

Abstract
Owing to their capacity for self-renewal and pluripotency, stem cells possess untold potential for revolutionizing the field of regenerative medicine through the development of novel therapeutic strategies for treating cancer, diabetes, cardiovascular and neurodegenerative diseases. Central to developing these strategies is improving our understanding of biological mechanisms responsible for governing stem cell fate and self-renewal. Increasing attention is being given to the significance of metabolism, through the production of energy and generation of small molecules, as a critical regulator of stem cell functioning. Rapid advances in the field of metabolomics now allow for in-depth profiling of stem cells both in vitro and in vivo, providing a systems perspective on key metabolic and molecular pathways which influence stem cell biology. Understanding the analytical platforms and techniques that are currently used to study stem cell metabolomics, as well as how new insights can be derived from this knowledge, will accelerate new research in the field and improve future efforts to expand our understanding of the interplay between metabolism and stem cell biology.
Keywords: stem cell, metabolism, NMR, mass spectrometry, MRS, flux analysis
Stem cells and metabolism
Stem cells are the foundation of all multi-cellular organisms. They have the ability to self-renew, maintain pluripotency, as well as differentiate to specific cellular lineages. Many factors, from transcriptional signaling to epigenetics, have been shown to contribute to fate determination; in addition, increasing attention is being given to the role of metabolism as a regulator of stem cell fate (Vacanti and Metallo, 2013; Ramm Sander et al., 2013; Ito and Suda, 2014). With new advancements in technology, we are now able to probe stem cell metabolism like never before (Fig. 1), leading to discoveries that are reshaping our traditional ideas regarding the role of metabolism in stem cell biology. The objective of this review is to provide a primer for entering the field of stem cell metabolomics with examples of frequently used methodologies and how they can be employed to uncover novel metabolic mechanisms important for the regulation of stem cell biology.
Figure 1.
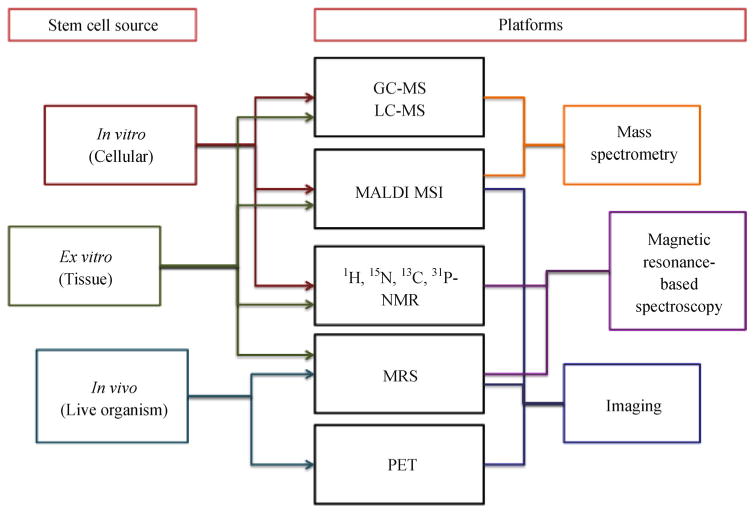
Platforms for studying stem cell metabolism.
From metabolism to metabolomics
Metabolism refers to all chemical reactions, essential for life, that occur in living organisms. These reactions involve metabolites (small molecules, less than 1kDa) and the enzymes that process them, and are organized in a set of pathways that allow cell growth, reproduction, and response to the environment. The complete set of metabolites within a cell or tissue is called “the metabolome.” The abundance of each metabolite within the metabolome depends on the specific physiological, developmental, and pathological state of a cell or tissue. Therefore, the metabolome reflects the phenotype of a cell or tissue, resulting in response to different genetic or environmental influences (Fiehn, 2002).
Traditionally, metabolism has been studied through biochemistry, focusing on one pathway at the time. From its inception in the 19th century (1833 discovery of amylase, Anselme Payen) to peak discoveries in the mid-20th century (glycolysis, Krebs cycle, electron transport chain), biochemistry has proven invaluable to our understanding of life. In addition, over the past decade, it has been increasingly important to grasp the whole metabolome in an unbiased way in order to understand how gene-environment interactions affect metabolic status of the cell. Consequently, studies of metabolism expanded toward a systems biology approach, metabolomics.
Metabolomics is the scientific study of global metabolite profiles of cells, tissues, and organisms, or “the metabolome” (Nicholson et al., 2012). The complexity of the metabolome has led to development of different analytical strategies in order to discern its details, such as targeted analysis and metabolomic profiling. While targeted analysis implies quantitation of a class of metabolites that comprise a specific metabolic pathway, metabolomic profiling provides the complete metabolite composition of a cell or a tissue. Metabolomic fingerprinting, a subcategory of the metabolomic profiling, represents a scan of a large number of intracellular metabolites, aiming to find a specific signature of a given tissue or a certain state of the tissue. In essence, it is a non-invasive and medically applicable technique for detection of low quantities of known metabolites and identification of unknown compounds (Griffin et al., 2002).
Unlike genomics, transcriptomics, and proteomics, metabolomics analysis is less complex because of fewer endpoints. It has been estimated that in the human body, there are more than 500 different histological cell types consisting of unique, dynamic cellular genomes, proteomes, and metabolomes (Nicholson et al., 2012). Nonetheless, the total number of human metabolites identified is relatively modest (the Human Metabolome Database (Wishart et al., 2007) currently has 41511 metabolite entries) compared with transcriptomics (∼85000) and proteomics (> 10000000), where more targets can be identified and quantified (Sreekumar et al., 2009; Shah et al., 2012). In addition, a major advantage of metabolomics over other ‘omics’ strategies is that metabolites are inherently linked to phenotypes (Fiehn, 2002). Metabolomics builds on more than 100 years of knowledge in biochemistry, which has thoroughly defined metabolic pathways enabling much faster translation of profile data than what is possible with other ‘omics’ fields. This has largely been mediated by publicly available pathway databases, such as KEGG (Kanehisa and Goto, 2000), Reactome (Milacic et al., 2012), and Biocarta (Nishimura, 2000), which enable one to map and consider metabolites of interest in systems-level context.
Despite advantages, metabolomics research has its own set of pitfalls. These primarily stem from an incredible physical and chemical complexity of the metabolites (de Graaf et al., 2010). This prevents identification and quantification of the whole metabolome by any one of the existing platforms. In general, when the goal of a study is to be as inclusive as possible, the method is termed untargeted, unbiased metabolomics. In contrast, when the goal is to be as accurate as possible on a known subset of the metabolome, the method is termed targeted metabolomics. Here, we outline in brief the major platforms for metabolomics studies, as well as considerations for sample preparation and data analysis.
Metabolomics: technology and techniques
Profiling the metabolome of stem cells requires the acquisition of high quality data on hundreds or thousands of unique molecules in the system. While there are many available options to achieve this goal, the two most common profiling platforms are mass spectrometry and nuclear magnetic resonance spectroscopy (Dunn et al., 2011). Further, metabolomics can be employed to study either steady-state metabolic differences between control and experimental conditions, which is valuable for biomarker discovery, or metabolic flux analysis, useful for assessing changes in the metabolic pathway utilization. When designing metabolomics experiments, careful consideration should be given to the pros and cons of each of these factors.
Technology
Mass spectrometry (MS) is an attractive platform in the pursuit of metabolomics data due to its high resolution and sensitivity. Briefly, MS is based on the concept of molecular separation by mass-to-charge (m/z) ratios. Every MS consists of 3 components: an ion source, a mass analyzer, and a detector (Dass, 2007). As the sample enters the instrument, molecules in the sample become charged via the ion source. The charged molecules are then accelerated and subjected to a magnetic or electric field which promotes separation based on each ion's m/z ratio. This separation promotes the detection of unique ionic species, allowing the MS to detect metabolites at very low concentrations within a sample. In metabolomics, mass spectrometry coupled to liquid chromatography (LC-MS) or gas chromatography (GC-MS) are commonly utilized as chromographic separation reduces matrix effects and the complexity of the sample (Gika et al., 2014). One major disadvantage of using MS is that samples are not recoverable after ionization; that is, MS is a destructive platform. Another disadvantage is that samples must be chemically prepared prior to analysis, possibly resulting in increased analytical variance and making absolute quantitative measurement challenging.
Conversely, Nuclear Magnetic Resonance Spectroscopy (NMR) is a platform that provides relatively low sensitivity but very accurate and reproducible quantitative measurements within a large dynamic range. NMR is a physical phenomenon whereby nuclei in a magnetic field absorb and re-emit electromagnetic energy at a specific resonance frequency depending on the strength of the magnetic field and the magnetic properties of the atoms (de Graaf, 2008). Mostly, NMR metabolomics utilizes proton (1H)-based spectroscopy, but analyses of other nuclei, such as 13C-, 31P-, and 15N-, are also relevant. Typically, a metabolite detected by the 1H-NMR contains one or more protons and each of the protons produces one or more peaks that can resonate at different chemical shifts. The pattern of NMR spectrum is also affected by scalar coupling (J coupling or spin-spin coupling), originating from the interactions among magnetic moments of nuclei. Such interactions can split resonances into several smaller ones. Consequently, the number of peaks and their resonances directly relate to the chemical structure of the molecule. Although each metabolite has a unique spectral pattern, one or more spectral peaks of different metabolites may overlap; therefore, the ability to resolve overlapping peaks is critical to any analytical method. To assist the identification of unknown metabolites a variety of 2D-and 3D-NMR approaches (COSY, TOCSY, high-resolution HSQC, HSQC-TOCSY) can be used. NMR is the basis for many applications, ranging from medical diagnostics (MRI scanners) and quantum computer design to high throughput metabolomics (Vandersypen et al., 2001; Mountford et al., 2010). One major advantage of NMR is that it requires little to no sample preparation prior to analysis, thus minimizing analytical variance and facilitating applications such as noninvasive metabolomic profiling. In addition, it does not require sample destruction for analysis, which is useful when sample quantities are limited as well as when the same sample needs to be utilized for other types of analyses. The primary disadvantage of NMR for metabolomic applications is its low sensitivity, which restricts the number of detectable molecular species. However, the high degree of connectivity within metabolic networks can somewhat reduce the problem of low sensitivity, because changes in low concentration metabolites may lead to indirect changes in higher concentration metabolites. In addition to analysis of biofIuids, cells, cell extracts, and tissue homogenates, NMR can be used for intact tissue analysis, using high resolution magic angle spinning (HR MAS). HR MAS has a high degree of reproducibility and a non-destructive nature, and thus, the same specimen can be assessed by histopathology, gene expression profiling or other methods after spectral analysis (DeFeo and Cheng, 2010). This allows direct comparisons between spectral and other features of the tissue, as well as integration of multitude of approaches for phenotypic characterization.
Recently, NMR and MS have started to be linked both instrumentally and experimentally. For analysis of a given sample, NMR is used first to provide quality control and basis content of untargeted metabolites, followed by MS for targeted analysis. The adoption of the combined approach is envisioned as a major accelerator of metabolomics field, not only for basic research but also for hands-on clinical applications. With new analytical methodologies, such as statistical heterospectroscopy (SHY), direct cross-correlation of chemical shifts (NMR) and m/z data (MS) can provide both structural and metabolic information (Coen et al., 2008), highly enhancing the utility of combined platforms for metabolomics research.
Ion mobility spectrometry (IMS) has long been considered an important analytical technique for detection of trace levels of analytes. In basic terms, IMS is a gas-phase electrophoretic separation of ions carried out in the presence of a neutral gas, under the influence of an electric field. In the past few decades, several research groups have coupled IMS to mass spectrometry (MS) to obtain a separation dimension based on the ion-neutral collision cross-section, which complements the m/z separation of MS. IM-MS provides at least four tangible benefits over conventional MS instrumentation: (i) improved dynamic range, in terms of both concentration and mass range, (ii) enhanced identification capabilities by differentiating analytes of different molecular class (e.g. nucleic acids, peptides, and lipids), (iii) in contrast with conventional scanning MS/MS approaches, IM-MS/MS can fragment all precursor ions simultaneously, and (iv) IM-MS provides further analytical dimensionality by increasing peak capacity and throughput through providing ultrafast (ms-ms) 2D separations in an infusion experiment or in concert with LC-MS methods (Sowell et al., 2004; Castro-Perez et al., 2011; Zinnel et al., 2012).
Capillary electrophoresis-mass spectrometry
Capillary electrophoresis (CE) when combined with QQQ or Q-TOF-based mass spectrometry, offers high separation efficiency high speed, and economy of sample size. The coupling of CE with MS combines the extremely high resolving power and structural information in one system. In CE-MS, analytes are identified both by their differential separation and their molecular masses and/or fragmentation patterns (Nevedomskaya et al., 2010; Takeuchi et al., 2013). This technology requires very small sample size, making it useful for applications such as cancer stem cell analysis or metabolo-mics of subsets of stem cells obtained from a given tissue.
Gas chromatography-flame ionization detection/mass spectrometry (GC-FID/MS) is another analytical method which shows promise in the field of metabolomics. In GC/FID-MS, samples are first separated on the GC via temperature gradient. Then, as analytes elute from the GC, they are simultaneously sent to the FID and MS instruments. FID provides an excellent means of precise quantification, which relies on the detection of ions formed during combustion where the abundance of ions is directly proportional to the concentration of the analyte coming from the GC. When coupled with the spectral information originating from the MS, this analytical method provides accurate identification and quantification. This platform has been useful in the quantification of lipid species found in biofiuids such as plasma and urine (Fancy et al., 2006; Zhang et al., 2011).
The “metabolomic phenotyping microarrays” are based on the premise that cells utilize metabolites in various biochemical pathways and generate reducing equivalents in the form of NADH or NADPH. These reducing equivalents can be quantified using a tetrazolium dye and reflect the biochemical reactome for the particular metabolite (Putluri et al., 2011). There are at least 14 different substrate plates available that can measure the flux through carbohydrate and amino acid/dipeptide pathways as well as utilization of ions (Bochner et al., 2011; Luo et al., 2012). Stem cells with various experimental conditions may be cultured with these small molecules, and the fluctuations in the cellular metabolism of each respective small molecule can be detected by chemometric changes with an energy-rich NADH redox dye. Although the Phenotype MicroArrays for mammalian cells system can screen through various metabolites, more targeted assays are also developed.
A Seahorse analyzer (XF 24) enables detailed analysis of mitochondrial biogenesis and mitochondrial dysfunction associated with oxidative stress and altered substrate utilization. It is a fully integrated, multi-well instrument that measures the oxygen consumption and H+ production of the cells in real-time, using disposable cartridge containing the probes and assay kits. It simultaneously measures oxygen consumption rate and extracellular acidification rate in as little as five minutes. With just a small number of cells, XF analyzer can measure the effects of up to four compounds on cellular metabolism, glycolysis, respiratory capacity, mitochondrial dysfunction, fatty acid oxidation and cell signaling. As mitochondrial changes underlie metabolic and cellular switches from pluripotent to differentiated states and vice versa, this technology may be strongly considered for specific studies of stem cell state transitions.
Technologies for studies of metabolism in vivo
The correlate of NMR is proton magnetic resonance spectroscopy (1H-MRS), used to detect and quantify a small number of metabolites in the living tissue (Soares and Law, 2009): N-acetylaspartate (NAA), a marker of neurons whose major peak resonates at 2.02 ppm; Creatine, resonating at 3.02 ppm, an energy metabolite considered to be stable and thus used as a house-keeping metabolite for normalization; Choline, resonating at 3.23 ppm and considered a marker of glial cells and membrane turnover, and Myoinositol, a marker of astrocytes which resonates at 3.56 ppm. Other metabolites commonly detected include alanine, lactate, glutamate, glutamine, glucose, GABA, and some macromolecular proteins and lipids (Soares and Law, 2009). The major issue with MRS is very low sensitivity, and its utility has been limited by analytical methods that focus on independently evaluated metabolites and require prior knowledge about which metabolites to examine.
Nevertheless, fatty acid moiety resonating at 1.28 ppm has been associated with NPCs in the human hippocampus (Manganas et al., 2007). Initially discovered in rodent NPCs, by high-field NMR (Manganas et al., 2007), the fatty acid enrichment appears to reflect increased amounts of mobile lipids necessary for the function of these cells (Knobloch et al., 2013). The identity of the 1.28 ppm metabolite remains unknown. Although it most likely contains a fatty acid component (Manganas et al., 2007), its exact molecular nature has not yet been determined and its functional significance for neurogenesis awaits further studies. Recent reports indicate that the 1.28 ppm and adjacent resonances may also be associated with apoptosis. A similar signal resonating at 1.30 ppm has been also reported in apoptotic ratgliomas in vivo (Liimatainen et al., 2008), and more recent studies have found that the 1.28 ppm signal in cultured NPCs increased during conditions that favored quiescence and apoptosis (Ramm et al., 2011). Apoptosis is common in the hippocampal neurogenic niche, as vast amounts of newborn cells die during critical periods of survival (Sierra et al., 2010). Thus, whether the 1.28 ppm peak detected in living brains originates from living or apoptotic NPCs remains to be determined and more research is necessary to unequivocally establish whether the 1.28 ppm spectral peak is a marker of neurogenesis with clinical value.
In addition to targeted MRS analysis, an untargeted, metabolomics type of analysis is also possible using MRS (Vingara et al., 2013). Metabolomic-type analysis can overcome signal distortions that can occur with MRS, providing previously unavailable information about living tissue, in vivo. Unlike other quantification tools, metabolomic analysis of the full resolution spectra has the advantage of not requiring a priori knowledge such as the line shapes of the metabolite resonances. Therefore, the resonances identified are not limited to the user's input criteria, and changes in small resonances can be extracted (Vingara et al., 2013). The untargeted metabolomics analysis can be used for comprehensive noninvasive tissue profiling of diseased and healthy tissue in vivo, as it captures in a single analysis metabolic alterations that otherwise require several independent studies. This analytical platform has not yet been applied for studies of stem cells. Nevertheless, in vivo metabolomics could be extended to studies of stem cells in any organ and particularly cancer stem cells, to model disease subtype, progression, or for treatment monitoring. In addition to being valuable for creating more patient-specific assessments such methodologies can also provide insight into the stem cell pathology.
Biochemical assays can theoretically be translated to in vivo studies. Fluorination of a metabolite of interest is used in studies involving Positron Emission Tomography (PET) (Buchsbaum and Hazlett, 1998). Such technique is limited by the metabolism of the small molecule in question and gives limited spatial information of 4 to 5 mm range. For stem cells, the utility of a particular technique is limited by the resolution, which requires resolution in a μm ranges. More advances in label-free microscopy methods of metabolic detection have given single cell resolution, which have allowed detection of stem cells in vivo. Using two-photon fluorescent microscopy, live rodent imaging can be done to distinguish metabolic markers of stem cells (Quinn et al., 2013; Stringari et al., 2015). As an example, two-photon excitation fluorescence can detect NADH and the second harmonic generation can detect collagen (Stringari et al., 2015). These two excitation parameters can be detected simultaneously using the same objective, thus giving single-cell resolution images. Using this method, the fluxes of NADH/NAD+ ratio can be determined, thus reflecting the glycolytic/oxidative phosphorylation ratio of various cell types. Since stem cells have more glycolytic characteristics, such technique is utilized to detect stem cells in vivo. In addition, Raman Scattering Spectroscopy is another label-free method that can be used in conjunction with other microscopy modalities to study lipid metabolism in stem cells. Recently developed modalities, such as Coherent Anti-Stokes Raman Scattering (CARS) microscopy and Stimulated Raman Scattering (SRS) microscopy, can be used for chemical imaging, as they allow visualization of certain classes of molecules such as lipids, at the sub-cellular level. These technologies are based on the vibration of a specific chemical group, which permits high-resolution imaging of individual molecules in vivo (Folick et al., 2011; Yu et al., 2014).
Metabolomics studies: Sample preparation
Appropriate collection, handling, and storage of the samples is critical to metabolomics analyses, as the methods are sensitive to small changes in the metabolite profile that may be introduced through poor sample handling procedures. With the exception of systems specifically equipped with a magic angle-spinning probe for tissue analysis (Duarte et al., 2009), all classic high resolution NMR as well as MS-based analytical methods require homogeneous liquid samples (Wu et al., 2008). Therefore, cell lysis and extraction is necessary to obtain samples adapted to liquid analytical spectroscopic techniques. These preparations are often the most labor intensive and rate-limiting steps in metabolomics as they require accuracy and reproducibility as well as robustness. There is a significant body of literature dedicated to optimizing metabolomic extraction methods (Mushtaq et al., 2014; Ser et al., 2015). A general extraction protocol will involve some form of quenching to cease metabolic activity, followed by metabolite extraction with a mixed solvent (i.e. methanol:chloroform:water). Depending on the source material (i.e cultured cells, tissue, biofluids, etc.) and types of metabolites to be investigated (i.e. lipids, amino acids, etc.), the sample extraction methods will differ, typically by varying the ratio of aqueous and organic solvents as well as pH of the buffer.
Sample preparation for MS-based examination of metabolome (unbiased and targeted)
Optimally, at least 25 mg of tissues or 5 million cells is necessary for the mass spectrometry-based metabolomic profiling. The process of metabolite extraction for these samples involves the introduction of an equimolar mixture of standard compounds followed by homogenization of the specimen. Subsequently, the metabolites in the homogenate are extracted using sequential application of aqueous (chilled water) and organic (chilled methanol and chloroform) solvents in the ratio 1:4:3:1 (water:methanol:chloroform:water) (Sana et al., 2008). The extract is deproteinized and the filtrate, containing metabolites, dried under vacuum and re-suspended in the injection solvent (Putluri et al., 2011). An equimolar mixture of the standard compounds and/or a characterized tissue sample (when examining cell line or tissue-based extracts) or a urine or plasma sample (when examining biofluids), is extracted and analyzed in tandem with the experimental samples. Each of the controls needs to be included multiple times in the randomization scheme to ensure that sample preparation and analytical variability are constantly monitored. Further, each sample needs to be followed by at least two blank runs to prevent any carryover of metabolites between samples. For LC-MS, the polar, mid-polar, and some of the non-polar compounds are separated using aqueous normal phase or reverse phase chromatographic separation. For GC-MS, the samples are re-dried under vacuum desiccation for a minimum of 24 h prior to being derivatized. Derivatization is a metabolite-dependent process and the modifying agent is selected based on the chemistry of the compound to be assessed. The quality control procedure prior to sample analysis involves locking the retention time using d27-myristic acid (Kind and Fiehn, 2009; Kind et al., 2009). For lipidomics studies, samples need to undergo additionally prepared because lipids represent a large class of molecules and as such, there are variations how to successfully prepare various classes of lipids. Identification of the individual species is based on their chromatographic, ion mobility drift times, and mass spectral characteristics and comparison to those of chemically defined standards.
Sample preparation for NMR-based examination of metabolome
NMR provides largely untargeted analysis of metabolites in given samples, using 1D-NMR and 1H-, 13C-, 31P-, and 15N-nuclei assessments. Samples are prepared based on the experimental design. The aqueous extracts are obtained in 500 uL of reconstitution buffer with lmM TSP as an internal standard. The organic extracts are reconstituted in 500 uL of CDCl3 with 0.03% TMS as an internal standard. The NMR acquisition then involves locking to the solvent and shimming to achieve optimal line-shape. Specific pulse-sequences are applied depending on the experimental hypothesis. Typically, ID 1H NMR includes zgpr, noesyprld (for single solvent suppression) and lclpnf2 (for double-solvent suppression. Typical 2D NMR comprises COSY and TOCSY sequences. Hetero nuclear experiments include 1H-13C HSQC and HMBC sequences. The obtained data are analyzed and the set of metabolites is determined. In dubious cases, the specific metabolites can be validated by spike-in experiments.
Metabolomics: Techniques
Biochemical perturbations can occur due to changes in steady-state levels of metabolites, alterations in the rate of pathway activity or both (Fig. 2). With this consideration, metabolomics applications are broadly divided into two branches of analysis: steady-state profiling and metabolic flux analysis.
Figure 2.
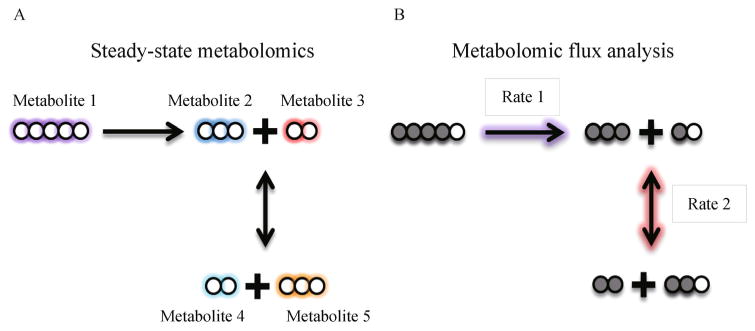
Metabolomics can be used to either (A) detect and quantify metabolites directly or (B) follow isotopically labeled carbons to determine metabolic pathway activity.
Steady-state profiling
Steady-state profiling yields a static snapshot of the relative abundances of individual metabolite pools between biologic states (i.e. differentiated vs undifferentiated stem cells). This can easily be applied to fresh or frozen tissue, and can yield quantitative or semiquantitative data. Although this is a relatively new area in the field of stem cell research, several recent findings highlight the potential for new discovery using these techniques. Notably, proliferating NPCs possess elevated mobile lipids (MLs) and a distinct lipogenic state important for neurogenesis, as demonstrated by both mass spectrometry and NMR metabolomics (Manganas et al., 2007; Knobloch et al., 2013). Additionally, an MS-based analysis has shown that the distribution of phosphatidylcholines and phosphatidylethanolamines, components of MLs, is significantly altered between embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), suggesting that differences in lipid metabolism, among others, may be linked to important regulatory differences underlying ESC and iPSC biology (Meissen et al., 2012). Interestingly, there have also been reports that MLs are elevated in glioblastoma cancer stem cells; however, as different groups have published conflicting results, it remains to be seen whether MLs are a marker of sternness or apoptosis (Ramm et al., 2011; Loewenbrück et al., 2011; Guidoni et al., 2014). In addition to lipogenesis, there are important studies outlining changes in energy metabolism between differentiated cells and stem cells (Fig. 3). Through the use of MS-based and NMR-based methods, ESCs, iPSCs, and long-term hematopoietic stem cells (LT-HSCs) have all been reported to shift away from oxidative phosphorylation toward glycolysis (Folmes et al., 2011; Panopoulos et al., 2012), a metabolic alteration akin to the Warburg Effect often observed in cancer (Warburg, 1956). Additionally, a large body of literature exists demonstrating that stem cells of all kinds show elevated levels of the amino acids glycine, alanine, and others (Urenjak et al., 1993; Griffin et al., 2002; Kulak et al., 2010), and it has been reported that mouse ESCs are highly dependent on threonine metabolism (Wang et al., 2009). MS-based metabolomics has identified that mouse ESCs possess more unsaturated molecules such as arachidonic acid and diacylglyercol compared to their differentiated progeny (Yanes et al., 2010). Knowing the differences in metabolic profiles as a function of stem cell type, potency, and state will be the key for determining how metabolic properties of stem cells, and particularly adult stem cells, are connected to quiescence and proliferation, differentiation capacity and age-related changes (Rando, 2006). Toward this goal, while steady-state profiling can be used to nominate which metabolic compartments are significantly altered in stem cells, it can be difficult to pinpoint the source of the metabolic alterations without kinetic data, such as those obtained using metabolic flux analysis.
Figure 3.
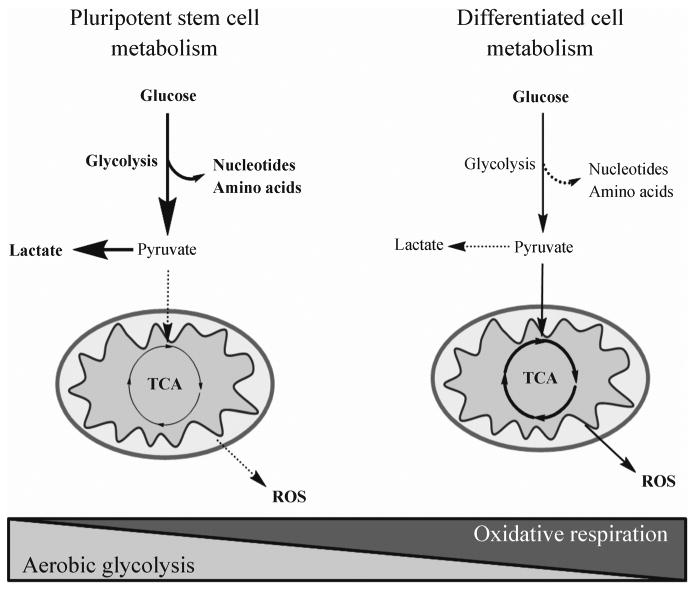
Metabolomic and flux analysis studies have demonstrated that pluripotent stem cells (PSCs) have increased dependence on glyolytic flux compared differentiated counterparts which exhibit increased mitochondrial oxidation. There are multiple hypotheses for why this occurs, such as PSCs minimize glucose oxidation in order to provide anabolic precursors to fuel self-renewal, or that ROS-induced stress may promote differentiation.
Metabolic flux analysis
Metabolic flux analysis is used to gather knowledge on metabolic pathway kinetics (Dass, 2007). With the high degree of interconnectivity between biochemical pathways, as well as the reversible nature of pathways or reactomes, a metabolite could participate in multiple pathways as well as function as a substrate or product within a pathway. The fate of the metabolite in each pathway and its nature of participation in different pathways define the flux or kinetics for the given metabolite. When put in a global perspective, individual metabolite fluxes together define the biochemical activity of a cell which in turn describes the physiological state of that cell. Hence, flux describes the dynamic nature of the biochemical pathway and is a key component in mechanistic underpinnings of cellular function.
Flux analysis can be accomplished by a range of technology platforms based on MS, NMR and non-MS-based methods (Dass, 2007). These define flux or pathway activity in cell lines, animal models and patients, and thus support basic and clinical translational research. Flux in pathways pertaining to a metabolite is a function of multitude of variables that include expression level of enzymes, affinity constants of the enzyme for the metabolite, rate of transport of the metabolite between various compartments of the cell and from outside, presence of inhibitors etc. Thus, the platform chosen for flux analysis needs to measure rates of synthesis, breakdown, utilization, and uptake of metabolite as well as corresponding rates of energy production, oxygen consumption etc. This is typically done through the use of isotopically-labeled tracer metabolites (e.g. glucose, glutamine, etc.) which are fed to cells in culture and allowed to be metabolized for a given time. When the cells are extracted, the relative amount of the isotopic label which became incorporated into downstream metabolites can be quantified and used to calculate the kinetic rate of the pathway, or flux. This data are then input into a model of the metabolic pathway to derive estimates of pathway activity (Antoniewicz et al., 2007; Zamboni et al., 2009).
At this time, there are very few published reports utilizing metabolic flux analyses to study stem cell metabolism (Turner et al., 2008; Sepúlveda et al., 2010; Yanes et al., 2010), apart to reinforce the importance of energy metabolism discovered via steady-state profiling. Given the observations of altered lipids and amino acids from steady-state profiling, there are many opportunities to apply metabolic flux analysis and phenotyping microarrays to further understand the role of metabolism in stem cell biology.
Single cell metabolomics
In addition to a common, population-based systems biology methods, single cell ‘omics’ studies are slowly starting to emerge (Zenobi, 2013). The need for such studies comes from a large heterogeneity of cell populations, particularly common confounder when studying stem cells. Namely, in any given preparation of a stem cell culture or a tissue enriched in stem cells, these cells can be found in a variety of different states – from quiescent, to any cell cycle stage, to different degrees of differentiating lineages. Thus, single cell investigations are very relevant to discern metabolic properties of each state of the cell. For single cell metabolomics, mass spectrometry is the platform of choice because of its detection sensitivity – the concentration of metabolites needs to be in attomole range (Amantonico et al., 2008; Heinemann and Zenobi, 2011). Several approaches are being developed, including nano-electrospray ionization, microfluidic chips and sample arrays for monodispersed sample droplets as sample sources for MS data acquisition (Urban et al., 2010). Detection of single cell metabolites is also a focus of emerging imaging mass spectrometry, which may achieve spatial resolution at the micron level (Secondary Ion Mass Spectrometry, SIMS) (Klerk et al., 2010). While this technology remains qualitative, it provides additional data relevant for molecular phenotyping in a variety of experi-mental conditions. These approaches may indeed prove invaluable for studies of stem cell metabolism, for detection of minute changes that ultimately lead to differentiation as well as those that lead to continuous proliferation as seen in cancer.
Metabolomics data analysis
Similar to other ‘omics’ data, such as genomics, transcrip-tomics, proteomics etc., metabolomic data pose special challenges for data analysis because they are high dimensional, acquired by multiple analytical methods, have some degree of missing data, and some degree of collinearity, nonlinearity, and non-normality, all of which need to be accounted for to achieve meaningful data interpretation. An additional challenge of metabolomics analysis is that the number and the identity of metabolites in the sample are unknown, which makes the power calculations very difficult.
In principle, the analysis of metabolomics data should start with an unsupervised method, such as Principal Component Analysis (PCA), that can identify outliers and the main source of variability. Since multivariate analysis methods such as PCA are based on variance, i.e., they seek out the greatest variance of the data, the variables need to be centered and scaled to reduce the bias placed on large variables (Craig et al., 2006). As with most analytical data, a large peak will exhibit a greater absolute variance than a small peak. To minimize this effect, the standard in metabolomics is to apply unit-variance or Pareto scaling. Once the outliers are identified and eliminated, supervised multivariate methods for pattern separation, such as Partial Least-Squares Regression (PLS) and Orthogonal PLS (OPLS), are employed to explore class differences and highlight explanatory spectral variables (Goodacre et al., 2004; Dunn et al., 2005; Weckwerth and Morgenthal, 2005; Coen et al., 2008; Allen and Maletic-Savatic, 2011). The dimensionality of the data can also be reduced to simple linear combinations that assist in identification of the metabolites that provide the largest variability (Allen et al., 2014). Variables that contribute to clustering of experimental groups are identified based on loadings plots. These variables then need to be annotated as the respective metabolites. SIMCA-P software (Umetrix, Malmö, Sweden) is commonly used for multivariate, pattern analysis in metabolomics, and AMIX package (Bruker, Inc.) provides similar paradigms. When using multivariate analyses, one needs to be aware that they suffer from over-fitting and thus, validation is obligatory. To build and validate the PLS or OPLS models for class discrimination and prediction, the data need to be randomly divided into a training set and a test set. The test set is excluded from model construction, and the model is used to predict class membership of the data in the test set. Typically, cross-validation approaches are used in which a proportion of the data (for example, every 10th sample) is removed, and the model is built with the remaining training set. This procedure is repeated many times until each sample has been in the test set exactly once. The accuracy of the model on the test samples gives an estimate of the predictive power and the robustness of the model to perturbations of the data (Castaldi et al., 2011; Ioannidis and Khoury, 2011).
Once multivariate analysis and statistical threshold (Bon-feronni, false discovery rate etc.) are done, the next step in analysis is metabolite identification and quantification. This part depends on the acquisition method used and can be done either as targeted analysis by mass spectrometry or by query of metabolomic databases for candidate metabolite identification, such as the Human Metabolome Database (HMDB; Wishart et al., 2007; Ulrich et al., 2008). In addition, several computational algorithms have been developed for deducing metabolite identities from NMR data. Statistical Total Correlation Spectroscopy (STOCSY), designed for NMR data analysis, explores correlations among the intensities of various peaks across the whole spectrum, and interprets the original data set as a two dimensional pseudo-spectrum. STOCSY algorithms improve not only the assignment of compounds in a biologic mixture, but also provide potential molecular connections for analyzing metabolic pathways. However, the interpretation of STOCSY data are not always straightforward (Blaise et al., 2010), and the existence of many different STOCSY algorithms indicates the scope of the challenge. Statistical diffusion-ordered spectroscopy (S-DOSY) uses signal intensity variations under different pulsed field gradient conditions to identify metabolites and requires sophisticated data filtering procedure when peak overlap exists in the sample (Smith et al., 2007). Iterative-STOCSY (I-STOCSY) calculates correlations from a “driver” peak and recursively finds correlated peaks to form nodes, the connectivity among which are then used to explore the inter- and intra-metabolite connections (Sands et al., 2011). However, parts of nodes generated in I-STOCSY algorithm to identify a specific metabolite can be missed due to peak overlap (Sands et al., 2011). Grouping procedures which utilize STOCSY are also numerous. Cluster analysis statistical spectroscopy (CLASSY) employs correlation matrix of peaks and an intersection matrix context to determine local clusters and consequently explore the intra and intermetabolite connections by hierarchical clustering of those local clusters (Robinette et al., 2009). Statistical recoupling of variables (SRV) groups variables by scanning the covariance/correlation landscape and combines the grouped variables (clusters) to superclusters according to the correlation strength among those clusters. STOCSY is then applied on the superclusters to help metabolite identification (Blaise et al., 2010). However, SRV clusters may be misled when overlapped peaks introduce variations that are not structural (Blaise et al., 2009). Recently published STOCSY-scaling method scales the spectra by designed functions to decrease the contribution of metabolites that have dominant intensities, such as glucose, to explore and identify metabolites that are covered at these regions (Maher et al., 2012). However, artificial correlation caused by peak overlap can also lead to undesired suppression of signals that actually should not be suppressed or lead to failing to suppress the targeted signals (Maher et al., 2012). As metabolite identification and quantification is mandatory for further analysis, many times it requires validation by experimental spike-in confirmation.
Finally, identified metabolites can be explored in the context of metabolic networks, through existing databases such as KEGG, Gene Spring, Ingenuity Pathway Analysis, and GeneGO, and freely-available software such as Gene Set Enrichment Analysis (GSEA), among others.
These approaches emerged as it was recognized that identifying a list of differentially expressed metabolites that may play a significant role in a biologic process most often fails to provide mechanistic insights into the underlying biology of the condition under study. Hence, attention has shifted from lists of molecules to sets of functionally related coordinated alterations that constitute pathways or bioprocesses, which in concert orchestrate the underlying biology at cellular or organismal level. In other words, this so called pathway-centric approach results in reduction of data dimensionality while preserving the interaction between the components within an experiment (Glazko and Emmert-Streib, 2009; Peterson et al., 2013). There are different approaches to define pathways using metabolomics data, some of which involve mapping metabolites to existing pathway maps and rely on enrichment methods, while others are much more sophisticated in that they explore enrichments across larger compendia of molecular processes assembled within databases without the prerequisite for pre-defined pathway maps. Eventually, the networks need to be visualized to allow the user to consider related pathways which cannot be readily inferred from the given profile. Network visualization can be achieved using Cytoscape with its app/plug-in KEGGscape (http://apps.cytoscape.org/apps/keggscape) (Nishida et al., 2014). Overall, the analytical path from data acquisition to network discovery in metabolomics involves numerous steps, computational and statistical analysis, as well as bioinfor-matics approaches necessary to produce accurate and biologically meaningful data.
Conclusion and future considerations
Undeniably, studies of metabolism of stem cells are critical for gaining insights into their fate and function. The metabolic phenotypes seen in stem cells and their progeny correlate to the energy demands for proliferation, lineage specification, and quiescence. Different cell states require specific metabolic programs to support the unique bioenergetics demands underlying their specialized functions. A variety of platforms and techniques are now available to delve deeper into the stem cell metabolism. The growing interest in metabolomics of stem cells as it pertains to both biology and pathology holds substantial promise for future discoveries in this relatively new field of science.
Acknowledgments
This work was supported by the NLM Training Program in Biomedical Informatics (T15LM007093), Developmental Biology Training Program (T32HD055200), and BCM Medical Scientist Training Program (W.T.C.); Susan Komen Foundation KG110818, NIH U01 CA179674-01, R21-CA185516-01, NSF DMS-1161759, RP120092, and Funds from the Alkek Center for Molecular Discovery (A.S.K.); and the Dana Foundation, McKnight Endowment for Science, and Nancy Chang Award for Excellence (M.M.-S.).
Footnotes
Compliance with ethics guidelines: Joseph M. Arnold, William T Choi, Arun Sreekumar and Mirjana Maletić-Savatić declare no conflicts of interest. This manuscript is a review article and does not involve a research protocol requiring approval by the relevant institutional review board or ethics committee.
References
- Allen GI, Maletić-Savatić M. Sparse non-negative generalized PCA with applications to metabolomics. Bioinformatics. 2011;27(21):3029–3035. doi: 10.1093/bioinformatics/btr522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JE, Saroya BS, Kunkel M, et al. Apoptotic circulating tumor cells (CTCs) in the peripheral blood of metastatic colorectal cancer patients are associated with liver metastasis but not CTCs. Oncotarget. 2014;5:1753–1760. doi: 10.18632/oncotarget.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amantonico A, Oh JY, Sobek J, Heinemann M, Zenobi R. Mass spectrometric method for analyzing metabolites in yeast with single cell sensitivity. Angew Chem Int Ed Engl. 2008;47(29):5382–5385. doi: 10.1002/anie.200705923. [DOI] [PubMed] [Google Scholar]
- Antoniewicz MR, Kelleher JK, Stephanopoulos G. Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metab Eng. 2007;9(1):68–86. doi: 10.1016/j.ymben.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaise BJ, Navratil V, Domange C, Shintu L, Dumas ME, Elena-Herrmann B, Emsley L, Toulhoat P. Two-dimensional statistical recoupling for the identification of perturbed metabolic networks from NMR spectroscopy. J Proteome Res. 2010;9(9):4513–4520. doi: 10.1021/pr1002615. [DOI] [PubMed] [Google Scholar]
- Blaise BJ, Shintu L, Elena B, Emsley L, Dumas ME, Toulhoat P. Statistical recoupling prior to significance testing in nuclear magnetic resonance based metabonomics. Anal Chem. 2009;81(15):6242–6251. doi: 10.1021/ac9007754. [DOI] [PubMed] [Google Scholar]
- Bochner BR, Siri M, Huang RH, Noble S, Lei XH, Clemons PA, Wagner BK. Assay of the multiple energy-producing pathways of mammalian cells. PLoS ONE. 2011;6(3):e18147. doi: 10.1371/journal.pone.0018147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsbaum MS, Hazlett EA. Positron emission tomography studies of abnormal glucose metabolism in schizophrenia. Schizophr Bull. 1998;24(3):343–364. doi: 10.1093/oxfordjournals.schbul.a033331. [DOI] [PubMed] [Google Scholar]
- Castaldi PJ, Dahabreh IJ, Ioannidis JP. An empirical assessment of validation practices for molecular classifiers. Brief Bioinform. 2011;12(3):189–202. doi: 10.1093/bib/bbq073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Perez J, Roddy TP, Nibbering NM, Shah V, McLaren DG, Previs S, Attygalle AB, Herath K, Chen Z, Wang SP, Mitnaul L, Hubbard BK, Vreeken RJ, Johns DG, Hankemeier T. Localization of fatty acyl and double bond positions in phosphatidylcholines using a dual stage CID fragmentation coupled with ion mobility mass spectrometry. J Am Soc Mass Spectrom. 2011;22(9):1552–1567. doi: 10.1007/s13361-011-0172-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen M, Holmes E, Lindon JC, Nicholson JK. NMR-based metabolic profiling and metabonomic approaches to problems in molecular toxicology. Chem Res Toxicol. 2008;21(1):9–27. doi: 10.1021/tx700335d. [DOI] [PubMed] [Google Scholar]
- Craig A, Cloarec O, Holmes E, Nicholson JK, Lindon JC. Scaling and normalization effects in NMR spectroscopic metabonomic data sets. Anal Chem. 2006;78(7):2262–2267. doi: 10.1021/ac0519312. [DOI] [PubMed] [Google Scholar]
- Dass C. Fundamentals of contemporary mass spectrometry. Hoboken, New Jersey: John Wiley. Sons, Inc.; 2007. [Google Scholar]
- de Graaf AA, Maathuis A, de Waard P, Deutz NE, Dijkema C, de Vos WM, Venema K. Profiling human gut bacterial metabolism and its kinetics using [U-13C]glucose and NMR. NMR Biomed. 2010;23(1):2–12. doi: 10.1002/nbm.1418. [DOI] [PubMed] [Google Scholar]
- de Graaf RA. In vivo NMR Spectroscopy: Principles and Techniques. New Jersey: John Wiley. Sons, Inc.; 2008. [Google Scholar]
- DeFeo EM, Cheng LL. Characterizing human cancer metabolomics with ex vivo 1H HRMAS MRS. Technol Cancer Res Treat. 2010;9(4):381–391. doi: 10.1177/153303461000900407. [DOI] [PubMed] [Google Scholar]
- Duarte IF, Lamego I, Rocha C, Gil AM. NMR metabonomics for mammalian cell metabolism studies. Bioanalysis. 2009;1(9):1597–1614. doi: 10.4155/bio.09.151. [DOI] [PubMed] [Google Scholar]
- Dunn WB, Bailey NJ, Johnson HE. Measuring the metabolome: current analytical technologies. Analyst (Lond) 2005;130(5):606–625. doi: 10.1039/b418288j. [DOI] [PubMed] [Google Scholar]
- Dunn WB, Broadhurst DI, Atherton HJ, Goodacre R, Griffin JL. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem Soc Rev. 2011;40(1):387–426. doi: 10.1039/b906712b. [DOI] [PubMed] [Google Scholar]
- Fancy SA, Beckonert O, Darbon G, Yabsley W, Walley R, Baker D, Perkins GL, Pullen FS, Rumpel K. Gas chromatography/flame ionisation detection mass spectrometry for the detection of endogenous urine metabolites for metabonomic studies and its use as a complementary tool to nuclear magnetic resonance spectroscopy. Rapid Commun Mass Spectrom. 2006;20(15):2271–2280. doi: 10.1002/rcm.2583. [DOI] [PubMed] [Google Scholar]
- Fiehn O. Metabolomics—the link between genotypes and phenotypes. Plant Mol Biol. 2002;48(1–2):155–171. [PubMed] [Google Scholar]
- Folick A, Min W, Wang MC. Label-free imaging of lipid dynamics using Coherent Anti-stokes Raman Scattering (CARS) and Stimulated Raman Scattering (SRS) microscopy. Curr Opin Genet Dev. 2011;21(5):585–590. doi: 10.1016/j.gde.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14(2):264–271. doi: 10.1016/j.cmet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gika HG, Theodoridis GA, Plumb RS, Wilson ID. Current practice of liquid chromatography-mass spectrometry in metabolomics and metabonomics. J Pharm Biomed Anal. 2014;87:12–25. doi: 10.1016/j.jpba.2013.06.032. [DOI] [PubMed] [Google Scholar]
- Glazko GV, Emmert-Streib F. Unite and conquer: univariate and multivariate approaches for finding differentially expressed gene sets. Bioinformatics. 2009;25(18):2348–2354. doi: 10.1093/bioinformatics/btp406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB. Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol. 2004;22(5):245–252. doi: 10.1016/j.tibtech.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Griffin JL, Bollard M, Nicholson JK, Bhakoo K. Spectral profiles of cultured neuronal and glial cells derived from HRMAS (1) H NMR spectroscopy. NMR Biomed. 2002;15(6):375–384. doi: 10.1002/nbm.792. [DOI] [PubMed] [Google Scholar]
- Guidoni L, Ricci-Vitiani L, Rosi A, Palma A, Grande S, Luciani AM, Pelacchi F, di Martino S, Colosimo C, Biffoni M, De Maria R, Pallini R, Viti V. 1H NMR detects different metabolic profiles in glioblastoma stem-like cells. NMR Biomed. 2014;27(2):129–145. doi: 10.1002/nbm.3044. [DOI] [PubMed] [Google Scholar]
- Heinemann M, Zenobi R. Single cell metabolomics. Curr Opin Biotechnol. 2011;22(1):26–31. doi: 10.1016/j.copbio.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Ioannidis JP, Khoury MJ. Improving validation practices in “omics” research. Science. 2011;334(6060):1230–1232. doi: 10.1126/science.1211811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15(4):243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind T, Fiehn O. What are the obstacles for an integrated system for comprehensive interpretation of cross-platform metabolic profile data? Bioanalysis. 2009;1(9):1511–1514. doi: 10.4155/bio.09.141. [DOI] [PubMed] [Google Scholar]
- Kind T, Wohlgemuth G, Lee Y, Lu Y, Palazoglu M, Shahbaz S, Fiehn O. FiehnLib: mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromato-graphy/mass spectrometry. Anal Chem. 2009;81(24):10038–10048. doi: 10.1021/ac9019522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klerk LA, Dankers PY, Popa ER, Bosnian AW, Sanders ME, Reedquist KA, Heeren RM. TOF-secondary ion mass spectrometry imaging of polymeric scaffolds with surrounding tissue after in vivo implantation. Anal Chem. 2010;82(11):4337–4343. doi: 10.1021/ac100837n. [DOI] [PubMed] [Google Scholar]
- Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Arauzo-Bravo MJ, Kovacs WJ, Karalay O, Suter U, Machado RA, Roccio M, Lutolf MP, Semenkovich CF, Jessberger S. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature. 2013;493(7431):226–230. doi: 10.1038/nature11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak A, Duarte JM, Do KQ, Gruetter R. Neurochemical profile of the developing mouse cortex determined by in vivo 1H NMR spectroscopy at 14.1 T and the effect of recurrent anaesthesia. J Neurochem. 2010;115(6):1466–1477. doi: 10.1111/j.1471-4159.2010.07051.x. [DOI] [PubMed] [Google Scholar]
- Liimatainen TJ, Erkkilä AT, Valonen P, Vidgren H, Lakso M, Wong G, Gröhn OH, Ylä-Herttuala S, Hakumäki JM. 1H MR spectroscopic imaging of phospholipase-mediated membrane lipid release in apoptotic rat glioma in vivo. Magn Reson Med. 2008;59(6):1232–1238. doi: 10.1002/mrm.21607. [DOI] [PubMed] [Google Scholar]
- Loewenbrück KF, Fuchs B, Hermann A, Brandt M, Werner A, Kirsch M, Schwarz S, Schwarz J, Schiller J, Starch A. Proton MR spectroscopy of neural stem cells: does the proton-NMR peak at 1.28 ppm function as a biomarker for cell type or state? Rejuvenation Res. 2011;14(4):371–381. doi: 10.1089/rej.2010.1102. [DOI] [PubMed] [Google Scholar]
- Luo J, Vijayasankaran N, Autsen J, Santuray R, Hudson T, Amanullah A, Li F. Comparative metabolite analysis to understand lactate metabolism shift in Chinese hamster ovary cell culture process. Biotechnol Bioeng. 2012;109(1):146–156. doi: 10.1002/bit.23291. [DOI] [PubMed] [Google Scholar]
- Maher AD, Fonville JM, Coen M, Lindon JC, Rae CD, Nicholson JK. Statistical total correlation spectroscopy scaling for enhancement of metabolic information recovery in biological NMR spectra. Anal Chem. 2012;84(2):1083–1091. doi: 10.1021/ac202720f. [DOI] [PubMed] [Google Scholar]
- Manganas LN, Zhang X, Li Y, Hazel RD, Smith SD, Wagshul ME, Henn F, Benveniste H, Djuric PM, Enikolopov G, Maletic-Savatic M. Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain. Science. 2007;318(5852):980–985. doi: 10.1126/science.1147851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissen JK, Yuen BT, Kind T, Riggs JW, Barupal DK, Knoeplfer PS, Fiehn O. Induced pluripotent stem cells show metabolomic differences to embryonic stem cells in polyunsaturated phosphatidylcholines and primary metabolism. PLoS ONE. 2012;7(10):e46770. doi: 10.1371/journal.pone.0046770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milacic M, Haw R, Rothfels K, Wu G, Croft D, Hermjakob H, D'Eustachio P, Stein L. Annotating cancer variants and anticancer therapeutics in reactome. Cancers (Basel) 2012;4(4):1180–1211. doi: 10.3390/cancers4041180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountford CE, Stanwell P, Lin A, Ramadan S, Ross B. Neurospectroscopy: the past, present and future. Chem Rev. 2010;110(5):3060–3086. doi: 10.1021/cr900250y. [DOI] [PubMed] [Google Scholar]
- Mushtaq MY, Choi YH, Verpoorte R, Wilson EG. Extraction for metabolomics: access to the metabolome. Phytochem Anal. 2014;25(4):291–306. doi: 10.1002/pca.2505. [DOI] [PubMed] [Google Scholar]
- Nevedomskaya E, Ramautar R, Derks R, Westbroek I, Zondag G, van der Pluijm I, Deelder AM, Mayboroda OA. CE-MS for metabolic profiling of volume-limited urine samples: application to accelerated aging TTD mice. J Proteome Res. 2010;9(9):4869–4874. doi: 10.1021/pr100634d. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross JM, Darzi AW, Takats Z, Lindon JC. Metabolic phenotyping in clinical and surgical environments. Nature. 2012;491(7424):384–392. doi: 10.1038/nature11708. [DOI] [PubMed] [Google Scholar]
- Nishida K, Ono K, Kanaya S, Takahashi K. KEGGscape: a Cytoscape app for pathway data integration. F1000Res. 2014;3:144. doi: 10.12688/f1000research.4524.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura D. Biotech software & Internet report. Larchmont, NY: Mary Ann Liebert, Inc.; 2000. [Google Scholar]
- Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, Herrerías A, Batchelder EM, Plongthongkum N, Lutz M, Berggren WT, Zhang K, Evans RM, Siuzdak G, Izpisua Belmonte JC. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22(1):168–177. doi: 10.1038/cr.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson C, Vannucci M, Karakas C, Choi W, Ma L, Maletić-Savatić M. Inferring metabolic networks using the Bayesian adaptive graphical lasso with informative priors. Stat Interface. 2013;6(4):547–558. doi: 10.4310/SII.2013.v6.n4.a12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putluri N, Shojaie A, Vasu VT, Vareed SK, Nalluri S, Putluri V, Thangjam GS, Panzitt K, Tallman CT, Butler C, Sana TR, Fischer SM, Sica G, Brat DJ, Shi H, Palapattu GS, Lotan Y, Weizer AZ, Terris MK, Shariat SF, Michailidis G, Sreekumar A. Metabolomic profiling reveals potential markers and bioprocesses altered in bladder cancer progression. Cancer Res. 2011;71(24):7376–7386. doi: 10.1158/0008-5472.CAN-11-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn KP, Sridharan GV, Hayden RS, Kaplan DL, Lee K, Georgakoudi I. Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci Rep. 2013;3:3432. doi: 10.1038/srep03432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramm P, Bettscheider M, Beier D, Kalbitzer HR, Kremer W, Bogdahn U, Hau P, Aigner L, Beier CP. 1H-nuclear magnetic resonance spectroscopy of glioblastoma cancer stem cells. Stem Cells Dev. 2011;20(12):2189–2195. doi: 10.1089/scd.2010.0567. [DOI] [PubMed] [Google Scholar]
- Ramm Sander P, Hau P, Koch S, Schütze K, Bogdahn U, Kalbitzer HR, Aigner L. Stem cell metabolic and spectroscopic profiling. Trends Biotechnol. 2013;31(3):204–213. doi: 10.1016/j.tibtech.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2006;441(7097):1080–1086. doi: 10.1038/nature04958. [DOI] [PubMed] [Google Scholar]
- Robinette SL, Veselkov KA, Bonus E, Coen M, Keun HC, Ebbels TM, Beckonert O, Holmes EC, Lindon JC, Nicholson JK. Cluster analysis statistical spectroscopy using nuclear magnetic resonance generated metabolic data sets from perturbed biological systems. Anal Chem. 2009;81(16):6581–6589. doi: 10.1021/ac901240j. [DOI] [PubMed] [Google Scholar]
- Sana TR, Waddell K, Fischer SM. A sample extraction and chromatographic strategy for increasing LC/MS detection coverage of the erythrocyte metabolome. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;871(2):314–321. doi: 10.1016/j.jchromb.2008.04.030. [DOI] [PubMed] [Google Scholar]
- Sands CJ, Coen M, Ebbels TM, Holmes E, Lindon JC, Nicholson JK. Data-driven approach for metabolite relationship recovery in biological 1H NMR data sets using iterative statistical total correlation spectroscopy. Anal Chem. 2011;83(6):2075–2082. doi: 10.1021/ac102870u. [DOI] [PubMed] [Google Scholar]
- Sepúlveda DE, Andrews BA, Papoutsakis ET, Asenjo JA. Metabolic flux analysis of embryonic stem cells using three distinct differentiation protocols and comparison to gene expression patterns. Biotechnol Prog. 2010;26(5):1222–1229. doi: 10.1002/btpr.448. [DOI] [PubMed] [Google Scholar]
- Ser Z, Liu X, Tang NN, Locasale JW. Extraction parameters for metabolomics from cultured cells. Anal Biochem. 2015;475:22–28. doi: 10.1016/j.ab.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SH, Kraus WE, Newgard CB. Metabolomic profiling for the identification of novel biomarkers and mechanisms related to common cardiovascular diseases: form and function. Circulation. 2012;126(9):1110–1120. doi: 10.1161/CIRCULATIONAHA.111.060368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Maher AD, Cloarec O, Rantalainen M, Tang H, Elliott P, Stamler J, Lindon JC, Holmes E, Nicholson JK. Statistical correlation and projection methods for improved information recovery from diffusion-edited NMR spectra of biological samples. Anal Chem. 2007;79(15):5682–5689. doi: 10.1021/ac0703754. [DOI] [PubMed] [Google Scholar]
- Soares DP, Law M. Magnetic resonance spectroscopy of the brain: review of metabolites and clinical applications. Clin Radiol. 2009;64(1):12–21. doi: 10.1016/j.crad.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Sowell RA, Koeniger SL, Valentine SJ, Moon MH, Clemmer DE. Nanoflow LC/IMS-MS and LC/IMS-CID/MS of protein mixtures. J Am Soc Mass Spectrom. 2004;15(9):1341–1353. doi: 10.1016/j.jasms.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, Nyati MK, Ahsan A, Kalyana-Sundaram S, Han B, Cao X, Byun J, Omenn GS, Ghosh D, Pennathur S, Alexander DC, Berger A, Shuster JR, Wei JT, Varambally S, Beecher C, Chinnaiyan AM. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457(7231):910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Stringari C, Wang H, Geyfman M, Crosignani V, Kumar V, Takahashi JS, Andersen B, Gratton E. In vivo single-cell detection of metabolic oscillations in stem cells. Cell Rep. 2015;10:1–7. doi: 10.1016/j.celrep.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Ohishi M, Ota S, Suzumura K, Naraoka H, Ohata T, Seki J, Miyamae Y, Honma M, Soga T. Metabolic profiling to identify potential serum biomarkers for gastric ulceration induced by nonsteroid anti-inflammatory drugs. J Proteome Res. 2013;12(3):1399–1407. doi: 10.1021/pr3010452. [DOI] [PubMed] [Google Scholar]
- Turner WS, Seagle C, Galanko JA, Favorov O, Prestwich GD, Macdonald JM, Reid LM. Nuclear magnetic resonance metabolomic footprinting of human hepatic stem cells and hepatoblasts cultured in hyaluronan-matrix hydrogels. Stem Cells. 2008;26(6):1547–1555. doi: 10.1634/stemcells.2007-0863. [DOI] [PubMed] [Google Scholar]
- Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Kent Wenger R, Yao H, Markley JL. BioMagResBank. Nucleic Acids Res. 2008;36(Database issue):D402–D408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban M, Enot DP, Dallmann G, Körner L, Forcher V, Enoh P, Koal T, Keller M, Deigner HP. Complexity and pitfalls of mass spectrometry-based targeted metabolomics in brain research. Anal Biochem. 2010;406(2):124–131. doi: 10.1016/j.ab.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Urenjak J, Williams SR, Gadian DG, Noble M. Proton nuclear magnetic resonance spectroscopy unambiguously identifies different neural cell types. J Neurosci. 1993;13(3):981–989. doi: 10.1523/JNEUROSCI.13-03-00981.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacanti NM, Metallo CM. Exploring metabolic pathways that contribute to the stem cell phenotype. Biochim Biophys Acta. 2013;1830(2):2361–2369. doi: 10.1016/j.bbagen.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Vandersypen LM, Steffen M, Breyta G, Yannoni CS, Sherwood MH, Chuang IL. Experimental realization of Shor's quantum factoring algorithm using nuclear magnetic resonance. Nature. 2001;414(6866):883–887. doi: 10.1038/414883a. [DOI] [PubMed] [Google Scholar]
- Vingara LK, Yu HJ, Wagshul ME, Serafin D, Christodoulou C, Pelczer I, Krupp LB, Maletić-Savatić M. Metabolomic approach to human brain spectroscopy identifies associations between clinical features and the frontal lobe metabolome in multiple sclerosis. V Neuroimage. 2013;82:586–594. doi: 10.1016/j.neuroimage.2013.05.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Alexander P, Wu L, Hammer R, Cleaver O, McKnight SL. Dependence of mouse embryonic stem cells on threonine catabolism. Science. 2009;325(5939):435–439. doi: 10.1126/science.1173288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Weckwerth W, Morgenthal K. Metabolomics: from pattern recognition to biological interpretation. Drug Discov Today. 2005;10(22):1551–1558. doi: 10.1016/S1359-6446(05)03609-3. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, Fung C, Nikolai L, Lewis M, Coutouly MA, Forsythe I, Tang P, Shrivastava S, Jeroncic K, Stothard P, Amegbey G, Block D, Hau DD, Wagner J, Miniaci J, Clements M, Gebremedhin M, Guo N, Zhang Y, Duggan GE, Macinnis GD, Weljie AM, Dowlatabadi R, Bamforth F, Clive D, Greiner R, Li L, Marrie T, Sykes BD, Vogel HJ, Querengesser L. HMDB: the Human Metabolome Database. Nucleic Acids Res. 2007;35(Database issue):D521–D526. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Southam AD, Hines A, Viant MR. High-throughput tissue extraction protocol for NMR- and MS-based metabolomics. Anal Biochem. 2008;372(2):204–212. doi: 10.1016/j.ab.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Yanes O, Clark J, Wong DM, Patti GJ, Sánchez-Ruiz A, Benton HP, Trauger SA, Desponts C, Ding S, Siuzdak G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat Chem Biol. 2010;6(6):411–417. doi: 10.1038/nchembio.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Ramachandran PV, Wang MC. Shedding new light on lipid functions with CARS and SRS microscopy. Biochim Biophys Acta. 2014;1841(8):1120–1129. doi: 10.1016/j.bbalip.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni N, Fendt SM, Rühl M, Sauer U. (13)C-based metabolic flux analysis. Nat Protoc. 2009;4(6):878–892. doi: 10.1038/nprot.2009.58. [DOI] [PubMed] [Google Scholar]
- Zenobi R. Single-cell metabolomics: analytical and biological perspectives. Science. 2013;342(6163) doi: 10.1126/science.1243259. 1243259. [DOI] [PubMed] [Google Scholar]
- Zhang X, Li M, Agrawal A, San KY. Efficient free fatty acid production in Escherichia coli using plant acyl-ACP thioesterases. Metab Eng. 2011;13(6):713–722. doi: 10.1016/j.ymben.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Zinnel NF, Pai PJ, Russell DH. Ion mobility-mass spectrometry (IM-MS) for top-down proteomics: increased dynamic range affords increased sequence coverage. Anal Chem. 2012;84:3390–3397. doi: 10.1021/ac300193s. [DOI] [PubMed] [Google Scholar]