

Abstract
Stroke is the leading cause of adult disability with ~80% being ischemic. Stem cell transplantation has been shown to improve functional recovery. However, the overall survival and differentiation of these cells is still low. The infarct cavity is an ideal location for transplantation as it is directly adjacent to the highly plastic peri-infarct region. Direct transplantation of cells near the infarct cavity has resulted in low cell viability. Here we deliver neural progenitor cells derived from induce pluripotent stem cells (iPS-NPC) to the infarct cavity of stroked mice encapsulated in a hyaluronic acid hydrogel matrix to protect the cells. To improve the overall viability of transplanted cells, each step of the transplantation process was optimized. Hydrogel mechanics and cell injection parameters were investigated to determine their effects on the inflammatory response of the brain and cell viability, respectively. Using parameters that balanced the desire to keep surgery invasiveness minimal and cell viability high, iPS-NPCs were transplanted to the stroke cavity of mice encapsulated in buffer or the hydrogel. While the hydrogel did not promote stem cell survival one week post-transplantation, it did promote differentiation of the neural progenitor cells to neuroblasts.
Keywords: Hydrogel, Hyaluronic acid, Induced pluripotent stem cells, Stroke
1. Introduction
Stroke is the leading cause of long-term adult disability and the number of stroke victims/survivors is expected to increase in the future. [1–2] Ischemic stroke occurs when there is a decrease in cerebral blood flow due to an embolus or local thrombosis. This results in tissue damage that includes the loss of neurons, astrocytes, oligodendrocytes, and endothelial cells. Recombinant tissue plasminogen activator (tPA) is the only approved therapy for stroke. This drug, which targets cell death in stroke, must be administered in the first 3 hours and is clinically applied to only 5% of stroke patients. There is no clinically approved therapy to promote stroke recovery. [3]
Studies have shown that transplanting stem cells can improve functional behavior in stroke models. [4–6] A variety of approaches ranging from delivering pluripotent cells adjacent to the infarct to delivering them directly into the infarct have been attempted. In the former strategy, one study showed approximately 8% of transplanted cells survived 4 weeks post-transplantation and this led to some functional recovery in Mongolian gerbils. [7] In the latter strategy, delivering cells into the infarct cavity resulted in approximately 4% survival 2 weeks post-transplantation. [8] This type of direct stem cell transplantation to the infarct results in much lower cell viability compared to cells grafted into the non-ischemic tissue. [9] This is likely due to the inflammatory environment and lack of blood vessels in and near the infarct. [10] However, the infarct cavity is an ideal site to directly inject stem cells because of its compartmentalized nature and close vicinity to the area of greatest neuroplasticity, the peri-infarct tissue. [11] Furthermore, the infarct represents a tissue cavity that can accept a stem cell injection without damage to normal or intact brain. An alternative to direct transplantation is to deliver the cells encapsulated in a protective scaffold. Previous studies have used Matrigel, particles and other scaffolds as matrices to support the survival and differentiation of progenitor cells in the infarct cavity. [8, 12–16] Delivering the cells via a scaffold significantly improved cell viability (two-fold) versus cell only transplantation controls. [8] Furthermore, this approach has been shown to reduce lesion volume, [15–16] and promote functional recovery. [14–16] Despite these improvements, the transplanted cell survival and subsequent differentiation is still low. Functional recovery correlates to the formation of new neuronal connections in the peri-infarct tissue [11, 17]. Thus, promoting differentiation of the precursor cells to neurons is a major aim in transplantation studies.
Proposed cell types for regeneration of the brain after stroke include the use of neural stem/progenitor cells (NPCs) [7, 18–19], immortalized neural cell lines [20–22], hematopoietic/endothelial progenitor and stromal cells from bone marrow and other cell sources. [23–24] The advantages and disadvantages of each cell type have been nicely chronicled in a recent review. [25] Some of the potential pitfalls range from the ethical issues of NPCs derived from embryonic stem cells to the potential of malignant transformation from immortalized cell lines. An alternative source of NPCs include induced pluripotent stem cells (iPSCs). [26–27] These cells are created by reprogramming somatic cells to a pluripotent state through the ectopic expression of specific transcription factors. First generated in 2006, iPSCs have promise for patient-specific cell therapies, but more characterization is needed to take full advantage of their therapeutic potential. [28] Herein, we report on a hyaluronic acid based hydrogel modified with the cell adhesion peptide RGD as a vehicle for iPS-NPCs after stroke, which induces minimal inflammation and promotes differentiation of the delivered cells in the infarct cavity.
2. Results and Discussion
2.1. Hydrogel Design
Hydrogel materials are ideal as a vehicle for stem cell transplantation in vivo. Hydrogels can provide a protective environment to enhance cell survival, while delivering bioactive signals to aid in transplanted stem cell differentiation and recruit endogenous cells to aid in regeneration. [29] In particular, for stem cell transplantation into the brain after ischemic stroke these hydrogels must fit several criteria. First, they must be injectable through a thin needle to cause the least amount of damage to the brain and to allow delivery into the potentially deep brain sites of ischemic stroke. Second, the hydrogel must gel slowly such that sufficient time is available for slow injection speeds (0.3 – 0.9 μl/min). Third, the hydrogel must not swell during or after gelation to prevent further damage to the brain, and finally these hydrogels must promote stem cell survival during injection and post transplantation. We propose to use hyaluronic acid hydrogel based hydrogels crosslinked through a Michael Addition reaction of acrylates present in the HA backbone with dicysteines present in a matrix metalloproteinase crosslinking peptide. Hyaluronic acid (HA) is a unique hydrogel candidate for neural applications due to its non-immunogenic properties and ability to hydrate. [30–31] It is a naturally occurring glycosaminoglycan and, when used as a polymer for hydrogels, has shown an ability to effect neural differentiation. [32–33] Furthermore, the hydrogel formulations from this system can be precisely controlled with reproducibility unseen in other systems like Matrigel. [34] We have previously shown that a HA-based hydrogel can support the growth of encapsulated stem cells and that hydrogel properties (HA%, crosslinking density, bioactive signal concentration/distribution) can be used to direct cell spreading, migration and proliferation. [35–36] For example, cells in stiffer gels (higher HA% and/or higher crosslinking density) had less spreading, migration and slower proliferation rates. Whereas increasing concentrations of RGD produced earlier and more abundant encapsulated cell spreading and migration. Michael addition was chosen because this chemistry has been extensively used for in situ cell encapsulation showing high cell viability and can have slow reaction kinetics to allow for sufficient time of injection. [37–39]
Hyaluronic acid based hydrogels were synthesized to contain the adhesion peptide, RGD, and were crosslinked with either matrix-metalloproteinase (MMP) degradable peptides or non-MMP degradable peptides (Figure 1A). The same hydrogel formulation could not be used with the two crosslinkers to obtain the same mechanical properties because of different reaction kinetics with the acrylated hyaluronic acid. The amount of each crosslinker type was varied until the hydrogels had similar storage modulus of ~300 Pa (d, nd. “soft”, Figure 1B, Figure S1). The hydrogels were specifically engineered for this modulus because that is the approximate stiffness of the brain. [40] A stiffer hydrogel was also made with the non-degradable peptide with a storage modulus greater than 1000 Pa. Hydrogel moduli were confirmed with a plate-to-plate rheometer using a constant strain of 1% between 0.1 and 10 Hz (Figure 1C). The storage moduli between gels “d” and “nd ‘soft’” were not statistically different (p>0.05).
Fig. 1.

(A) The hydrogel contains acrylated hyaluronic acid, peptide crosslinker, and an RGD-containing peptide. The crosslinker is MMP-degradable (d.) or MMP-nondegradable (nd.). The hydrogel is made by pre-reacting the HA with the RGD peptide and then mixing in the cross-linker and cells. (B) Three hydrogel formulations were made with (C) rheology confirming their mechanical properties.
2.2. Inflammatory Response to Hydrogel
A large inflammatory response to the initial stroke is one of the biggest problems facing stroke patients. The lack of blood flow from the stroke ultimately leads to the activation and infiltration of leukocytes, microglia/macrophages, and astrocytes. [41] These inflammatory cells can further damage brain tissue directly and secrete factors that stimulate an even larger response. [42–44] Our approach is to deliver a cell containing hydrogel directly to the stroke cavity when the acute reaction begins to subside to give the cells the best chance of survival and differentiation. To ensure that the injection of our hyaluronic acid does not itself induce an inflammatory reaction that could further aggravate the already injured stroked brain, our hydrogel formulations were injected into the brain and the inflammatory response around the implant analyzed.
The three hydrogel conditions tested enabled us to isolate the effect of crosslinker degradability (d. vs. nd. “soft”) and the effect of mechanical stiffness (nd. “soft” vs. nd. “stiff”) on the mouse’s inflammatory response. This response to different hydrogel formulations was determined by transplanting the hydrogels into the striatum of naïve (non-stroked) C57BL/6 mice. While downstream in vivo experiments with encapsulated cells include the RGD-containing adhesion peptide, these inflammatory response experiments did not. The adhesion peptide has been shown to be mildly immunogenic and could potentially cloud the effect of crosslinker degradability and mechanical stiffness. [45] The main buffer component of the hydrogel (0.3 M HEPES) was injected as a comparative control. Two weeks after transplantation, the mice were sacrificed and the brains fixed and sectioned for analysis. Parallel series of sections spanning the transplantation zone were stained for reactive astrocytes (GFAP), a scar forming cell in the adult brain, and microglia (IBA1), the brain’s only endogenous inflammatory cell (Figure 2A). Three images of the peri-infarct tissue were taken for each section as well as one image for the contralateral striatum of the section (Figure 2B–E). The GFAP and IBA1 ipsilateral signal from each image was normalized to the contralateral image for each corresponding section. [46] The reactive astroctye signal in the nd. “stiff” condition was found to be statistically higher than the HEPES control condition (p<0.01), the d. condition (p<0.001), and the nd. “soft” condition (p<0.05, Figure 2J.). No significant IBA1 signal was observed between any of the 4 injected conditions. These results indicate that specific hyaluronic acid hydrogel formulations do not cause more inflammation than buffer alone. While the degradability of the crosslinker does not have an effect on the inflammatory response in the brain the mechanical properties do increase the inflammatory response to the hydrogel. However, the naïve brain has significantly less protease and hyaluronidase expression than the stroke cavity and thus the inflammatory response could be different when more significant gel degradation occurred. [47]
Fig. 2.

Four different conditions were injected into the striatum of naïve or stroked mice: HEPES buffer, d. hydrogel, nd. “soft” hydrogel and nd. “stiff” hydrogel. (A) Overview of entire brain section showing contralateral and ipsilateral regions of interest. (B–E) Images were taken on the contralateral side of the brain as well as the area (B′–E′) directly adjacent to the injection site (ipsilateral) in naïve mice. (J) GFAP and (K) IBA1 signal was quantified. The same four conditions were then injected into the infarct cavity of stroked mice. Images were taken on the (F–I) contralateral and (F′–I′) ipsilateral of the brain for (L) GFAP and (M) IBA1 quantification. Scale bar = 50 μm
To further investigate the inflammatory response to hydrogel implantation, mice were given a cortical photothrombotic stroke and the three different hydrogel formulations, along with the HEPES control, were injected into the stroke cavity one week later. The site of injection as well as the time of injection was chosen in accordance with the proposed stem cell transplantation site and time. The infarct cavity is a good location candidate for stem cell/biomaterial delivery because it contains loose tissue and is adjacent to the area of greatest neuroplasticity, the peri-infarct tissue. [11] While previous studies have transplanted their cell/hydrogel constructs at various time points after stroke ( i.e. 1 day, 1 week, 3 weeks), we chose to transplant one week after stroke induction. [14–16] Not only does the strong post-stroke inflammatory response begin to subside, but brain tissue repair in the form of angiogenesis and neurogenesis peaks at this one week time point. [48–50] The mice were sacrificed two weeks after implantation and the brains fixed, sectioned and stained for GFAP and IBA1 for further analysis. Three images of the peri-infarct tissue and one contralateral image were taken of each section (Figure 2F–I). The contralateral images were used to normalize ipsilateral images for each section. There was no significant difference in reactive astrocyte signals between conditions (Figure 2L). The microglia signal was significantly higher in the nd. “stiff” condition versus the HEPES control (p<0.01), the d. condition (p<0.001), and the nd. “soft” condition (p<0.05, Figure 2M). Whereas the nd. “stiff” hydrogel had an increased GFAP signal in the naïve model, it had an increased IBA1 signal in the stroke model. These results indicate that hydrogels with a storage modulus of ~300 Pa is more suitable for the brain.
The brain’s response to injury and disease is complex and involves multiple cell types. Reactive astrocytes play a key role in several processes ranging from forming scars around the region of high inflammation to inhibiting axon regeneration to protecting existing neurons and neural function. [51–52] The reactive astrocytes seen in the naïve model injections are only due to the hydrogels transplanted. Our results show that the “stiff” hydrogel condition resulted in an increase in reactive astrocytes. Cell mechanosensitivity plays a key role in cell pathology. Thus, implants that do not have similar mechanical properties to the native tissue may invoke a greater inflammatory response. [53] This finding agrees with other published reports on the effect of mechanical stiffness on implants in the central nervous system. [54] However, the GFAP signal seen in the stroke model is due to both the transplantation and the stroke itself. The effect of the different hydrogel formulations on the astrocyte response was likely masked by the extreme provocation of reactive astrocytosis from the initial stroke stimulus. Thus, the hydrogel effect on astrocyte activation was more pronounced in the naïve stroke model.
Activated by brain ischemia, microglia are phagocytes that remove damaged cells. [41, 55] Once activated, the microglia level diminishes to control levels by 2–3 weeks. However, microglia can be sensitized and have a molecular memory of this original stimulation event. [56–57] The hydrogels in the stroke model were superimposed on these sensitized microglia. Thus, the effect of the hydrogels was more pronounced in the stroke model vs. the naïve model and we observed the increased microglia signal in the stiffest hydrogel condition. The results from the naïve and stroke model experiments both confirm that the mechanical properties of the “d.” and nd. “soft” hydrogels induced a smaller inflammatory response.
2.3. Cell viability
Transplanting stem cells directly to the infarct cavity via a hydrogel carrier is a promising therapy, but a relatively new approach. Improving transplanted cell viability is one of the main motivations behind delivering cells via a biomaterial. Existing studies using this approach have primarily used the middle cerebral artery occlusion stroke model in rats. [13–16] A variety of materials (PLGA particles, Matrigel, Collagen) have been utilized and show that biomaterials improve transplanted cell survival. Another study utilizing a photothrombotic stroke model in mice reports a two-fold increase in transplanted cell survival. All of these studies focus on the biomaterial aspect of the therapy. However, beyond the type of hydrogel carrier, previous research has also shown that the injection process itself can have a large impact on cell viability.[58] Ultimately, both of these variables can play a role in the cell viability seen in vivo. These parameters vary from group to group and the effect on cell viability is likely biomaterial dependent as well. Rheological properties and the viscous nature of the materials can provide different protective effects against the sheer stress experiences by the cells during the injection through the needle. Thus, we wanted to thoroughly examine this injection process in vitro with our hydrogel system to look for additional ways of improving the transplanted cell viability.
The injection parameters should balance the desire to keep the surgery as minimally invasive as possible with the desire to deliver as many viable cells as possible. [59] While high needle gauges and fast infusion speeds can increase the sheer stress experienced by the cells as they pass through the needle, a viscous hydrogel can provide a protective effect. Thus, a variety of injection parameters (needle gauge, infusion speed, cell density) relevant to our in vivo injection setup were investigated in vitro to determine their effect on iPS-NPC viability. Each parameter was tested by suspending the cells in buffer or the hydrogel solution “d.” (300 Pa). The same Hamilton syringe/syringe pump setup that was used for the in vivo hydrogel injections described in 2.2. was used for these studies (Figure 3A). Viability was measured by quantifying live/dead cells immediately after the injection process through Trypan Blue staining. While keeping the cell density (33k/μL) and needle gauge (30 gauge) constant, there was no significant difference among the infusion speeds tested (0.3, 0.6, 0.9 μL/min) for cells injected with or without the hydrogel (Figure 3B). Additionally, encapsulating cells in the viscous hydrogel solution did not improve viability at any of the tested infusion speeds.
Fig. 3.
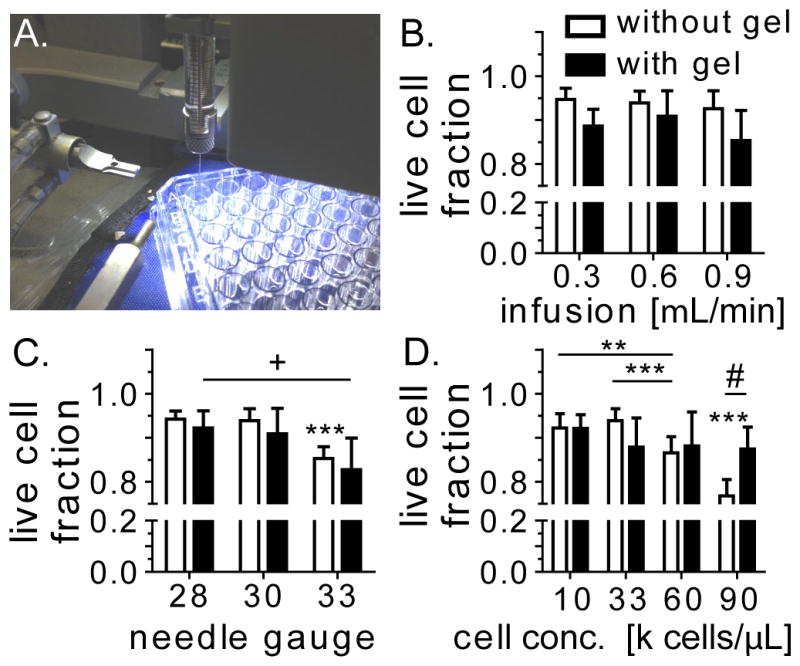
(A) The effect of the injection process on iPS-NPC viability was tested using the same setup used on in vivo experiments. Viability of cells encapsulated in media or hydrogel “d.” were tested at different (B) infusion speeds, (C) needle gauge, and (D) cell concentration. +: between w/gel, * between w/o gel, # between w/ and w/o gel
There was no significant difference between 28 and 30 gauge needles for iPS-NPCs (33k cells/μL) injected in buffer at an infusion speed of 0.6 μL/min (Figure 3C). However, a 33 gauge needle significantly reduced cell viability compared to both other needle types (p<0.001), whereas cells encapsulated inside the hydrogel only had decreased viability between the 28 and 33 gauge needle (p<0.05).
Cell density was also varied, while keeping the infusion speed and needle gauge constant at 0.6 μL/min and 30 gauge, respectively. For cells suspended in buffer, there was a significant difference in cell viability between the 10k cells/μL condition and the 60k cells/μL condition (p<0.01) as well as the 90k cells/μL condition (p<0.001). Additionally, there was a significant decrease in viability between 33k cells/μL and 60k cells/μL (p<0.001) and 60k cells/μL and 90k cells/μL (p<0.001). However, there was no significant viability decrease across cell densities when cells were encapsulated in a hydrogel. Additionally, the hydrogel did significantly increase cell viability at 90k cells/μL versus cells suspended in the buffer (p<0.05). The viscous nature of the hydrogel could be shielding the cells and the reason behind the improved viability observed at the higher needle gauge and cell densities. Based on these results, an injection protocol utilizing a 30 gauge needle and 0.6 μL/min infusion speed with 33,000 cells/μL in the hydrogel was chosen to keep cell viability >90% with as minimally invasive a procedure as possible.
With the hydrogel type (condition “d.”) and injection parameters (30 gauge, 0.6 mL/min, 33k cells. uL) finalized, immune-compromised mice were chosen to complete the cell viability studies. Alternative mouse models require immunosuppression via drugs like cyclosporine. However, the brain levels of these drugs are variable due to the blood brain barrier and may have an effect on the stroke. [60–61] The NSG mice were given a cortical stroke and 100,000 iPS-NPCs were delivered to the stroke cavity in 3 μL of buffer or “d.” hydrogel with 300 μM RGD one week later. The hydrogel mechanical properties do not significantly change with the addition of cells (Figure S2). Thus, the same “d.” hydrogel formulation used in the naïve model injections were used in these cell viability studies. The cells were transplanted using a 30 gauge needle at an infusion speed of 0.6 μL/min. One week after transplantation, the mice were sacrificed and the brain isolated for further analysis. Serial sections covering the transplantation zone were mounted onto slides and stained for GFAP and IBA1. Three ipsilateral images of the peri-infarct tissue and one contralateral image were taken for each section (Figure 4A–B). GFAP and IBA1 ipsilateral image signal was normalized to the contralateral image of each section. While there was no statistical significance in GFAP signal between the cell only or cell + hydrogel condition, there was a decrease in IBA1 signal for the cell + hydrogel condition (p<0.01, Figure 4C–D). This indicates that the hydrogel decreases the inflammatory response to the transplantation by acting to shield the endogenous tissue from recognizing the transplanted human cells. This could play a potential role in promoting transplanted cell viability.
Fig. 4.

100,000 iPS-NPCs injected into the infarct cavity of stroked NSG mice suspended in buffer or hydrogel “d.” (A–B) The inflammatory response of the peri-infarct tissue was quantified and normalized to the contralateral side for (C) GFAP and (D) IBA1. (E–G) Human nuclei were stained and (H) quantified for both conditions. Scale bar = 50 μm
Sections spanning the transplantation zone were stained for human nuclei to investigate transplanted iPS-NPC viability (Figure 4E–G). Nuclei in every 10 sections were manually counted and extrapolated to determine the total number of viable cells. In the cell only group, an average of 30,000 cells survived one week after transplantation compared to 38,000 for the cell + hydrogel condition (Figure 4H). There was no statistical difference between the two groups (p>0.05). The nature of the animal setup, NSG mice, could have played a role in masking the protective capabilities of transplanting iPS-NPCs encapsulated in a hydrogel matrix. The superior xenografting ability of this immunodeficient mouse strain may have clouded the protective effects of the hydrogel that have been previously reported. [62–63] While it was understood that the cell viability in both conditions would be improved in immunodeficient mice versus immunosuppressed mice, we thought the protective effect of the hydrogel would still be shown. Previous studies comparing the effect of a polymer matrix to directly transplanted cells used regular immunosuppressed mice and rats. [8, 14–16] Additionally, the distribution of transplanted cells was different between conditions. Qualitatively, cells transplanted in buffer were more compacted and denser, whereas cells were more scattered throughout the transplantation area in the cell + hydrogel condition. While the hydrogel did not promote statistically significant increase in transplanted cell viability, this cell distribution could potentially play a role in cell differentiation.
2.4. Stem cell differentiation post transplantation
In addition to keeping transplanted cells alive, we aim to promote the differentiation of the NPCs to neurons in an effort to help replace the damaged tissue. This would produce a positive impact in stroke recovery through several mechanisms. First, it would mean that the transplanted cells are integrating into the host tissue. Second, the exogenous cells could help promote endogenous repair like axonal sprouting [64] and angiogenesis. [65] Various time points post-transplantation, ranging from one to eight weeks, have been utilized to look at transplanted cell differentiation in the infarct cavity. The majority of NPCs delivered into the infarct via a collagen matrix began to adopt a neuronal fate one week post-transplantation. [14] Another study, utilizing Matrigel and an eight week timepoint, observed transplanted NPCs positive for neuroblasts (DOUBLECORTIN) and a mature neuron marker (MAP2). [16]
We were interested in looking at the differentiation of the viable transplanted cells at the one week time point used for viability. Sections from both conditions were stained with three different markers: SOX2, DOUBLECORTIN (DCX), and NF200 to assess the extent of differentiation. SOX2 is a transcription factor associated with the multipotent progenitor fate. [66] DCX is a microtubule associated protein that is found in immature neurons and NF200 is an epitope on the neurofilament protein in mature neurons. [67–68] Images were taken in the middle of the transplantation zone for further analysis. In the cell only condition, transplanted cells were positive for SOX2 and DOUBLECORTIN and negative for NF200 (Figure 5). Similar images were taken for the cell + hydrogel condition and transplanted cells were also positive for SOX2 and DOUBLECORTIN while being negative for NF200 (Figure 5). To quantify these stains, the level of signal in the desired stain (SOX2 or DCX) was normalized to the number of human nuclei. The number of human nuclei in the image was determined by looking at the DAPI stained images, as human nuclei and mouse nuclei have a different nuclear morphology in this stain. While there was no difference in SOX2 signal between the two conditions (Figure 5Y, p>0.05), there was a significant increase in DOUBLECORTIN signal in the cell + hydrogel condition (Figure 5Z, p<0.05). Colocalization analysis was done to determine whether the DOUBLECORTIN signal observed was from our transplanted GFP-positive cells or from the native tissue (Figure 6A–C). The Manders coefficient of colocalization was significantly higher for the cell + hydrogel condition (p<0.0001), which indicates several things. First, the DCX signal observed from the cell only condition is not from the transplanted NPC’s but from the mouse itself. Conversely, the majority of the DOUBLECORTIN positive signal observed in the cell + hydrogel condition is from the transplanted NPC’s differentiating to neuroblasts. This is important because the transplanted cells are not only able to survive, but differentiate toward a neuronal phenotype. It was easier for endogenous cells to migrate into the cell only infarct/transplantation area because that condition did not have the physical presence of the hydrogel. As the hydrogel degrades over time, more endogenous cell infiltration could potentially occur. Secondly, the increase in transplanted cell differentiation in the hydrogel condition could be due to the mechanical support that the polymer matrix gives the cells. It has been previously shown that mechanics play a large role in the differentiation of progenitor cells. [40, 69–71] In vitro, our hydrogel system promotes the differentiation of iPS-NPC to a neuronal phenotype compared to cells plated in two dimensions (Figure S3). Additionally, the distribution of cells in the implant zone, as seen in Figure 4, could play a role in the increased DOUBLECORTIN signal. The transplanted cells have more room to spread due to the increased space provided within the hydrogel.
Fig. 5.

Transplantation zone was stained for markers of the GFP-labeled (A–H) iPS-NPCs (SOX2), (I–P) neuroblasts (DOUBLECORTIN, DCX), and (Q–X) mature neurons (NF200). (Y) SOX2 and (Z) DCX signal was quantified and with an increased DOUBLECORTIN signal seen in the cell + hydrogel condition. Scale bar = 100 μm
Fig. 6.
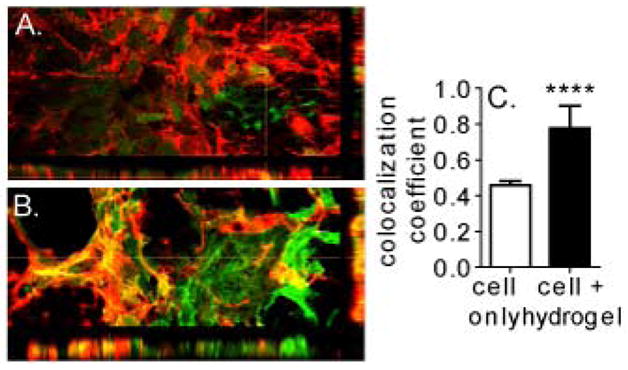
3-dimensional reconstruction of (A) cell only and (B) cell + hydrogel sections stained for GFP-labeled transplanted cells and DOUBLECORTIN. (C) Colocalization analysis shows that the majority of DCX positive signal seen in cell + hydrogel condition is from transplanted cell differentiation.
3. Conclusion
In this study, we investigated the different components involved in transplanting neural progenitor cells into the brain post-stroke in an effort to improve transplanted cell viability and differentiation. First, we looked at the inflammatory response of the brain due to the hydrogel itself and found that the mechanical properties can affect the response. Second, the actual injection process was studied with large needle gauges and high cell concentrations decreasing cell viability. After picking parameters that balanced the desires to have a minimally invasive surgery with maximum cell viability, we injected iPS-NPCs into the infarct cavity of stroked NSG mice with or without a hydrogel. While the hydrogel did not promote increased cell viability one week after transplantation, it did promote the differentiation of the viable cells. Our results indicate that the hydrogel can play a role in promoting the neuronal differentiation of transplanted NPCs in the brain of stroked mice.
4. Experimental Section
4.1. Cell culture
hiPS-NPCs were cultured as previously described. [72] A self-inactivating lentivirus with GFP under the CMV promoter was used to transduce the cells. [46] The cells were maintained in Dulbecco’s modified eagle’s medium:F12 (DMEM:F12, Sigma-Aldrich) supplemented with 1x B27 (Sigma-Aldrich, St. Louis, MO), 1x N2 (Sigma-Aldrich), epidermal growth factor (50 ng/mL, EGF, Sigma-Aldrich), basic fibroblast growth factor (20 ng/mL, bFGF, Sigma-Aldrich), 1% penicillin/streptomycin (Invitrogen, Grand Island, NY), and 0.1% primocin (InVivoGen, San Diego, CA). They were cultured at 370C with 5% CO2 using standard protocols.
4.2. Hyaluronic acid modification and hydrogel gelation
Hyaluronic acid (60,000 Da, Genzyme, Cambridge, MA) was functionalized with an acrylate group as previously described. [35] After dissolving the HA (2.0 g, 5.28 mmol) in water, it was reacted with adipic dihydrazide (ADH, 18.0 g, 105.5 mmol) in the presence of 1-ethyl-3-(dimethylaminopropyl) carbodiimide hydrochloride (EDC, 4.0 g, 20 mmol) overnight at a pH of 4.75. The hydrazide-modified hyaluronic acid (HA-ADH) was purified with decreasing amounts of NaCl (100, 75, 50, 25 mmol) for 4 hours each via dialysis (8,000 MWCO). After 2 days purifying against deionized water, the HA-ADH was lyophilized. The HA-ADH was re-suspended in 4-(2-hydroxyethyl)-1-piperazine ethane-sulfonic acid (HEPES) buffer (10 mM HEPES, 150 mM NaCl, 10 mM EDTA, pH 7.4) and reacted with N-acryloxysuccinimide (NHS-AC), 1.33 g, 4.4 mmol) overnight. After purifying the acrylated HA (HA-AC) via dialysis as described earlier via dialysis, it was lyophilized. The acrylate modification was found to be 12.19% via 1H NMR (D2O) by dividing the multplet peak at δ = 6.2 (cis and trans acrylate hydrogens) by the singlet peak of δ = 1.6 (singlet peak of acetyl methyl protons in HA).
The hydrogel was made by dissolving the lyophilized HA-AC in 0.3 M HEPES buffer for 15 minutes at 37°C. Studies with the immune-compromised mice contained 500 μM of the adhesion peptide Ac-GCGYGRGDSPG-NH2 (RGD, Genscript, Piscataway, NJ). The peptide was dissolved in 0.3 M HEPES and the appropriate amount was added to the dissolved HA-AC and reacted for 20 minutes. To crosslink the gels, an aliquot of the desired crosslinker (Ac-GCREGPQGIWGQERCG-NH2, MMP-degradable or Ac-GCREGDQGIAGFERCG-NH2, MMP-nondegradable) was dissolved in 0.3 M HEPES and added to the gel precursor solution. For rheometry, 40 μL of the hydrogel solution was pipetted onto, and sandwiched between two Sigmacote (Sigma-Aldrich) functionalized glass coverslips and allowed to gel at 37°C for 30 minutes. For viability and animal injections, the precursor was loaded into the Hamilton syringe directly after mixing in the desired crosslinking peptide.
4.3. Rheometry
Hydrogels were made without cells and allowed to swell overnight before being cut to size using an 8.0 mm biopsy punch. An 8 mm plate-to-plate rheometer (Physica MCR 301, Anton Paar, Ashland, VA) with an evaporation blocker system was used to measure the modulus with a frequency range of 0.1–10 rad/s under a 1% constrain at 37°C.
4.4. iPS-NPC viability injection
The iPS-NPCs were concentrated to the desired final injection concentration (ie 10k, 33k, 60k, 90k cells/μL) in media. For hydrogel conditions, the desired number of cells was encapsulated in 5 μL of the hydrogel “d.” precursor solution. The solution was loaded into a 25 μL Hamilton syringe (Hamilton, Reno, NV) with a 28 gauge needle (Hamilton, Reno, NV) and connected to a syringe pump. After swapping in the desired gauge needle (28, 30, 33), 3 μL of the solution was injected onto a 96 well non-tissue culture treated plate at the desired infusion speed (0.3, 0.6, 0.9 μL/min). 50 μL of media was added to the well to keep the cells/hydrogel hydrated. To quantify the cell viability, the solution was incubated in Trypan Blue and live/dead cells tabulated on a standard hemocytometer. For the hydrogel conditions, the material was incubated in 1500 U/mL of collagenase I (Worthington, Lakewood, NJ) for 15 minutes at 37°C to degrade the gel before Trypan Blue incubation. Each condition was performed in triplicate with each sample counted three times.
4.5. Mouse models
All procedures performed follow National Institutes of Health Animal Protection Guidelines and are approved by the UCLA Chancellor’as Animal Research Committee. Two different mouse models were used in this study: naïve mice and a cortical photothrombotic stroke model. For the naïve model, mice were initially anesthetized with 5% isoflurane and placed in a stereotactic setup. The mice were kept at 2.5% isoflurane in N2O:O2 for the duration of the surgery. A midline skin incision was made and a burr hole drilled through the skull at 1.5 mm anterior and 2 mm lateral left of the bregma. 5 μL of the hydrogel mixture was loaded into a 25 μL Hamilton syringe and 3 μL was injected in liquid form at a depth of 2.8 mm at a rate of 0.6 μL/min. The needle was withdrawn from the mouse 5 minutes after the injection was complete.
For the stroke model, the mice were anesthetized and loaded onto a stereotactic setup as described above. A midline incision was made and Rose Bengal (10 mg/mL, Sigma-Aldrich) was injected intraperitoneally at 10 μL/g of mouse body weight. After 5 minutes, a 2-mm diameter cold fiberoptic light source was centered at 0 mm anterior/1.5 mm lateral left of the bregma for 15 minutes. [73–74] One week post-stroke, 100,000 iPS-NPCs were transplanted into the stroke cavity with or without the hydrogel as described above. Two different types of mice were used for the stroke models: C57BL/6 mice (Charles River Laboratories, Wilmington, MA) and NSG immune compromised mice (Jackson Laboratories, Bar Harbor, ME). All mice were given sulfamethoxazole and trimethoprim oral suspension (TMS, 303 mL TMS/250 mL H20, Amityville, NY) every 5 days for the entire length of the experiment.
4.6. Mouse tissue processing and immunohistochemistry
Mice were sacrificed one week after transplantation as described in Zhong, et al. [8] Briefly, each mouse was deeply anesthetized with isoflurane and perfused with 50 mL of phosphate-buffered saline (PBS) and 20 mL of 4% paraformaldehyde (PFA). After isolation, the brain was post-fixed in 4% PFA for 2 hours and then placed in 30% sucrose for 48 hours. Next, the brains were cut in parallel series at a thickness of 14 μm and mounted onto coverglass. Slides not immediately stained were kept at −20°C.
To begin staining, each slide was rinsed with 1x PBS for 10 minutes at room temperature. Next, they were incubated in a blocking solution containing 1x PBS + 0.1% Triton X-100 and 2% normal donkey serum for one hour at room temperature. The slides were then incubated in the primary antibody at the appropriate dilution in 1x PBS + 0.1% Triton X-100 overnight at room temperature. After 3x 10 minute washes in 1x PBS, the slides were incubated in the appropriate secondary antibodies for 2 hours at room temperature. The slides were then counterstained with the nuclear marker 4′, 6-diamidino-2-phenylindole (DAPI, 1:500, Invitrogen) for 15 minutes at room temperature. After 3x 10 minute washes in 1x PBS, the slides were dehydrated in ascending ethanol baths, incubated in xylene and coverslipped. Primary antibodies were used as follows: goat anti-green fluorescent protein (GFP, 1:500, gift of Dr. Nathaniel Heinz, Rockefeller University), rat anti-glial fibrillary acidic protein (GFAP, 1:500, Life Sciences), rabbit anti-IBA1 (1:500, Wako), rabbit anti-SOX2 (1:300, Cell Signaling), guinea pig anti-DOUBLECORTIN (DCX, 1:2000, Milipore), rabbit anti-NF200 (1:500, Sigma), and mouse anti-human nuclei (1:500, Milipore). Secondary antibodies, matching the desired primary antibody host, conjugated to cyanine 2, cyanine 3, and cyanine 5 (1:200, Jackson Immuno Research, West Grove, PA) were used. To stain viable transplanted cells, the same immunohistochemistry protocol as described above was used, except for the following changes. Prior to the serum blocking step, the slides were incubated in an avidin and biotin block (Thermo Scientific) for 15 minutes each at room temperature with a 3x 10 minute 1xPBS wash in between. Following PBS washes after primary incubation, a Vectastain Elite ABC kit (Vector Laboratories) was used. This composed of a biotinylated secondary antibody incubation for 30 minutes, 3x 10 minute 1x PBS wash, ABC reagent incubation for 30 minutes, and a 3x 10 minute 1x PBS wash. Finally, the slides were incubated in a ImmPACT Dab chromogen working solution for 10 minutes (ImmPACT DAB Substrate kit, Vector Laboratories). To stain the biotynilated hyaluronic acid hydrogel, a tyramide signal amplification kit (Molecular Probes) was used.
4.7. Microscopy and quantification
The numbers of positively stained human nuclei were stained and manually counted. Six to twelve sections spanning the entire transplantation zone were quantified per mouse. For immune response quantification, three images around the peri-infarct area were imaged per slide section. An additional contralateral image was taken per section as well. Six sections spanning the entire transplantation zone were quantified per mouse. ImageJ was used to quantify the amount of positive signal by 1) converting the image to an 8 bit image, 2) thresholding the positive signal and 3) measuring the area fraction of positive signal. Each image was normalized to the corresponding thresholded contralateral image of that section. A similar thresholding technique was used to analyze differentiation stain images. Each image was normalized to the number of human nuclei in the image. [46]
4.8. Statistics
Statistical analysis was performed using Prism (GraphPad, San Diego, CA). Data was analyzed using either a 2-sample t test or a one way analysis of variance (ANOVA) test with a Tukey-Kramer post-test and a 95% confidence interval.
Supplementary Material
Acknowledgments
The authors would like to thank the California Institute for Regenerative Medicine (CIRM RT2-01881) and the National Insitutes of Health (NIH-NINDS R01NS079691, NIH-NHLBI R01HL110592, NIH-NIGMS 5P01GM099134-4) for funding. JL would like to thank the NIH funded Biotechnology Training Grant (T32 GM067555) for a predoctoral fellowship. JL would also like to thank Lina Nih for helpful discussions and advice as well as members of the Lowry Lab (Jessica Cinkornpumin and Soheila Azghadi) for preparing the iPS-NPCs and members of the Carmichael Lab (Andrew Brumm and Pouria Moshayedi) for their guidance and help with the animal experiments.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Taylor TN, Davis PH, Torner JC, Holmes J, Meyer JW, Jacobson MF. Stroke. 1996;27:1459. doi: 10.1161/01.str.27.9.1459. [DOI] [PubMed] [Google Scholar]
- 2.Broderick JP. Stroke. 2004;35:205. doi: 10.1161/01.STR.0000106160.34316.19. [DOI] [PubMed] [Google Scholar]
- 3.Donnan GA, Fisher M, Macleod M, Davis SM. Lancet. 2008;371:1612. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- 4.Buhnemann C, Scholz A, Bernreuther C, Malik CY, Braun H, Schachner M, Reymann KG, Dihne M. Brain. 2006;129:3238. doi: 10.1093/brain/awl261. [DOI] [PubMed] [Google Scholar]
- 5.Hicks AU, Lappalainen RS, Narkilahti S, Suuronen R, Corbett D, Sivenius J, Hovatta O, Jolkkonen J. European Journal of Neuroscience. 2009;29:562. doi: 10.1111/j.1460-9568.2008.06599.x. [DOI] [PubMed] [Google Scholar]
- 6.Daadi MM, Lee SH, Arac A, Grueter BA, Bhatnagar R, Maag AL, Schaar B, Malenka RC, Palmer TD, Steinberg GK. Cell Transplant. 2009;18:815. doi: 10.3727/096368909X470829. [DOI] [PubMed] [Google Scholar]
- 7.Mizusawa H, Ishibashi S, Sakaguchi M, Kuroiwa T, Yamasaki M, Kanemura Y, Shizuko I, Shimazaki T, Onodera M, Okano H. Journal of Neuroscience Research. 2004;78:215. doi: 10.1002/jnr.20246. [DOI] [PubMed] [Google Scholar]
- 8.Zhong J, Chan A, Morad L, Kornblum HI, Fan GP, Carmichael ST. Neurorehab Neural Re. 2010;24:636. doi: 10.1177/1545968310361958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly S, Bliss TM, Shah AK, Sun GH, Ma M, Foo WC, Masel J, Yenari MA, Weissman IL, Uchida N, Palmer T, Steinberg GK. P Natl Acad Sci USA. 2004;101:11839. doi: 10.1073/pnas.0404474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grabowski M, Johansson BB, Brundin P. Exp Neurol. 1994;127:126. doi: 10.1006/exnr.1994.1086. [DOI] [PubMed] [Google Scholar]
- 11.Carmichael ST. Annals of Neurology. 2006;59:735. doi: 10.1002/ana.20845. [DOI] [PubMed] [Google Scholar]
- 12.Uemura M, Refaat MM, Shinoyama M, Hayashi H, Hashimoto N, Takahashi J. Journal of Neuroscience Research. 2010;88:542. doi: 10.1002/jnr.22223. [DOI] [PubMed] [Google Scholar]
- 13.Bible E, Chau DYS, Alexander MR, Price J, Shakesheff KM, Modo M. Biomaterials. 2009;30:2985. [Google Scholar]
- 14.Yu HW, Cao B, Feng MY, Zhou Q, Sun XD, Wu SL, Jin SZ, Liu HW, Jin LH. Anatomical Record-Advances in Integrative Anatomy and Evolutionary Biology. 2010;293:911. doi: 10.1002/ar.20941. [DOI] [PubMed] [Google Scholar]
- 15.Matsuse D, Kitada M, Ogura F, Wakao S, Kohama M, Kira J, Tabata Y, Dezawa M. Tissue Eng Pt A. 2011;17:1993. doi: 10.1089/ten.TEA.2010.0585. [DOI] [PubMed] [Google Scholar]
- 16.Jin KL, Mao XO, Xie L, Galvan V, Lai B, Wang YM, Gorostiza O, Wang XM, Greenberg DA. J Cerebr Blood F Met. 2010;30:534. doi: 10.1038/jcbfm.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chopp M, Zhang RL, Zhang ZG. Neuroscientist. 2005;11:408. doi: 10.1177/1073858405278865. [DOI] [PubMed] [Google Scholar]
- 18.Steinberg GK, Bliss T, Guzman R, Daadi M. Stroke. 2007;38:817. doi: 10.1161/01.STR.0000247888.25985.62. [DOI] [PubMed] [Google Scholar]
- 19.Steinberg GK, Kelly S, Bliss TM, Shah AK, Sun GH, Ma M, Foo WC, Masel J, Yenari MA, Weissman IL, Uchida N, Palmer T. P Natl Acad Sci USA. 2004;101:11839. doi: 10.1073/pnas.0404474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andrews PW, Damjanov I, Simon D, Banting GS, Carlin C, Dracopoli NC, Fogh J. Laboratory Investigation. 1984;50:147. [PubMed] [Google Scholar]
- 21.Trojanowski JQ, Mantione JR, Lee JH, Seid DP, You T, Inge LJ, Lee VMY. Exp Neurol. 1993;122:283. doi: 10.1006/exnr.1993.1128. [DOI] [PubMed] [Google Scholar]
- 22.Pleasure SJ, Lee VMY. Journal of Neuroscience Research. 1993;35:585. doi: 10.1002/jnr.490350603. [DOI] [PubMed] [Google Scholar]
- 23.Chopp M, Li Y, Chen J, Chen XG, Wang L, Gautam SC, Xu YX, Katakowski M, Zhang LJ, Lu M, Janakiraman N. Neurology. 2002;59:514. doi: 10.1212/wnl.59.4.514. [DOI] [PubMed] [Google Scholar]
- 24.Low WC, Xiao J, Nan ZH, Motooka Y. Stem Cells Dev. 2005;14:722. doi: 10.1089/scd.2005.14.722. [DOI] [PubMed] [Google Scholar]
- 25.Bliss TM, Andres RH, Steinberg GK. Neurobiol Dis. 2010;37:275. doi: 10.1016/j.nbd.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Cell. 2007;131:861. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 27.Patterson M, Chan DN, Ha I, Case D, Cui YY, Van Handel B, Mikkola HKA, Lowry WE. Cell Research. 2012;22:178. doi: 10.1038/cr.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamanaka S, Takahashi K. Cell. 2006;126:663. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 29.Chai C, Leong KW. Mol Ther. 2007;15:467. doi: 10.1038/sj.mt.6300084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allison DD, Grande-Allen KJ. Tissue Eng. 2006;12:2131. doi: 10.1089/ten.2006.12.2131. [DOI] [PubMed] [Google Scholar]
- 31.Fraser JRE, Laurent TC, Laurent UBG. J Intern Med. 1997;242:27. doi: 10.1046/j.1365-2796.1997.00170.x. [DOI] [PubMed] [Google Scholar]
- 32.Hou SP, Xu QY, Tian WM, Cui FZ, Cai Q, Ma J, Lee IS. J Neurosci Meth. 2005;148:60. doi: 10.1016/j.jneumeth.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 33.Wang YF, Lapitsky Y, Kang CE, Shoichet MS. Journal of Controlled Release. 2009;140:218. doi: 10.1016/j.jconrel.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 34.Kleinman HK, Martin GR. Seminars in Cancer Biology. 2005;15:378. doi: 10.1016/j.semcancer.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Lei YG, Gojgini S, Lam J, Segura T. Biomaterials. 2011;32:39. doi: 10.1016/j.biomaterials.2010.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam J, Segura T. Biomaterials. 2013;34:3938. doi: 10.1016/j.biomaterials.2013.01.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, Garcia AJ. Adv Mater. 2012;24:64. doi: 10.1002/adma.201103574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hassan W, Dong YX, Wang WX. Stem Cell Res Ther. 2013;4 doi: 10.1186/scrt182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hiemstra C, van der Aa LJ, Zhong ZY, Dijkstra PJ, Feijen J. Biomacromolecules. 2007;8:1548. doi: 10.1021/bm061191m. [DOI] [PubMed] [Google Scholar]
- 40.Georges PC, Miller WJ, Meaney DF, Sawyer ES, Janmey PA. Biophysical Journal. 2006;90:3012. doi: 10.1529/biophysj.105.073114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Q, Tang XN, Yenari MA. J Neuroimmunol. 2007;184:53. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nadareishvili ZG, Li H, Wright V, Maric D, Warach S, Hallenbeck JM, Dambrosia J, Barker JL, Baird AE. Neurology. 2004;63:1446. doi: 10.1212/01.wnl.0000142260.61443.7c. [DOI] [PubMed] [Google Scholar]
- 43.Giulian D, Corpuz M, Chapman S, Mansouri M, Robertson C. Journal of Neuroscience Research. 1993;36:681. doi: 10.1002/jnr.490360609. [DOI] [PubMed] [Google Scholar]
- 44.Saas P, Boucraut J, Walker PR, Quiquerez AL, Billot M, Desplat-Jego S, Chicheportiche Y, Dietrich PY. Glia. 2000;32:102. [PubMed] [Google Scholar]
- 45.Yano A, Onozuka A, Matin K, Imai S, Hanada N, Nisizawa T. Vaccine. 2003;22:237. doi: 10.1016/s0264-410x(03)00561-9. [DOI] [PubMed] [Google Scholar]
- 46.Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, Dinov ID, Toga AW, Carmichael ST. Proc Natl Acad Sci U S A. 2012;109:E2230. doi: 10.1073/pnas.1204386109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Al’Qteishat A, Gaffney J, Krupinski J, Rubio F, West D, Kumar S, Kumar P, Mitsios N, Slevin M. Brain. 2006;129:2158. doi: 10.1093/brain/awl139. [DOI] [PubMed] [Google Scholar]
- 48.Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Stroke. 2009;40:1849. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- 49.Wei L, Erinjeri JP, Rovainen CM, Woolsey TA. Stroke. 2001;32:2179. doi: 10.1161/hs0901.094282. [DOI] [PubMed] [Google Scholar]
- 50.Zhang RL, Zhang ZG, Wang L, Wang Y, Gousev A, Zhang L, Ho KL, Morshead C, Chopp M. J Cerebr Blood F Met. 2004;24:441. doi: 10.1097/00004647-200404000-00009. [DOI] [PubMed] [Google Scholar]
- 51.Sofroniew MV. Neuroscientist. 2005;11:400. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- 52.Sofroniew MV, Vinters HV. Acta Neuropathologica. 2010;119:7. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blakney AK, Swartzlander MD, Bryant SJ. J Biomed Mater Res A. 2012;100A:1375. doi: 10.1002/jbm.a.34104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moshayedi P, Ng G, Kwok JC, Yeo GS, Bryant CE, Fawcett JW, Franze K, Guck J. Biomaterials. 2014;35:3919. doi: 10.1016/j.biomaterials.2014.01.038. [DOI] [PubMed] [Google Scholar]
- 55.Yenari MA, Kauppinen TM, Swanson RA. Neurotherapeutics. 2010;7:378. doi: 10.1016/j.nurt.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gomez-Nicola D, Fransen NL, Suzzi S, Perry VH. J Neurosci. 2013;33:2481. doi: 10.1523/JNEUROSCI.4440-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perry VH, Cunningham C, Holmes C. Nature Reviews Immunology. 2007;7:161. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- 58.Aguado BA, Mulyasasmita W, Su J, Lampe KJ, Heilshorn SC. Tissue Eng Pt A. 2012;18:806. doi: 10.1089/ten.tea.2011.0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carmichael ST. Stroke. 2008;39:1380. doi: 10.1161/STROKEAHA.107.499962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Okonkwo DO, Melon DE, Pellicane AJ, Mutlu LK, Rubin DG, Stone JR, Helm GA. Neuroreport. 2003;14:463. doi: 10.1097/00001756-200303030-00033. [DOI] [PubMed] [Google Scholar]
- 61.Osman MM, Lulic D, Glover L, Stahl CE, Lau T, van Loveren H, Borlongan CV. Neuropeptides. 2011;45:359. doi: 10.1016/j.npep.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 62.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen XH, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, Greiner DL, Handgretinger R. Journal of Immunology. 2005;174:6477. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 63.Shultz LD, Ishikawa F, Greiner DL. Nature Reviews Immunology. 2007;7:118. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 64.Shen LH, Li Y, Chen J, Zhang J, Vanguri P, Borneman J, Chopp M. Neuroscience. 2006;137:393. doi: 10.1016/j.neuroscience.2005.08.092. [DOI] [PubMed] [Google Scholar]
- 65.Chen JL, Zhang ZG, Li Y, Wang L, Xu YX, Gautam SC, Lu M, Zhu Z, Chopp M. Circulation Research. 2003;92:692. doi: 10.1161/01.RES.0000063425.51108.8D. [DOI] [PubMed] [Google Scholar]
- 66.Uwanogho D, Rex M, Cartwright EJ, Pearl G, Healy C, Scotting PJ, Sharpe PT. Mech Develop. 1995;49:23. doi: 10.1016/0925-4773(94)00299-3. [DOI] [PubMed] [Google Scholar]
- 67.Rao MS, Shetty AK. European Journal of Neuroscience. 2004;19:234. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- 68.Ruscheweyh R, Forsthuber L, Schoffnegger D, Sandkuhler J. J Comp Neurol. 2007;502:325. doi: 10.1002/cne.21311. [DOI] [PubMed] [Google Scholar]
- 69.Discher DE, Sweeney L, Sen S, Engler A. Biophysical Journal. 2007:32a. [Google Scholar]
- 70.Lee J, Abdeen AA, Zhang D, Kilian KA. Biomaterials. 2013;34:8140. doi: 10.1016/j.biomaterials.2013.07.074. [DOI] [PubMed] [Google Scholar]
- 71.Saha K, Keung AJ, Irwin EF, Li Y, Little L, Schaffer DV, Healy KE. Biophysical Journal. 2008;95:4426. doi: 10.1529/biophysj.108.132217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R, Clark AT, Plath K. Proc Natl Acad Sci U S A. 2008;105:2883. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Clarkson AN, Huang BS, MacIsaac SE, Mody I, Carmichael ST. Nature. 2010;468:305. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clarkson AN, Lopez-Valdes HE, Overman JJ, Charles AC, Brennan KC, Carmichael ST. J Cerebr Blood F Met. 2013;33:716. doi: 10.1038/jcbfm.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.