell movement but did not image single cells from multiple cell populations simultaneously. Live imaging of differentiating pancreatic cells at such high resolution could reveal basic cellular aspects of development, permit single-cell analysis of phenotypes not measurable in fixed sections, and enhance studies of Neurog3-dependent phenotypes. Here, we present approaches to studying pancreas development that combine somatic gene alterations, mutant analysis, genetic complementation or suppression, live cell imaging, and single-cell analysis to assess the regulation of endocrine cell morphogenesis and differentiation in the developing pancreas.
Research Design and Methods
Animals
All animal studies were performed in accordance with Stanford University Institutional Animal Care and Use Committee guidelines. Mouse Insulin1 promoter–green fluorescent protein (GFP) transgenic mice were obtained from The Jackson Laboratory (33). Neurog3gfp knock-in mice were a gift from Klaus Kaestner (University of Pennsylvania) (34). NOD-SCID mice for transplantation experiments were purchased from The Jackson Laboratory.
The Neurog3-tdTomatomosaic transgenic line expresses tdTomato with a bovine growth hormone polyadenylation sequence under the control of a 6.5-kb fragment of the mouse Neurog3 promoter. No growth hormone coding sequence is present in the transgene construct. The line was generated in the Stanford Transgenic Mouse Research Facility. After construct injection, 23 founders were obtained and were crossed to CD1 females; two lines were maintained. The line reported here exhibits mosaic expression, whereas the transgene reported previously (35) is expressed in all Neurog3-positive cells. Genotyping was performed using primers to detect the Tomato coding sequence.
For growth of human pancreatic tissue in the renal subcapsular space, NOD-SCID mice were anesthetized with ketamine/xylazine and tissue was transplanted into the left renal capsule. Kidneys were harvested after 2 weeks for histological analysis.
Immunofluorescence and RNA Analysis
Tissue was fixed in 4% paraformaldehyde and embedded in optimal cutting temperature for cryosectioning. Sections (10 μm) were prepared for immunofluorescence analysis. Primary antibodies used are indicated in Table 1. Alexa-Fluor–conjugated secondary antibodies (Life Technologies and Jackson Immunoresearch) were used for detection.
Table 1.
Antibodies used in this study
| Antigen and host | Source | Dilution |
|---|---|---|
| Neurog3, rabbit polyclonal | Seung Kim Laboratory | 1:500 |
| Insulin, guinea pig polyclonal | Sigma | 1:500 |
| Glucagon, guinea pig polyclonal | Linco | 1:500 |
| Chromogranin A, rabbit polyclonal | Immunostar | 1:500 |
| Mucin, hamster monoclonal | Neomarkers | 1:250 |
| Carboxypeptidase A1, rabbit polyclonal | Chemicon | 1:1,000 |
| Dolichos biflorus agglutinin lectin, biotinylated | Vector Laboratories | 1:1,000 |
| GFP, chicken polyclonal | Abcam | 1:500 |
RNA was purified from organ cultures using the standard TRIzol protocol (Invitrogen). cDNA was synthesized using the RetroScript kit (Ambion) and was analyzed by quantitative PCR with commercially available TaqMan probes (Applied Biosystems) for genes indicated in the text. Analysis of sorted cell populations by FACS was performed as previously described using transgenes indicated in the text (36). Adenovirally expressed human NEUROG3 was detected using custom TaqMan probes: 6-FAM/CTGGGAAGGTGGGCAGGACA/TAMRA-Sp (probe), CTCAACTCGGCACTGGAC (primer), and AGATGTAGTTGTGGGCGAAG (primer). Expression levels were normalized to ActB, and results were shown as levels relative to control samples indicated in the text. At least three independent organs were analyzed for each condition.
Organ Culture and Imaging
Fetal pancreata were dissected on ice from mice at embryonic day (E) 13.5 for culture and imaging. Epithelium and mesenchyme from dorsal and ventral remained visibly intact. Organ rudiments were visually screened for the expression of Ins1-GFP, Neurog3-tdTomatom, or Neurog3gfp alleles with a Leica epifluorescence dissecting microscope. Neurog3gfp/gfp organs were distinguished from Neurog3gfp/+ organs visually by morphology and GFP intensity. Genotypes were confirmed retrospectively by PCR for the presence of GFP and the wild-type Neurog3 allele.
After dissection, organ rudiments were transferred to a chambered cover slip (Nunc Laboratory-Tek) or a 60-mm tissue culture dish and embedded in type 1 collagen gel (Wako). Organs were cultured in phenol red-free DMEM/F12 (Invitrogen) supplemented with 10% FBS (HyClone), 1% penicillin streptomycin, and 1% insulin/transferrin/selenium (Sigma). Media was refreshed every 2 days of culture. Vascular labeling was accomplished by injecting fluorescein isothiocyanate–conjugated Lycopersicum esculentum lectin (Vector Laboratories) into the umbilical vein of the E13.5 fetus before separation from the placenta.
Imaging was performed using Zeiss LSM510 and Leica SP5 confocal microscopes with ×20 water-immersion objectives and environmental-control chambers to maintain an imaging environment at 37°C with 5% CO2. Images were acquired at 1,024 × 1,024 resolution with 4 μm separating Z sections and 20 min separating Z-stack acquisitions. Data were saved in LSM (Zeiss) or LIF (Leica) formats. ImageJ (National Institutes of Health) or Volocity (PerkinElmer) was used to compile Z stacks into XYT datasets, which were exported in TIFF format for analysis or AVI and MOV formats for viewing.
Analysis of Imaging Data
Movies were analyzed in ImageJ. For cell shape analysis, individual cells were manually traced and shape factor was calculated at each time point using the “Circularity” function. Shape factor = 4π(area)/(perimeter2). Cell shape traces over time were manually generated in ImageJ and analyzed using Microsoft Excel. Shape factor of single cells was traced from the first time point at which a cell was clearly detected until the last time point at which it could be distinguished from other cells. Simple averaging of single-cell shape traces did not indicate a stereotyped pattern of deformation for either Neurog3-tdTomato or Ins1-GFP cells. Normalization to the mean shape factor of an epithelial cell (E-cadherinhigh pancreatic epithelial cells) followed by integration of the single-cell shape factor curve generated a plot of single-cell accumulated shape factor over time, a measure of a single cell’s accumulated deviation from epithelioid shape. This path-independent measure of deformation allowed measurement of cell deformation undergone over time by defined cell populations. For statistical comparisons, area under the curve was computed for each individual sample, and biological groups were compared using a t test.
Clustering was analyzed by computing mean object size at each time point using the “Analyze Particles” function in ImageJ (32). Clustering values were normalized to the value calculated at t = 0 and plotted over time. Data were analyzed in Microsoft Excel.
GFPhigh cells in Neurog3gfp/+, Neurog3gfp/gfp, and Ad-NEUROG3 rescues were counted from Z stacks 50–70 μm thick acquired at single time points at the beginning of live imaging data sets. At least three organs per condition were analyzed.
Human Tissue
Experiments involving use of human tissue were approved by the Stanford University Institutional Review Board. Human pancreatic tissue between gestational days 76 and 110 was obtained through the Birth Defects Research Laboratory, University of Washington (Seattle, WA), and all donors provided informed consent.
Identification of Human NEUROG3P39X Mutation
The study protocol was approved by the University of California, Los Angeles, institutional review board, and pertinent medical history, pathology, and DNA samples were obtained from the subject. The subject presented clinically with severe malabsorptive diarrhea as a neonate, and like others with enteric anendocrinosis was managed with parenteral nutrition (16). At 3 years of age, diabetes was diagnosed, and a C-peptide level of 3.6 ng/mL was measured (M.G.M., unpublished data). An Oragene saliva kit was used to obtain DNA, and exon 2 of NEUROG3 was PCR amplified and sequenced using oligonucleotides (5′CAATCGAATGCACGACCTCAAC-3′ [sense] and 5′-AGATTATGGGGTGGTGGCAGAG-3′ [antisense]). Sequencing revealed a homozygous single base deletion in codon 39 (c.117delC; p.P39PfsX38), leading to a frame-shift mutation and 38 additional amino acids that are out of frame prior to a premature transcriptional termination sequence (M.G.M., unpublished data).
Adenovirus Construction
Adenoviruses were generated by cloning cDNAs of interest into the pDual-RFP-CCM or pDual-H2B:mCherry shuttle vectors, which express a fluorescent protein and the gene of interest under the control of separate cytomegalovirus promoters. The pDual-H2B:mCherry vector was constructed by amplification of the H2B:mCherry cDNA sequence by PCR from pcDNA3-H2B:mCherry (Addgene Plasmid 20972) and replacing the red fluorescent protein (RFP) sequence in pDual-RFP (Vector Biolabs, Philadelphia, PA). Custom high-titer adenoviruses (≥1 × 1010 plaque-forming units/mL) were generated by Vector Biolabs for all viruses except Ad-NEUROD1 and Ad-PAX6, which were purchased premade from Vector Biolabs. All adenovirus work was performed in compliance with regulations from the Stanford University Administrative Panel on Biosafety.
Adenovirus Microinjection
Pancreatic rudiments were dissected from E13.5 embryos, preserving both dorsal and ventral pancreatic epithelia and mesenchyme. Intact pancreas rudiments were separated from surrounding intestinal and vascular structures. Organs were embedded in Type 1 Collagen gel (see above), and gel was allowed to polymerize for 1 h at 37°C before overlaying with cell culture media. Organs were incubated at 37°C for at least 1 h before beginning the injection procedure.
For injections, needles were produced by pulling 25 μL glass microcapillary tubes (no filament, cat. no. 53432-761; VWR) using a Flaming/Brown micropipette puller (model P-87; Sutter Instrument Co.) with the following settings: pressure 400, heat 580, pull 75, velocity 60, time 100. Needles were mounted on a brass straight-arm needle holder (MINJ-4; Tritech Research) attached to a standard manual control micromanipulator (640056; Harvard Apparatus). For preparation of needles, tips were broken with forceps, and viral solution (1 × 106 plaque-forming units/μL) was loaded by drawing into the needle through the tip. A syringe and tubing filled with mineral oil were used to control pressure for delivery of the injection solutions.
Cultured pancreatic rudiments were visualized under a stereomicroscope, and epithelial structures were visually distinguished from mesenchyme: mesenchyme appears opaque under reflected light illumination, whereas epithelial structures appear translucent. Less than 2 μL virus solution was injected into the epithelium: larger volumes caused distension of the organ and could rupture the tissue. Efficiency of labeling could be increased by entering the epithelium at multiple locations throughout the dorsal and ventral pancreas, but doing so increased the risk of transducing nonepithelial cells. Injection solution was delivered slowly to minimize distension of the epithelial network. Viral labeling of pancreatic epithelial cells occurred in essentially all organs attempted, and epithelial restriction occurred in >80% of rudiments (n > 100). Fluorescence from a cytomegalovirus-RFP reporter transgene was detectable by the day after injection and increased through at least 5 days of culture. Addition of adenovirus only to the organ culture media, without injection, primarily resulted in infection of mesenchymal cells along the surface of the organ (data not shown).
Statistical Analysis
A two-tailed Student t test was used for statistical comparisons of gene expression, cell counts, and live-cell phenotypes. P values <0.05 were considered significant. Data are presented as means ± SEM.
Results
Restoration of Islet Development in Neurog3 Mutants by Targeted Gene Transduction
Assessment of pancreas developmental genetics, especially transcriptional regulators of epithelial development, could be advanced by tests of gene function in a developmentally relevant context. For example, such assays could permit complementation or suppression analysis. To achieve this goal, we tested whether Neurog3 mutant phenotypes could be rescued by exploiting the accessibility of pancreatic epithelium to microinjection in organ cultures. We embedded E13.5 organs in collagen gel and overlaid with culture media (37). Over 5 days of culture, we observed substantial growth and multilineage differentiation. After identifying the fetal pancreatic duct, we injected recombinant adenoviral vectors expressing a transgene encoding RFP (mRFP) into the lumen of cultured Neurog3gfp/gfp pancreatic rudiments (Fig. 1A). The Neurog3gfp allele replaces the coding sequence of Neurog3 with GFP, creating a null allele (34). The viral injection solution appeared restricted to the ductal epithelial tree. Consistent with this, immunostaining of rudiments after 5 days of culture showed that expression of mRFP was predominantly restricted to pancreatic epithelial cells (Fig. 1B and C). Quantification revealed that 31 ± 4% of pancreatic E-cadherin+ epithelial cells coexpressed RFP, indicating a high efficiency of infection and gene transduction (n = 4) (Fig. 1B and C). Thus, genetic transduction of fetal pancreatic epithelium was feasible in our system.
Figure 1.

Transduction of Neurog3 mutant fetal pancreatic cells with human NEUROG3 by adenovirus microinjection restores islet differentiation and morphogenesis. A–C: Microinjection of adenovirus into the fetal pancreatic epithelium permits transduction of epithelial cells. D: Rescue of islet-specific gene expression in Neurog3gfp/gfp organ rudiments by expression of wild-type human NEUROG3 and assessment of enteric anendocrinosis patient-derived allele function. E: Histological evidence of islet differentiation and morphogenesis. Ad-NEUROG3 efficiently restores islet gene expression and formation of islet cell clusters. The R107S patient allele restores a limited amount of differentiation, consistent with hypomorphic function. P39X and N89D (RFP) alleles fail to induce any changes in islet gene expression or morphogenesis, consistent with complete lack of function. Gene expression measurements were normalized to control Neurog3gfp/+;Ad-RFP cDNA. *P < 0.05, Student t test.
To assess the capacity for genetic complementation, we next injected homozygous mutant Neurog3gfp/gfp rudiments with adenovirus coexpressing wild-type human NEUROG3 and RFP or control virus expressing RFP alone. Human mouse Neurog3 share 75% identity at the protein level. At baseline, Neurog3 mutants fail to form islet cells, express markers of islet differentiation, or form islet-like cell clusters (5,6), phenotypes recapitulated in our organ cultures (see below). In addition, GFP expression is very low in Neurog3gfp/gfp organs relative to Neurog3gfp/+ controls, likely reflecting a lack of positive autoregulation in Neurog3-null pancreata (38). After transduction with wild-type NEUROG3, quantitative PCR (qPCR) analysis revealed that NEUROG3 rescue resulted in a significant restoration of islet markers, with expression of Neurog3 target genes comparable with Neurog3gfp/+ controls (Fig. 1D and Supplementary Fig. 1). For example, we observed restored expression of several direct targets of Neurog3, including NeuroD1 and Rfx6, as well as expression of genes encoding islet hormones, including Ins2 and Gcg (Fig. 1D). Expression levels of Neurog3 target genes approached levels in Neurog3gfp/+ littermates but did not reach those observed in Neurog3+/+ controls, indicating that the level of NEUROG3 rescue was partial (Supplementary Fig. 1). Pdx1, a gene expressed independently of Neurog3 in pancreatic epithelial progenitors, remained unchanged as expected. Immunostaining of Neurog3gfp/gfp organs injected with Ad-NEUROG3 confirmed restoration of insulin and glucagon protein expression and demonstrated the formation of large endocrine cell clusters resembling control islets (Fig. 1E). We also observed a marked increase in GFP intensity, expressed from the endogenous Neurog3 locus, throughout Neurog3gfp/gfp organ rudiments injected with Ad-NEUROG3 (Fig. 1E) (see below), consistent with NEUROG3 autoregulatory activity (38). In control Neurog3gfp/+ organs injected with Ad-RFP, we observed 220 ± 27 GFPhigh cells per field, while in Neurog3gfp/gfp;Ad-RFP control organs we observed only 6 ± 1 GFPhigh cells per field (P = 0.001) (Supplementary Fig. 1). (See research design and methods.) Thus, infection with virus, per se, did not induce GFP expression. Expression of wild-type NEUROG3 restored GFPhigh cell counts to 212 ± 17 cells per field in Neurog3gfp/gfp (P = 0.001 relative to Neurog3gfp/gfp, P = 0.52 relative to Neurog3gfp/+). These findings provide additional unique evidence that NEUROG3 is capable of positive autoregulation (38). All rescued cells, as indicated by elevated GFP expression or expression of islet markers including ChgA, also coexpressed the RFP reporter, suggesting that the rescue observed was cell autonomous (Supplementary Fig. 1). In summary, our organ culture and microinjection system revealed the capacity for genetic complementation studies in mouse Neurog3 mutant pancreata.
Complementation Analysis of Human NEUROG3 Alleles
Mutations in NEUROG3 have been linked to human congenital diarrhea and intestinal anendocrinosis with diabetes of variable onset and severity (14–16). It remains unclear, however, whether diabetes severity reflects a range of allele strength because current methods used to test NEUROG3 alleles rely on in vitro cell line assays or ectopic expression in prepancreatic endoderm of nonmammalian model organisms. These systems may not recapitulate the appropriate pancreatic developmental context and do not yet permit analysis of dynamic phenotypes regulated by NEUROG3.
To test the function of three mutant human NEUROG3 alleles in native fetal pancreatic epithelial cells, we microinjected adenoviruses into the Neurog3gfp/gfp epithelium and cultured organs for 5 days, assaying the ability of each allele to restore endocrine gene expression and features of islet morphogenesis. We tested the NEUROG3R107S patient allele (16), the synthetic null mutant NEUROG3N89D (17), and NEUROG3P39X, a new patient-derived allele (M.G.M., unpublished observations). qPCR probes that recognize wild-type and mutant forms of NEUROG3 revealed comparable levels of corresponding transgenic NEUROG3 mRNA after infection (Supplementary Fig. 1). Expression of NEUROG3R107S restored GFPhigh expression in a significant fraction of cells (48 ± 5 cells per field, P = 0.001 compared with 6 ± 1 in Ad-RFP control, Supplementary Fig. 1). Consistent with this finding, we also observed that expression of mRNAs encoding pancreatic endocrine factors and hormones was partially restored by NEUROG3R107S (Fig. 1D). Hormone production was also detected by immunohistology in rudiments rescued by NEUROG3R107S (Fig. 1E). By contrast, we detected no increase in GFP fluorescence after delivery of the NEUROG3 P39X allele compared with Ad-RFP and Ad-N89D negative controls (7 ± 1 vs. 6 ± 1 cells per field, P = 0.29 compared with Ad-RFP control, Supplementary Fig. 1). Together these data support prior studies suggesting that NEUROG3R107S is a hypomorphic allele (17) and provide evidence that NEUROG3P39X is a null allele.
We postulated that our gene transduction system based on organ culture and subsequent analysis could be adapted to study human fetal pancreas development. We developed a sequential strategy for microinjection of adenoviral vectors into the ductal lumen of human fetal pancreatic tissue to modify gene expression, followed by transplantation in NOD-SCID mice to permit vascularization and development (39,40). We then tested whether misexpression of NEUROG3 was sufficient to induce endocrine differentiation in developing human pancreatic tissue. All adenoviruses coexpressed RFP under control of a second promoter to permit identification of transduced cells. Upon misexpression of wild-type NEUROG3, we observed clear induction of the “pan-endocrine” marker protein Chromogranin A (CHGA) within many transduced cells and establishment of large RFP-expressing cell clusters (Supplementary Fig. 2). CHGA expression was observed in 67 ± 2% of cells overexpressing wild-type NEUROG3. In contrast, we observed CHGA expression in only 12 ± 3% of RFP+ transduced cells in organs infected with the control NEUROG3N89D mutant, which lacks DNA-binding activity (P = 1.3 × 10−4, n = 3) (Supplementary Fig. 2). Thus, NEUROG3 misexpression was sufficient to direct endocrine differentiation and to promote islet cell cluster formation in developing human pancreatic cells.
Genetic Suppression of Neurog3 Mutant Phenotypes
Classical mutant suppression analysis can reveal or clarify functions of genetic regulators but to our knowledge has not previously been achieved with pancreas developmental mutants. Targets of Neurog3 include NeuroD1, Rfx6, and Pax4 (18,19,22,23). We used adenoviral vectors to express human orthologs of these factors or the islet transcriptional regulator PAX6 to assess their ability to suppress phenotypes in Neurog3gfp/gfp mutant rudiments.
We observed that NEUROD1 overexpression partially restored expression of mRNA encoding chromogranin A and islet hormones compared with undetectable expression in controls infected with Ad-RFP (Fig. 2A). Moreover, we observed ChgA protein expression by GFPhigh cells in small clusters adjacent to major ductal structures (Fig. 2B and C). Restoration of hormone and ChgA mRNA expression in these studies indicates successful suppression or bypass of the Neurog3 mutation, although the magnitude of effect was less than that achieved by NEUROG3 complementation. By contrast, genetic suppression of Neurog3gfp/gfp pancreas development using other transcriptional regulators was less robust. Ad-RFX6 infection led to a small but significant induction of ChgA (5.7% of control; P = 0.03) (Fig. 2A) but not other markers of islet development. PAX6 significantly induced mRNA encoding glucagon (P = 2 × 10−8) (Supplementary Fig. 3) but not other markers, consistent with known roles for Pax6 in activating α-cell–specific gene expression and α-cell differentiation (41–43). Infection with adenovirus encoding Pax4 resulted in no detectable changes of mRNA levels relative to Ad-RFP controls for genes including all islet hormones, Neurod1, and ChgA (Supplementary Fig. 3). In combination, NEUROD1 and RFX6 induced both ChgA and Ghrelin mRNA to levels greater than achieved by NEUROD1 alone (Fig. 2A and Supplementary Fig. 3) (P = 9 × 10−4 and P = 0.03, respectively) and generated small multicellular clusters that expressed ChgA protein. Multiple other factors or combinations did not result in additional restoration of islet differentiation (Supplementary Fig. 3). Together, these data suggest that RFX6 and NEUROD1 may collaborate to induce endocrine differentiation and islet cell aggregation without NEUROG3. Collectively, these results also show that genetic suppression analysis is feasible in our system.
Figure 2.

Suppression of Neurog3gfp/gfp phenotypes by downstream targets of NEUROG3. A: Injection of Neurog3gfp/gfp fetal pancreatic epithelia with adenoviruses carrying single and combinations of factors partially restores expression of markers of endocrine differentiation. Expression of NEUROD1 induced small increases in expression of all hormones and of ChgA. Expression of RFX6 alone caused small but significant increases in ChgA expression. Combination of NEUROD1 and RFX6 induced expression of Ghrelin (see Supplementary Fig. 1) and ChgA but not expression of other hormones. B–E: Histological analysis of organs injected with factors downstream of NEUROG3 reveals ChgA-expressing cells as singlets or small clusters exiting the pancreatic epithelium in Neurog3gfp/gfp;Ad-NEUROD1 organs and fails to show any ChgA protein expression in Neurog3gfp/gfp;Ad-RFX6 organs. Organs transduced with RFX6 and NEUROD1 viruses in combination formed small clusters of ChgA-expressing cells separated from the epithelium. Scale bars: 25 μm. Gene expression measurements were normalized to Neurog3gfp/+;Ad-RFP controls. *P < 0.05, Student t test.
Two-Color Live Imaging at Single-Cell Resolution Reveals Cell Movement, Deformation, Differentiation, and Morphogenesis by Developing Fetal Pancreatic Cells
Tools to study living islet precursors at single-cell resolution could enhance studies of islet development, including assessment of Neurog3 gene functions. To image and assess distinct live cell types in pancreas development, including our genetic complementation studies, we first labeled endocrine precursor cells by generating transgenic mice expressing the RFP tdTomato (44) under the control of a 6.5-kb fragment of the mouse Neurog3 promoter (35). Immunostaining and FACS purification of pancreatic cells from this Neurog3-tdTomato transgenic line confirmed tdTomato labeling in fetal Neurog3-expressing pancreatic cells (Supplementary Fig. 4). Because of the perdurance of tdTomato labeling, we observed tdTomato expression in differentiated hormone+ islet cell progeny of Neurog3-expressing cells (Supplementary Fig. 4). FACS and immunofluorescence analysis demonstrated that tdTomato transgene expression was mosaic in the mouse line used for these studies, labeling approximately half the Neurog3-expressing pancreatic cells marked by a Neurog3gfp knock-in allele (34). This level of mosaicism facilitated subsequent single-cell tracking (see below) and distinguishes this line from one without mosaicism previously reported by our group (35). To label both endocrine precursors and hormone-expressing β-cells, we bred Neurog3-tdTomato mice with Insulin1-GFP transgenic mice (33). Resulting bitransgenic Neurog3-tdTomato;Ins1-GFP embryos enabled simultaneous labeling and tracking of Neurog3-expressing endocrine precursors and nascent fetal insulin-expressing β-cells in the same cultured pancreas rudiments. Thus, Neurog3-tdTomato transgenic mice provide a tool for analyzing a subset of living endocrine progenitor cells.
Islet cell differentiation and morphogenesis after birth of Neurog3+ cells requires several days; thus, to permit imaging experiments of sufficient duration, we next established a stable system for fetal pancreatic culture, development, and growth that minimized artifactual tissue movement. We used the same collagen-embedded organ culture scheme described above for rescue experiments, in which growth and differentiation occur as assessed by histology and qPCR (Supplementary Figs. 5 and 6). Immunohistology also revealed islet cell clustering after 5 days of culture (Supplementary Fig. 6). Thus, multiple criteria indicated that development of the three major cell lineages in the fetal pancreas occurred and that fundamental features of islet morphogenesis were reproduced in this culture system. To establish that our frame of reference for live imaging was stable, we labeled the vasculature by injecting fluorescein isothiocyanate–conjugated tomato lectin through the umbilical vein of mice prior to pancreas extraction; imaging of cells relative to the static vascular “reference” label indicated that our imaging field was stable (Supplementary Fig. 5 and Supplementary Movie 1). To confirm that we analyzed single cells, we labeled nuclei with a histone H2B:GFP transgene (45) and endocrine precursors with Neurog3-tdTomato and calculated the mean two-dimensional area of single cells. We used that area, 104 ± 7 μm2, as a reference during our analysis to identify single cells for tracking (Supplementary Fig. 5).
A wave of pancreatic endocrine differentiation begins in fetal mice at approximately E13.5 (1). Thus, we cultured organs isolated at E13.5 and imaged at 20-min time intervals for 48 h to assess islet cell dynamics. Live imaging revealed several phenotypes in pancreata of Neurog3-tdTomato;Ins1-GFP mice (Fig. 3A–D and Supplementary Movie 2). We directly observed differentiation of individual red Neurog3-tdTomato+ endocrine precursor cells, as they initiated expression of Ins1-GFP and became progressively yellow and then green (Fig. 3E–H). Not all Neurog3-tdTomato–expressing cells generated Ins1-GFP–expressing cells during our time-lapse studies, consistent with prior studies showing that Neurog3+ cells generate β-cells and non-β-cells and with immunostaining data indicating that Tomato+ cells also expressed glucagon or insulin after organ culture experiments (Supplementary Fig. 6). Moreover, not all Ins1-GFP cells were derived from Neurog3-tdTomato+ cells, consistent with transgene mosaicism in this line. Second, we observed movement of Neurog3-tdTomato+ and Ins1-GFP+ single cells into multicellular clusters (Fig. 3A–D and Supplementary Movie 2). At the end of time-lapse imaging, green Ins1-GFP+ multicellular clusters were a dominant feature. Thus, our studies permitted observation of endocrine differentiation and multicellular morphogenesis in living tissue at single-cell resolution. Third, imaging revealed that Neurog3-tdTomato and Ins1-GFP cells achieved often-dramatic morphological deformation, involving extension and retraction of cellular processes resembling filopodia, while moving into multicellular nascent islets (Fig. 3I–L). For example, in some cases, cells extended processes up to three times longer than the cell body diameter (Fig. 3I–L), a degree of deformation not previously reported to our knowledge. Thus, we have developed an organ culture and imaging system enabling discovery and measurement of dynamic cellular phenotypes during pancreatic endocrine differentiation and islet development.
Figure 3.
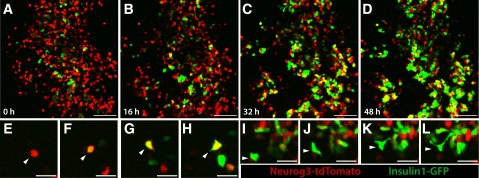
Time-lapse confocal microscopy of developing islet cells reveals dynamic single-cell phenotypes. Endocrine precursors at E13.5 were labeled with a mosaic Neurog3-tdTomato transgene, and insulin-expressing cells were labeled with an Insulin1-GFP transgene. Cell movement and deformation are visible in both populations, and clustering and differentiation of β-cells are observed throughout the movie. A–D: Frames from a 48-h movie. E–H: Single Neurog3-tdTomato–expressing cells differentiate into β-cells, initiating expression of the Ins1-GFP transgene. I–L: Extensive cell deformation and extension of processes are observed during cell movement and clustering. Scale bars: 200 μm (A–D) and 25 μm (E–L).
Quantitative Analysis of Live-Cell Phenotypes in Pancreas Development
The single-cell resolution afforded by our live imaging system permitted quantification of dynamic cellular phenotypes like shape changes, movement, and clustering during endocrine differentiation. To quantify cell deformation, we calculated the shape factor of cells, which relates cell perimeter and area. A shape factor of zero corresponds to a straight line, whereas a shape factor of 1.0 corresponds to a perfect circle (Fig. 4A and B). We found that the mean shape factor of a fetal pancreatic ductal epithelial cell after immunostaining with an E-cadherin antibody (see research design and methods) was 0.83 ± 0.01 (Supplementary Fig. 6). Prior studies have applied shape factor measurements to analyze cell shape in islet development but relied on single or instantaneous measurements, which may obscure dynamic single-cell phenotypes over time (46,47). We took advantage of our ability to track single cells over many hours of development and integrated the accumulated deformation of a cell over time. Tracking the shape factor of single cells during our time-lapse studies yielded continuous single-cell shape traces (Fig. 4A and B, green line). We normalized to epithelial shape, integrated shape factor curves with respect to time, and calculated the accumulated deviation from epithelioid shape of individual Neurog3-tdTomato+ cells and motile Ins1-GFP+ cells not yet merged with islets (Fig. 4A and B, blue line). We observed no significant difference in the shape factor between Neurog3-tdTomato+ and Ins1-GFP+ cells (P = 0.61) (Fig. 4C). This similarity of cumulative cell shape changes is therefore consistent with our observation that differentiating pancreatic endocrine cells begin deformation and movement at the Neurog3-expressing stage and maintain those phenotypes during the initial stages of Ins2 expression in moving β-cells.
Figure 4.
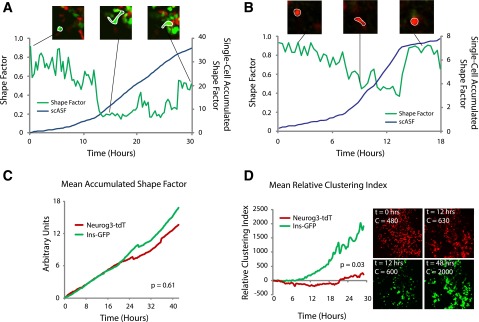
Quantitative analysis of dynamic cell phenotypes. Single-cell resolution live cell imaging enables quantification of phenotypes including deformation and clustering. Shape factor, a measure of deformation, was calculated over time for a single β-cell (A) and a single Neurog3-tdTomato endocrine precursor (B). A shape factor value of one corresponds to a perfect circle, and a value of zero corresponds to a straight line. Integration of the single-cell shape factor curve (green lines in A and B) with respect to time provides the single-cell accumulated shape factor (scASF), a path-independent measurement corresponding to the amount of deformation undergone by a single cell (blue lines in A and B). C: Calculation of mean accumulated shape factors for the Neurog3-tdTomato and Ins1-GFP cell populations demonstrates no quantitative difference in deformation as observed by time-lapse microscopy. D: Calculation of the clustering index for the two cell populations indicates that the Ins1-GFP population tends to cluster more than the Neurog3-tdTomato population. Hrs, hours.
To quantify Neurog3-tdTomato+ and Ins1-GFP+ cell clustering in nascent islets, we adapted a clustering index previously used to assess pancreatic cell aggregation (32). This measurement calculates “object” size in image data: thus, assuming consistent cell size, increased object size corresponds to increased clustering of fetal islet cells. We normalized the clustering index to its value at t = 0 h for each time-lapse study to facilitate statistical comparisons of changes in clustering across multiple movies. We observed a transient decrease in the clustering index of Neurog3-tdTomato+ cells, consistent with the observation that these cells originate from fetal pancreatic epithelium and disperse during migration toward islet cell clusters (Fig. 4D). By contrast, Ins1-GFP+ cell aggregation was reflected by a large increase in their clustering index (P = 0.03) (Fig. 4D). These live-cell measurements should be useful for describing experimental alterations in islet cell migratory patterns and clustering, including the analysis of phenotypes in mutant pancreatic development (see below).
Neurog3 Is Required for Cell Shape Changes and Migration Into Nascent Islet Clusters
Prior studies suggest that Neurog3 is required for the mobilization of islet progenitors from fetal epithelium, although the evidence for this has been indirectly inferred from fixed tissue analyses (9,26). For example, lineage tracing of Neurog3-null cells suggests that these cells remain within the fetal pancreatic epithelium, where they redirect their fate toward ductal or acinar cell lineages (13,48). To test directly the requirement for Neurog3 in islet cell deformation and movement, we imaged Neurog3gfp/gfp organs at E13.5. In heterozygote controls, imaging revealed highly dynamic Neurog3gfp/+ cells that deformed and extended processes, moved, and eventually clustered in nascent islets (Fig. 5A–C and Supplementary Movie 3). By contrast, homozygous Neurog3gfp/gfp cells failed to initiate movement and remained in epithelial tubes (Fig. 5D–F and Supplementary Movie 4). Quantification of the shape factor for homozygous mutant Neurog3gfp/gfp cells revealed a significant reduction in cell deformation compared with control Neurog3gfp/+ cells (P = 0.006) (Fig. 5G). The lack of cell aggregation or islet differentiation in Neurog3gfp/gfp organs precluded quantification of clustering. Thus, single-cell imaging methods identified defects in pancreatic cell migration in mice lacking Neurog3.
Figure 5.
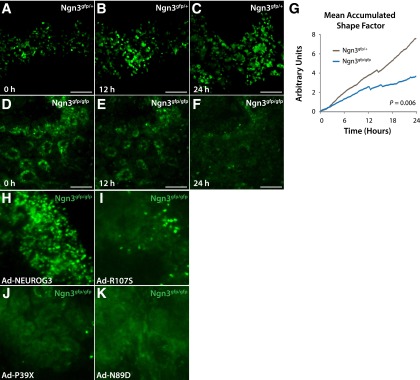
Time-lapse imaging of cells lacking Neurog3 reveals a failure to undergo morphological changes and cell migration. A–C: Cells heterozygous for Neurog3gfp migrate and cluster, whereas Neurog3gfp/gfp cells (D–F) fail to leave epithelial tubules. G: A significant quantitative reduction in cell deformation was detected. H–K: Evaluation of the capacity for human NEUROG3 alleles to induce islet cell deformation. Wild-type NEUROG3 restores cell deformation and movement, NEUROG3R107S has a limited capacity for rescue and restores motility in only a few scattered cells, and NEUROG3P39X and NEUROG3N89D have no detectable effect on islet cell movement phenotypes. Scale bars: 100 μm. P value from Student t test.
To test whether rescue of Neurog3 mutant phenotypes by human NEUROG3 alleles extended to live-cell deformation phenotypes, we generated time-lapse movies of Neurog3gfp/gfp organ rudiments injected with adenoviruses expressing human NEUROG3 alleles. In contrast to Ad-RFP or Ad-NEUROG3N89D injection, Ad-NEUROG3 in Neurog3gfp/gfp organs restored cell movement, shape changes, and islet morphogenesis. Any GFPhigh cells detected in Neurog3gfp/gfp;Ad-NEUROG3R107S organs also underwent deformation, suggesting a binary output in which cells with high Neurog3 activity undergo deformation, whereas cells with low Neurog3 activity do not (Fig. 5H–K and Supplementary Movies 5–7). Consistent with the lack of activity detected for NEUROG3P39X, we did not detect GFPhigh cells or cell deformation in Neurog3gfp/gfp;Ad-NEUROG3P39X organs (Fig. 5J and Supplementary Movie 8). Together with molecular and immunohistological data above, these data provide evidence for rescue of multiple dynamic phenotypes by functional human NEUROG3 alleles.
Discussion
Detailed assessment of cell and developmental phenotypes in normal and mutant living tissue provides powerful opportunities to understand gene function in organogenesis. Here, we describe a combination of fetal organ culture, live imaging, and genetic methods to dissect functions of Neurog3, a gene crucial for endocrine cell ontogeny and islet morphogenesis in the pancreas. By creating platforms for unique gene complementation and suppression studies in Neurog3 mutant tissue, we evaluated the function of human disease-associated Neurog3 mutations and assessed the capacity for Neurog3-independent islet differentiation. Quantification of dynamic cell phenotypes in distinct endocrine progeny with single-cell resolution in the fetal pancreas permitted us to measure and show that Neurog3 is required for initiating multiple steps in islet morphogenesis, including cell deformation, movement, differentiation into hormone-expressing progeny, and cell aggregation to form nascent islets. The dissection of gene functions and cell dynamics using approaches detailed here should accelerate discovery of cell and genetic mechanisms underlying the mechanisms that coordinate islet formation.
Development of methods for transgenic alterations in cultured pancreatic tissue developing ex vivo permitted us to efficiently assess the capacity for patient-derived human alleles of NEUROG3 to induce islet differentiation and formation. In these studies, we were able to study the activity of these alleles in a developmentally relevant context in pancreatic cells at an embryonic stage appropriate for Neurog3 expression. Prior assessments of activity of these alleles has relied on in vitro assays in cell lines that are not known to have capacity for islet differentiation or on assays using chick embryonic models of endodermal development in Neurog3 wild-type backgrounds (14–17). In these other systems, autoregulation by endogenous Neurog3 could complicate interpretation of results. By using embryonic Neurog3-null mouse pancreatic tissue and assessing multiple Neurog3-dependent phenotypes—including autoregulation, morphological changes, and activation of islet gene expression programs—we provide unique evidence that while at least one NEUROG3 allele associated with enteric anendocrinosis is hypomorphic, the newly identified NEUROG3P39X allele has no detectable function and is likely a null allele.
The proband homozygous for the NEUROG3P39X allele did not develop neonatal insulin-dependent diabetes and had detectable serum C-peptide, suggesting β-cell development and function in this patient (M.G.M., unpublished observations). Thus, to the extent that NEUROG3P39X is a null allele, our studies raise the possibility that NEUROG3 function may not be required for β-cell development to the same degree as in mice, a possibility consistent with prior human studies (14,15). In some vertebrates, including pigs and zebrafish, a clear ortholog of NEUROG3 has not been identified. Thus, we speculate that other transcription factors could substitute for Neurog3 in regulating islet cell development in human islet development, as is the case for Ascl1b and Neurod1 in zebrafish (49). Consistent with this possibility, here we observed that NEUROD1 overexpression was sufficient for partial bypass of the requirement for Neurog3 in mouse islet development in cultured fetal pancreata. Establishment of NEUROG3P39X as a null allele suggests that a NEUROG3-independent gene network may permit formation of a limited population of islet cells in human development.
The gene delivery methods described here should enable testing of genetic hypotheses using gain-of-function methods, including dissection of pathways regulating islet differentiation downstream of Neurog3. For example, delivery of direct Neurog3 target genes including NeuroD1 was sufficient to restore some islet differentiation in Neurog3-null pancreas. Without using in vitro gene delivery methods, similar genetic suppression experiments might require generation of multiple new mouse transgenic or knock-in lines. In addition, we demonstrate that similar viral methods could be applied to analyze human pancreatic development, enabling future studies relevant to human biology. We recognize limitations to viral expression of transgenes, which may include failure to appropriately regulate transcript levels and background labeling of nonepithelial cell types. In principle, these approaches permitting genetic complementation should be readily adaptable to study the activity of other genes and alleles in pancreas development or any other fetal organ accessible to in vitro culture and viral gene transduction.
Dual-color confocal imaging permitted assessment of transient or dynamic phenotypes like cell deformation and movement across multiple stages of islet development. Compared with prior efforts at live cell analysis (32,47,50), we present data using single cells from multiple cell populations tracked simultaneously. These studies demonstrate the capacity for directly observing differentiation and tracking morphological dynamics of single cells. The cell shape changes and cellular extensions observed in our video data were not detected in fixed sections, possibly reflecting limitations of histological approaches to preserve fine or transient subcellular morphologies (51). Applying quantitative methods, we described the maintenance of migratory phenotypes as islet cells differentiate from hormone-negative endocrine precursors into hormone-expressing fetal β-cells. Thus, our work provides a foundation for analysis of disrupted islet cell development and morphogenesis in mutant pancreatic development.
To develop methods for genetic studies of living tissue, we analyzed Neurog3 mutants—which have severe disruptions of cell movement and clustering—and optimized methods for quantifying and analyzing these phenotypes. Although indirect evidence suggested that Neurog3 controls the acquisition of cell deformation or migratory phenotypes (9,48), here we have provided direct evidence for a failure by islet progenitor cells to initiate cell shape changes or movement in the absence of Neurog3. Future experiments could dissect whether acquisition of migratory phenotypes is invariably linked to islet differentiation or whether one process could occur in the absence of the other. We also applied live-imaging methods to visualize directly the failure of pancreatic cells to elevate Neurog3 promoter activity in Neurog3 knockout cells, corroborating prior findings supporting a positive autoregulatory loop driving fetal pancreatic Neurog3 expression (38). Consistent with the results from our genetic complementation experiments, we found that NEUROG3 alleles could restore morphogenetic movements, with the number of motile cells observed in each organ rudiment proportional to allele strength. Overall, the imaging methods we have developed could allow tracking of the behavior of multiple cell populations in the context of genetic or chemical interventions in pancreatic development.
In summary, we have assessed human allele function in a developmentally relevant context, evaluated the capacity for Neurog3 targets to restore islet differentiation in the absence of Neurog3, and measured live cell phenotypes in normal and mutant islet development. The methods described here should be useful for dissecting genetic networks and cell biological events controlling pancreatic islet development.
Article Information
Acknowledgments. The authors thank members of the Seung Kim Laboratory, including Jonghyeob Lee, for helpful discussion and comments; Jon Mulholland and Kitty Lee of the Stanford Cell Sciences Imaging Facility for assistance with confocal time lapse microscopy; and the Stanford Neuroscience Microscopy Service.
Funding. P.T.P. was supported by the Howard Hughes Medical Institute (HHMI) Medical Fellows program, the Stanford Medical Scientist Training Program, and the U.S. National Institutes of Health (F30DK102301). S.E.S. was supported by a Stanford University Vice President for Undergraduate Education fellowship. Work in the Kim laboratory was supported by the JDRF, HHMI, and HL Snyder and Elser Foundations. S.K.K. is an investigator of the HHMI.
The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. P.T.P. and S.K.K. designed experiments and wrote the manuscript. P.T.P., T.S., S.E.S., G.W.M., and J.W. performed experiments. M.G.M. identified and characterized human patients with NEUROG3P39X mutations. All authors reviewed and revised the manuscript. S.K.K. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db15-0042/-/DC1.
References
- 1.Benitez CM, Goodyer WR, Kim SK. Deconstructing pancreas developmental biology. Cold Spring Harb Perspect Biol. 1 June 2012 [Epub ahead of print]. DOI: 10.1101/cshperspect.a012401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim SK, MacDonald RJ. Signaling and transcriptional control of pancreatic organogenesis. Curr Opin Genet Dev 2002;12:540–547 [DOI] [PubMed] [Google Scholar]
- 3.Puri S, Hebrok M. Cellular plasticity within the pancreas—lessons learned from development. Dev Cell 2010;18:342–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang S, Hecksher-Sorensen J, Xu Y, et al. Myt1 and Ngn3 form a feed-forward expression loop to promote endocrine islet cell differentiation. Dev Biol 2008;317:531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A 2000;97:1607–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Apelqvist A, Li H, Sommer L, et al. Notch signalling controls pancreatic cell differentiation. Nature 1999;400:877–881 [DOI] [PubMed] [Google Scholar]
- 7.Schwitzgebel VM, Scheel DW, Conners JR, et al. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 2000;127:3533–3542 [DOI] [PubMed] [Google Scholar]
- 8.Gasa R, Mrejen C, Lynn FC, et al. Induction of pancreatic islet cell differentiation by the neurogenin-neuroD cascade. Differentiation 2008;76:381–391 [DOI] [PubMed] [Google Scholar]
- 9.Gouzi M, Kim YH, Katsumoto K, Johansson K, Grapin-Botton A. Neurogenin3 initiates stepwise delamination of differentiating endocrine cells during pancreas development. Dev Dyn 2011;240:589–604 [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg LC, Lafon ML, Pedersen JK, et al. The transcriptional activity of Neurog3 affects migration and differentiation of ectopic endocrine cells in chicken endoderm. Dev Dyn 2010;239:1950–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith SB, Watada H, German MS. Neurogenin3 activates the islet differentiation program while repressing its own expression. Mol Endocrinol 2004;18:142–149 [DOI] [PubMed] [Google Scholar]
- 12.Wang S, Jensen JN, Seymour PA, et al. Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proc Natl Acad Sci U S A 2009;106:9715–9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S, Yan J, Anderson DA, et al. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Dev Biol 2010;339:26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinney SE, Oliver-Krasinski J, Ernst L, et al. Neonatal diabetes and congenital malabsorptive diarrhea attributable to a novel mutation in the human neurogenin-3 gene coding sequence. J Clin Endocrinol Metab 2011;96:1960–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubio-Cabezas O, Jensen JN, Hodgson MI, et al. Permanent Neonatal Diabetes and Enteric Anendocrinosis Associated With Biallelic Mutations in NEUROG3. Diabetes 2011;60:1349–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Cortina G, Wu SV, et al. Mutant neurogenin-3 in congenital malabsorptive diarrhea. N Engl J Med 2006;355:270–280 [DOI] [PubMed] [Google Scholar]
- 17.Jensen JN, Rosenberg LC, Hecksher-Sørensen J, Serup P. Mutant neurogenin-3 in congenital malabsorptive diarrhea. N Engl J Med 2007;356:1781–1782 [DOI] [PubMed]
- 18.Huang HP, Liu M, El-Hodiri HM, Chu K, Jamrich M, Tsai MJ. Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol Cell Biol 2000;20:3292–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith SB, Gasa R, Watada H, Wang J, Griffen SC, German MS. Neurogenin3 and hepatic nuclear factor 1 cooperate in activating pancreatic expression of Pax4. J Biol Chem 2003;278:38254–38259 [DOI] [PubMed] [Google Scholar]
- 20.Mellitzer G, Bonné S, Luco RF, et al. IA1 is NGN3-dependent and essential for differentiation of the endocrine pancreas. EMBO J 2006;25:1344–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson KR, Torres CA, Solomon K, et al. Cooperative transcriptional regulation of the essential pancreatic islet gene NeuroD1 (beta2) by Nkx2.2 and neurogenin 3. J Biol Chem 2009;284:31236–31248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soyer J, Flasse L, Raffelsberger W, et al. Rfx6 is an Ngn3-dependent winged helix transcription factor required for pancreatic islet cell development. Development 2010;137:203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith SB, Qu H-Q, Taleb N, et al. Rfx6 directs islet formation and insulin production in mice and humans. Nature 2010;463:775–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arda HE, Benitez CM, Kim SK. Gene regulatory networks governing pancreas development. Dev Cell 2013;25:5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benitez CM, Qu K, Sugiyama T, et al. An integrated cell purification and genomics strategy reveals multiple regulators of pancreas development. PLoS Genet 2014;10:e1004645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rukstalis JM, Habener JF. Snail2, a mediator of epithelial-mesenchymal transitions, expressed in progenitor cells of the developing endocrine pancreas. Gene Expr Patterns 2007;7:471–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo J, Kilimnik G, Kim A, Guo C, Periwal V, Hara M. Formation of pancreatic islets involves coordinated expansion of small islets and fission of large interconnected islet-like structures. Biophys J 2011;101:565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jo J, Hörnblad A, Kilimnik G, Hara M, Ahlgren U, Periwal V. The fractal spatial distribution of pancreatic islets in three dimensions: a self-avoiding growth model. Phys Biol 2013;10:036009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kilimnik G, Kim A, Jo J, Miller K, Hara M. Quantification of pancreatic islet distribution in situ in mice. Am J Physiol Endocrinol Metab 2009;297:E1331–E1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller K, Kim A, Kilimnik G, et al. Islet formation during the neonatal development in mice. PLoS ONE 2009;4:e7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gasa R, Mrejen C, Leachman N, et al. Proendocrine genes coordinate the pancreatic islet differentiation program in vitro. Proc Natl Acad Sci U S A 2004;101:13245–13250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puri S, Hebrok M. Dynamics of embryonic pancreas development using real-time imaging. Dev Biol 2007;306:82–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hara M, Wang X, Kawamura T, et al. Transgenic mice with green fluorescent protein-labeled pancreatic beta -cells. Am J Physiol Endocrinol Metab 2003;284:E177–E183 [DOI] [PubMed] [Google Scholar]
- 34.Lee CS, Perreault N, Brestelli JE, Kaestner KH. Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev 2002;16:1488–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugiyama T, Benitez CM, Ghodasara A, et al. Reconstituting pancreas development from purified progenitor cells reveals genes essential for islet differentiation. Proc Natl Acad Sci U S A 2013;110:12691–12696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugiyama T, Rodriguez RT, McLean GW, Kim SK. Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proc Natl Acad Sci U S A 2007;104:175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gittes GK, Galante PE, Hanahan D, Rutter WJ, Debase HT. Lineage-specific morphogenesis in the developing pancreas: role of mesenchymal factors. Development 1996;122:439–447 [DOI] [PubMed] [Google Scholar]
- 38.Ejarque M, Cervantes S, Pujadas G, Tutusaus A, Sanchez L, Gasa R. Neurogenin3 cooperates with Foxa2 to autoactivate its own expression. J Biol Chem 2013;288:11705–11717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castaing M, Péault B, Basmaciogullari A, Casal I, Czernichow P, Scharfmann R. Blood glucose normalization upon transplantation of human embryonic pancreas into beta-cell-deficient SCID mice. Diabetologia 2001;44:2066–2076 [DOI] [PubMed] [Google Scholar]
- 40.Scharfmann R, Xiao X, Heimberg H, Mallet J, Ravassard P. Beta cells within single human islets originate from multiple progenitors. PLoS ONE 2008;3:e3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gosmain Y, Marthinet E, Cheyssac C, et al. Pax6 controls the expression of critical genes involved in pancreatic alpha cell differentiation and function. J Biol Chem 2010;285:33381–33393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sander M, Neubüser A, Kalamaras J, Ee HC, Martin GR, German MS. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev 1997;11:1662–1673 [DOI] [PubMed] [Google Scholar]
- 43.St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature 1997;387:406–409 [DOI] [PubMed] [Google Scholar]
- 44.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods 2005;2:905–909 [DOI] [PubMed] [Google Scholar]
- 45.Hadjantonakis A-K, Papaioannou VE. Dynamic in vivo imaging and cell tracking using a histone fluorescent protein fusion in mice. BMC Biotechnol 2004;4:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borden P, Houtz J, Leach SD, Kuruvilla R. Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Reports 2013;4:287–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kesavan G, Lieven O, Mamidi A, et al. Cdc42/N-WASP signaling links actin dynamics to pancreatic β cell delamination and differentiation. Development 2014;141:685–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyatsuka T, Kosaka Y, Kim H, German MS. Neurogenin3 inhibits proliferation in endocrine progenitors by inducing Cdkn1a. Proc Natl Acad Sci U S A 2011;108:185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flasse LC, Pirson JL, Stern DG, et al. Ascl1b and Neurod1, instead of Neurog3, control pancreatic endocrine cell fate in zebrafish. BMC Biol 2013;11:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mellitzer G, Martín M, Sidhoum-Jenny M, et al. Pancreatic islet progenitor cells in neurogenin 3-yellow fluorescent protein knock-add-on mice. Mol Endocrinol 2004;18:2765–2776 [DOI] [PubMed] [Google Scholar]
- 51.Sanders TA, Llagostera E, Barna M. Specialized filopodia direct long-range transport of SHH during vertebrate tissue patterning. Nature 2013;497:628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]